

A high-affinity mutant CD8 (haCD8) has been developed with the aim of developing a therapeutic immunosuppressor. In order to fully understand the nature of the haCD8 interaction, this protein was crystallized using the sitting-drop vapour-diffusion method.
Keywords: CD8αα, high affinity, structure-based design, immunosuppressors
Abstract
The class I CD8 positive T-cell response is involved in a number of conditions in which artificial down-regulation and control would be therapeutically beneficial. Such conditions include a number of autoimmune diseases and graft rejection in transplant patients. Although the CD8 T-cell response is dominated by the TCR–pMHC interaction, activation of T cells is in most cases also dependent on a number of associated signalling molecules. Previous work has demonstrated the ability of one such molecule (CD8) to act as an antagonist to T-cell activation if added in soluble form. Therefore, a high-affinity mutant CD8 (haCD8) has been developed with the aim of developing a therapeutic immunosuppressor. In order to fully understand the nature of the haCD8 interaction, this protein was crystallized using the sitting-drop vapour-diffusion method. Single haCD8 crystals were cryocooled and used for data collection. These crystals belonged to space group P6422 (assumed by similarity to the wild type), with unit-cell parameters a = 101.08, c = 56.54 Å. V M calculations indicated one molecule per asymmetric unit. A 2 Å data set was collected and the structure is currently being determined using molecular replacement.
1. Introduction
CD8 T-cell responses occur through the interaction between the T-cell receptor (TCR), the major histocompatibility complexes bound to peptide (pMHC) and various signalling molecules such as the CD8 co-receptor and the CD3 complex (Delon et al., 1998 ▶; Dembic et al., 1987 ▶; Sgro, 1995 ▶). This interaction is responsible for controlling antigenic specificity through the recognition of a pMHC complex by a specific TCR (Davis & Bjorkman, 1988 ▶) and a CD8/CD3-mediated signalling cascade leading to T-cell activation and subsequent cytolytic events (reviewed in Janeway, 1992 ▶). Although this pathway is responsible for host defences against viral infections etc., there are also reports of CD8 T-cell mediated auto-reactivity, for example type 1 diabetes (Amrani et al., 2000 ▶; Anderson et al., 1999 ▶), rheumatoid arthritis (Gringhuis et al., 2000 ▶; Prinz et al., 2002 ▶), multiple sclerosis (Babbe et al., 2000 ▶), intestinal autoimmunity (Prinz et al., 2002 ▶), autoimmune neuronal degeneration (Albert et al., 1998 ▶) and autoimmune thyroiditis (Gringhuis et al., 2000 ▶; Kong et al., 1989 ▶; Vladutiu & Rose, 1971 ▶). CD8 T-cell responses involved in conditions such as chronic heart rejection in transplant patients have also been reported (Fischbein et al., 2002 ▶).
The CD8 co-receptor has been shown to be critical for the activation of most CD8 T cells (Meuer et al., 1982 ▶; Swain, 1981 ▶). It binds to pMHC as a homodimer, referred to here as CD8αα. Structural studies of CD8αα–pMHC have alluded to the importance of the interaction between the CD8α, the α3 and, to a lesser extent, the α2 and β2m domains of pMHC (Gao et al., 1997 ▶). Of particular importance is the interaction between residues 223–228 of the pMHC α3 domain and residues 51–55 of the CDR-like loop of the CD8α chain. Mutation of these sites has been shown to disrupt CD8–pMHC interactions (Giblin et al., 1994 ▶). Furthermore, soluble CD8 has been shown to act as a strong antagonist to CD8 T-cell interactions, with approximately five- to tenfold higher efficacy than anti-CD8 antibodies (Sewell et al., 1999 ▶). Therefore, we have used our knowledge of the wild-type CD8–pMHC crystal structure to design a high-affinity CD8αα (haCD8) molecule with threefold greater affinity than the wild-type CD8 (Cole et al., unpublished work). We achieved this by mutating residue Ser53 on the CD8α chain to Asn. This mutagenesis was carried out to improve the electrostatic interaction between residue Asp227 on the α3 domain of pMHC and residue Asn53 on the CDR-like loop of haCD8 owing to the bulkier and more electron-dense nature of the Asn side chain (Dawson et al., 1986 ▶). In an attempt to improve refolding and to reduce the likelihood of non-specific intermolecular disulfide bonds forming between CD8α chains, the unpaired Cys33 was mutated to Ala.
A pGMT7 Escherichia coli expression system (Rosenburg et al., 1987 ▶) was used to make inclusion bodies (IBs) which were refolded to produce soluble haCD8, which was in turn used to prepare crystals. Crystallographic structural analysis would be the first step in better understanding the interaction in the haCD8–pMHC complex and should aid the design of even higher affinity CD8 mutants suitable for therapeutic applications.
2. Materials and methods
2.1. Protein expression
The plasmid stock containing the sequence for amino acids 1–120 of haCD8 (Gao et al., 1998 ▶) was transformed into E. coli Rosetta DE3 competent cells and used to inoculate 1 l TYP media. 0.5 mM IPTG was used to induce expression and the resulting inclusion bodies were harvested by lysis (10 mM Tris, 10 mM MgCl2, 150 mM NaCl, 10% glycerol), sonication and treatment with Triton wash buffer (0.5% Triton X-100, 50 mM Tris, 100 mM NaCl, 10 mM EDTA). Washed inclusion bodies were then dissolved in guanidine buffer (6 M guanidine, 50 mM Tris, 2 mM EDTA, 100 mM NaCl) and purity was measured using 10% Bis-Tris SDS–PAGE. Protein concentration was estimated using a Bradford reagent kit (Sigma–Aldrich) with BSA as a standard.
2.2. Protein refolding and purification
This was carried out as previously described (Gao et al., 1998 ▶). Briefly, 60 mg l−1 CD8α chain was incubated at 310 K for 15 min with 10 mM DTT and then added to cold refolding buffer (100 mM Tris base, 76 mM Tris–HCl, 1 mM EDTA, 0.6 M l-arginine, 6 mM β-mercaptoethylamine and 4 mM cysteine). Dialysis was carried out in 10 mM MES pH 8 buffer for 2 d (changing the dialysis buffer once). The mixture was then filtered and diluted by half with 10 mM MES pH6. The pH was adjusted to 6 using saturated MES solution. The haCD8 protein was purified by cation exchange using a Poros 50HS column followed by gel filtration using a Superdex200 HR.
2.3. Crystallization
The haCD8 was concentrated to 20 mg ml−1 in 10 mM MES, 10 mM NaCl buffer. Crystal screens were initiated using Crystal Screens 1, 2 and Cryo 1 (Hampton Research) buffers 1–48 with 0.5 µl drops of protein and 0.5 µl drops of crystallization buffer using the hanging-drop method. Plates were incubated at 291 K and analysed after 24 h, 48 h and one week. Needle-shaped crystals were observed in Crystal Screen Cryo buffer 15 (25.5% PEG 8000, 0.085 M sodium cacodylate pH 6.5, 0.17 M ammonium sulfate, 15% glycerol) after 24 h. Prismatic long needle-shaped crystals (typical dimensions 50 × 50 × 2000 µm; Fig. 1 ▶) were obtained from the sitting-drop method after extensive optimization using 1 µl 20 mg ml−1 haCD8 and 1 µl buffer [25.5% PEG 8000, 0.085 M sodium cacodylate pH 6.5, 0.17 M ammonium sulfate, 15% glycerol, 3%(v/v) ethylene glycol]. Crystals were flash-cooled in liquid nitrogen in cryoloops.
Figure 1.

Typical appearance of crystals in the hanging drops. Individual needles were separated for cryocooling.
2.4. Data collection and processing
Data were collected with the rotation method at station 14.2 of the Synchrotron Radiation Source (SRS), Daresbury, UK using an ADSC Quantum 4 CCD-detector system. The wavelength (λ) was set to 0.978 Å. The total number of frames recorded was 100, each covering 1° of rotation. The crystal was maintained at 100 K in an Oxford Cryostream. Reflection intensities were estimated with the MOSFLM package (Leslie, 1992 ▶) and the data were scaled, reduced and analysed with the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶). Crystal data and relevant statistics are given in Table 1 ▶.
Table 1. Diffraction data statistics.
Values in parentheses refer to the highest resolution shell.
| Space group | P6422 |
| Unit-cell parameters (Å) | a = 101.08, c = 56.54 |
| Wavelength (Å) | 0.978 |
| Resolution (Å) | 29–2.0 (2.11–2.0) |
| Measured reflections | 110349 (12964) |
| Unique reflections | 11946 (1693) |
| Completeness (%) | 99.9 (99.6) |
| Multiplicity | 9.2 (7.7) |
| I/σ(I) | 9.9 (0.8) |
| Rmerge† (%) | 20.9 (89.3) |
R
merge = 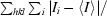
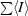 , where I
i is the intensity of a particular reflection in the set of symmetry equivalents for any hkl index and 〈I〉 is the average intensity of all the equivalents of that index.
, where I
i is the intensity of a particular reflection in the set of symmetry equivalents for any hkl index and 〈I〉 is the average intensity of all the equivalents of that index.
3. Results and discussion
The haCD8 crystals were analysed by X-ray diffraction and found to belong to space group P6422, with unit-cell parameters a = b = 101.08, c = 56.54 Å (Table 1 ▶). The enantiomeric space group P6222 was discounted at this stage by virtue of the wild-type crystals showing the former symmetry. One molecule (MW = 12 kDa) was present per asymmetric unit, resulting in a V M value of 3.5 Å3 Da−1 and a solvent content of 64%. A full data set was collected to 2 Å resolution (Fig. 2 ▶). A total of 110 349 observations were measured including 11 946 unique reflections. The completeness was 99.9%, with a multiplicity of 9.2 and an R sym of 20.9%.
Figure 2.

A typical diffraction pattern, showing diffraction beyond the 2.1 Å ring. The image on the right is an expansion, with change in contrast, of the top part of the pattern on the left.
The high-resolution cutoff was extended to 2.0 Å despite the apparently poor statistics in the outermost shell. This was to afford the opportunity of using as much information as possible for discerning fine features of the structure during refinement. The multiplicity is high and although 〈I/σ(I)〉 had fallen below 1.0, there are likely to be many individual reflections that are reliable and should be included in the refinement. This has been debated at length on specialist bulletin boards (e.g. ccp4bb@dl.ac.uk), where the consensus was that it is beneficial to retain these reflections in refinement based on maximum-likelihood methods, e.g. as encoded in REFMAC5 from the CCP4 package of crystallographic software.
Structure determination and refinement is currently under way and will be compared with the wild-type CD8αα homodimer (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1cd8).
Acknowledgments
We wish to thank the staff at the SRS for providing technical support, the UK Research Councils for providing the beamtime and Avidex Ltd for providing resources, research and technical support for the duration of this project. DKC is supported by an MRC studentship.
References
- Albert, M. L., Darnell, J. C., Bender, A., Francisco, L. M., Bhardwaj, N. & Darnell, R. B. (1998). Nature Med.4, 1321–1324. [DOI] [PubMed] [Google Scholar]
- Amrani, A., Verdaguer, J., Serra, P., Tafuro, S., Tan, R. & Santamaria, P. (2000). Nature (London), 406, 739–742. [DOI] [PubMed] [Google Scholar]
- Anderson, B., Park, B. J., Verdaguer, J., Amrani, A. & Santamaria, P. (1999). Proc. Natl Acad. Sci. USA, 96, 9311–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbe, H., Roers, A., Waisman, A., Lassmann, H., Goebels, N., Hohlfeld, R., Friese, M., Schroder, R., Deckert, M., Schmidt, S., Ravid, R. & Rajewsky, K. (2000). J. Exp. Med.192, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Davis, M. M. & Bjorkman, P. J. (1988). Nature (London), 334, 395–402. [DOI] [PubMed] [Google Scholar]
- Dawson, R. M. C., Elliott, D. C., Elliott, W. H. & Jones, K. M. (1986). Editors. Data for Biochemical Research, 3rd ed., pp. 1–31. Oxford: Clarendon Press.
- Delon, J., Gregoire, C., Malissen, B., Darche, S., Lemaitre, F., Kourilsky, P., Abastado, J. P. & Trautmann, A. (1998). Immunity, 9, 467–473. [DOI] [PubMed] [Google Scholar]
- Dembic, Z., Haas, W., Zamoyska, R., Parnes, J., Steinmetz, M. & von Boehmer, H. (1987). Nature (London), 326, 510–511. [DOI] [PubMed] [Google Scholar]
- Fischbein, M. P., Yun, J., Laks, H., Irie, Y., Fishbein, M. C., Bonavida, B. & Ardehali, A. (2002). J. Thorac. Cardiovasc. Surg.123, 803–809. [DOI] [PubMed] [Google Scholar]
- Gao, G. F., Gerth, U. C., Wyer, J. R., Willcox, B. E., O’Callaghan, C. A., Zhang, Z., Jones, E. Y., Bell, J. I. & Jakobsen, B. K. (1998). Protein Sci.7, 1245–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, G. F., Tormo, J., Gerth, U. C., Wyer, J. R., McMichael, A. J., Stuart, D. I., Bell, J. I., Jones, E. Y. & Jakobsen, B. K. (1997). Nature (London), 387, 630–634. [DOI] [PubMed] [Google Scholar]
- Giblin, P. A., Leahy, D. J., Mennone, J. & Kavathas, P. B. (1994). Proc. Natl Acad. Sci. USA, 91, 1716–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gringhuis, S. I., Leow, A., Papendrecht-Van Der Voort, E. A., Remans, P. H., Breedveld, F. C. & Verweij, C. L. (2000). J. Immunol.164, 2170–2179. [DOI] [PubMed] [Google Scholar]
- Janeway, C. A. Jr (1992). Annu. Rev. Immunol.10, 645–674. [DOI] [PubMed] [Google Scholar]
- Kong, Y. M., Waldmann, H., Cobbold, S., Giraldo, A. A., Fuller, B. E. & Simon, L. L. (1989). Clin. Exp. Immunol.77, 428–433. [PMC free article] [PubMed] [Google Scholar]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Meuer, S. C., Hussey, R. E., Hodgdon, J. C., Hercend, T., Schlossman, S. F. & Reinherz, E. L. (1982). Science, 218, 471–473. [DOI] [PubMed] [Google Scholar]
- Prinz, I., Zerrahn, J., Kaufmann, S. H. & Steinhoff, U. (2002). J. Autoimmun.18, 281–287. [DOI] [PubMed] [Google Scholar]
- Rosenburg, A. H., Lade, B. N., Chui, D. S., Lin, S. W., Dunn, J. J. & Studier, F. W. (1987). Gene, 56, 125–135. [DOI] [PubMed] [Google Scholar]
- Sewell, A. K., Gerth, U. C., Price, D. A., Purbhoo, M. A., Boulter, J. M., Gao, G. F., Bell, J. I., Phillips, R. E. & Jakobsen, B. K. (1999). Nature Med.5, 399–404. [DOI] [PubMed] [Google Scholar]
- Sgro, C. (1995). Toxicology, 105, 23–29. [DOI] [PubMed] [Google Scholar]
- Swain, S. L. (1981). Proc. Natl Acad. Sci. USA, 78, 7101–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladutiu, A. O. & Rose, N. R. (1971). Science, 174, 1137–1139. [DOI] [PubMed] [Google Scholar]