

The structure-based fragment-assembly approach described in the present study facilitated the rapid identification and subsequent synthesis of caspase-1 inhibitors with cell-based activity and provided insights into the enzyme’s ability to accommodate different inhibitor-binding modes in the active site.
Keywords: caspase-1, Tethering, structure-based drug design
Abstract
Caspase-1 is a key endopeptidase responsible for the post-translational processing of the IL-1β and IL-18 cytokines and small-molecule inhibitors that modulate the activity of this enzyme are predicted to be important therapeutic treatments for many inflammatory diseases. A fragment-assembly approach, accompanied by structural analysis, was employed to generate caspase-1 inhibitors. With the aid of Tethering® with extenders (small molecules that bind to the active-site cysteine and contain a free thiol), two novel fragments that bound to the active site and made a disulfide bond with the extender were identified by mass spectrometry. Direct linking of each fragment to the extender generated submicromolar reversible inhibitors that significantly reduced secretion of IL-1β but not IL-6 from human peripheral blood mononuclear cells. Thus, Tethering with extenders facilitated rapid identification and synthesis of caspase-1 inhibitors with cell-based activity and subsequent structural analyses provided insights into the enzyme’s ability to accommodate different inhibitor-binding modes in the active site.
1. Introduction
Caspases form a family of aspartate-specific cysteine endopeptidases. To date, 14 mammalian caspases have been identified, of which 11 have human homologs (Riedl & Shi, 2004 ▶). Many of the caspase-family members are activated during programmed cell death and their proteolytic activity is a central biochemical feature of the apoptotic process (Solary et al., 1998 ▶). Extensive studies of caspases in inflammation and apoptosis validated the enzymes as attractive drug targets, the inhibition of which could alleviate a variety of human ailments (O’Brien & Lee, 2004 ▶).
Caspase-1 (also known as interleukin 1β-converting enzyme or ICE) was originally identified as a monocyte-specific endopeptidase responsible for the post-translational processing of the proinflammatory cytokine interleukin 1β (IL-1β; Cerretti et al., 1992 ▶; Howard et al., 1991 ▶; Thornberry et al., 1992 ▶), a major mediator of inflammatory diseases such as rheumatoid arthritis. Soon thereafter, caspase-1 was shown to process interleukin-18 (IL-18), a cytokine structurally similar to IL-1 that is involved in T-cell activation (Dinarello, 1998 ▶).
Caspase-1 is expressed as a procaspase-1 zymogen that is processed into a catalytically competent form through autoproteolysis induced by protein oligomerization in vitro (Yang et al., 1998 ▶), but may require caspase-5 for efficient activation in vivo (Kang et al., 2000 ▶; Martinon et al., 2002 ▶; Wang et al., 1998 ▶). The cleavage reaction removes the 119-residue propeptide and an 18-residue sequence that separates the large (p20; residues 120–298) and small (p10; residues 317–404) subunits of the mature enzyme. The functional form of caspase-1 is thought to be a tetramer composed of two p20–p10 heterodimers (Gu et al., 1995 ▶; Wilson et al., 1994 ▶).
Given the importance of caspases in inflammatory and apoptotic processes, there has been an intense effort to characterize the family members structurally. Most of the structures presented to date show proteins bound to cofactors or inhibitors (reviewed in Romanowski et al., 2004 ▶), with the majority of inhibitors covalently interacting with the active-site cysteine residue. Idun Pharmaceuticals is currently conducting phase II clinical trials with IDN-6556, an irreversible caspase inhibitor. Recently, the structure of caspase-1 in complex with a non-covalent malonate ion was reported (Romanowski et al., 2004 ▶). The structure revealed the protein in its open (active-site ligand-bound) conformation in which malonate reproduces the hydrogen-bonding network observed in structures with covalent inhibitors. In comparison, the structure of a ligand-free caspase-1 demonstrated a significantly different active-site conformation.
We applied a structure-based fragment-assembly process utilizing Tethering as a discovery tool for rapid identification of small-molecule caspase-1 inhibitors (Erlanson et al., 2004 ▶; Erlanson & Hansen, 2004 ▶). Tethering identifies small-molecular-weight fragments (with potencies typically >100 µM) that bind in non-overlapping sites on the protein. The potency of the fragments can then be improved by either direct optimization or by linking to other identified fragments to generate more complex molecules that combine the recognition features of both fragments. Here, we apply a variation of this approach to circumvent the difficulty of linking two fragments with unknown binding modes by directly connecting an identified fragment with a known fragment (Erlanson et al., 2003 ▶). In this case, one of the fragments (an extender) is a known small molecule that (i) covalently modifies a surface thiol on the target protein, (ii) incorporates a protected thiol group and (iii) incorporates recognition elements for the target (see Fig. 1 ▶ a). The choice of an extender that integrates features of the cognate substrate, specifically in its P1 position, is a critically important step for successful Tethering in the active site of caspase-1; we have recently shown that caspase-1 exists predominantly in a closed-active-site form in the absence of a substrate or an inhibitor and that aspartic acid or its mimic is necessary and sufficient to open up the active site and poise the enzyme for catalysis (Romanowski et al., 2004 ▶). Once the extender has been irreversibly coupled to the protein and the thiol group deprotected, the protein–extender complex is screened against a library of disulfide-containing fragments. Subsequently, hits can be combined with binding elements from the extender to generate reversible inhibitors. The advantage of this approach is that companion fragments can be selected and linked to the first fragment (the extender) in a single step. Here, we use this approach to identify nanomolar caspase-1 inhibitors that display cell-based activity and, as revealed by X-ray crystallography, exhibit different binding modes depending upon the composition of the linker. Additionally, we demonstrate that secretion of IL-1β but not IL-6 from human peripheral blood mononuclear cells has been inhibited by the newly generated compounds.
Figure 1.

Tethering with extenders as applied to caspase-1. (a) Overview of the use of Tethering with an extender for caspase-1. (b) Tethering identified fragments 1 and 2 as specifically modifying the caspase-1–extender complex. (c) Crystal structure of caspase-1 modified with the extender (compound 1) solved at 2.3 Å resolution. Hydrogen bonds between the extender and the protein are displayed as dashed yellow lines. with their associated distances shown in black. Selected residues comprising the active site are also shown. (d) A 2F o − F c electron-density map within a 4 Å distance of the extender, contoured at the 1σ level, documenting a covalent bond between the molecule and the active-site Cys285.
2. Materials and methods
2.1. Cloning and expression of caspase-1
DNA sequences of the large (p20; Asn120–Asp297; MW = 19 843.8 Da) and small (p10; Ala317–His404; MW = 10 243.7 Da) subunit of caspase-1 (Genbank accession No. NM_033292) were produced by RT-PCR (Stratagene ProSTAR HF) from RNA purified from THP-1 cells (ATCC TIB-202). The large and small subunits were separately subcloned into the EcoRI–NdeI site of pRSET B (Invitrogen) and were transformed into BL21 Codon Plus cells. After induction with 1 mM IPTG for 4 h at 310 K, cells were resuspended in a buffer composed of 10 mM Tris pH 8.0, 1 mM EDTA (TE buffer) and 2 mM DTT and microfluidized twice. Inclusion bodies were isolated as described previously (Romanowski et al., 2004 ▶).
2.2. Caspase refolding, modification and screening
Refolding of caspase-1 essentially followed published protocols (Ramage et al., 1995 ▶). The extender, compound 1 (20 µM), was added to the renaturation buffer and stirred at room temperature overnight. Samples were dialyzed twice overnight against a buffer composed of 20 mM HEPES pH 7.0, 100 mM NaCl, 5% glycerol and 2 mM DTT. Covalent addition of compound 1 to the large subunit was verified by mass spectrometry (M r = 20 178). Compound 1 was deacetylated during refolding and modification.
The protein was exchanged into TE buffer with Nap-5 columns (Amersham Pharmacia Biotech). General conditions for Tethering were 2 µM extender-modified caspase-1, 300 µM 2-mercaptoethanol and 300 µM disulfide-library pool (typically containing a mixture of ten compounds) in a total volume of 35 µl. Reactions were allowed to reach equilibrium for a minimum of 1 h at room temperature before analysis.
2.3. Compound synthesis
Detailed protocols for compound synthesis will be published elsewhere (Fahr et al., in preparation).
2.4. Caspase activity assays
The activities of human caspase-1 and caspase-5 were measured using a fluorometric assay (Choong et al., 2002 ▶). All 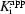 values were calculated using the Morrison tight-binding equation (Kuzmic et al., 2000 ▶) in GraphPad Prism.
values were calculated using the Morrison tight-binding equation (Kuzmic et al., 2000 ▶) in GraphPad Prism.
2.5. Cell-based assays
Peripheral blood mononuclear cells (PBMCs; Stanford University Blood Bank) were separated from buffy coats of healthy volunteer donors by density-gradient centrifugation using Ficoll-Plaque Plus (Pharmacia Biotech 17–1440-02) and stored in 90% FBS, 10% DMSO in liquid nitrogen. Aliquots were thawed, washed and resuspended in RPMI 1640 containing 10% FBS, 10 mM HEPES, 2 mM l-glutamine, 50 mg ml−1 penicillin G sodium, 50 U ml−1 streptomycin sulfate and 0.125 mg ml−1 amphotericin B. Cells were plated on a 96-well plate at a density of ∼1.2 × 106 cells ml−1 and incubated for 3–4 h at 310 K/5% CO2. Nonadherent cells were discarded and adherent cells were treated with the inhibitor compounds (final DMSO concentration of 0.2%) in duplicate for 18 h in the presence of 0.5 ng ml−1 lipopolysccharide (Sigma L4391). IL-1β and IL-6 levels in the supernatant were quantified by ELISA using Maxisorp plates with a primary monoclonal anti-human antibody [R&D Systems; MAB601(IL-1β), MAB206(IL-6)] and a biotinylated anti-human detection antibody [R&D Systems; BAF201(IL-1β), BAF206(IL-6)].
2.6. Crystallization, data collection and structure determination
Crystals of caspase-1 in complex with inhibitors were obtained by hanging-drop vapour diffusion at 277 K against a reservoir of 0.1 M HEPES pH 7.0, 2 M (NH4)2SO4 and 25 mM DTT (compound 1), 0.1 M PIPES pH 6.0, 200 mM Li2SO4, 25% PEG 2000 MME, 10 mM DTT, 3 mM NaN3 and 2 mM MgCl2 (compounds 4 and 6) or 0.1 M PIPES pH 6.0, 175 mM (NH4)2SO4, 25% PEG 2000 MME, 10 mM DTT, 3 mM NaN3 and 2 mM MgCl2 (compounds 5 and 8). All crystals for data collection were cryoprotected in mother liquor supplemented with 22%(v/v) glycerol for 1–2 min and immersion in liquid nitrogen.
Diffraction data were collected under standard cryogenic conditions on beamline 7.1 at the Stanford Synchrotron Research Laboratory using a mar345 image-plate detector (compounds 1, 5 and 6) or a Rigaku RU-3R rotating-anode generator and an R-AXIS IV detector (compounds 4 and 8) and were processed and scaled with CrystalClear from Rigaku/Molecular Structure Corporation (Pflugrath, 1999 ▶). The structures were determined from single-wavelength native diffraction experiments by molecular replacement with AMoRe (Navaza, 2001 ▶) using a search model from a previously determined structure (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1ice). The refinement of the initial solutions with REFMAC (Murshudov et al., 1997 ▶, 1999 ▶; Pannu et al., 1998 ▶) yielded experimental electron-density maps suitable for model building with O (Jones et al., 1991 ▶). Residues 120–124 were not visible in the electron-density maps for any of the protein–inhibitor complexes and were omitted from refinement of the final atomic models. PROCHECK (Laskowski et al., 1993 ▶) revealed no disallowed (ϕ, ψ) combinations and excellent stereochemistry (see Table 1 ▶ for a summary of X-ray data and refinement statistics). All proteins and small-molecule inhibitors in the figures were rendered with PyMOL (DeLano, 2002 ▶).
Table 1. Crystallographic data and refinement statistics.
Values in parentheses are for high-resolution shells: compound 1, 2.38–2.3 Å; compound 4, 2.8–2.7 Å; compound 5, 2.05–2.00 Å; compound 6, 2.17–2.1 Å; compound 8, 2.28–2.2 Å.
| Compound | 1 | 4 | 5 | 6 | 8 |
|---|---|---|---|---|---|
| PDB code | http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1rwk | http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1rwm | http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1rwn | http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1rwo | http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1rwp |
| Space group | P43212 | P43212 | P43212 | P43212 | P43212 |
| Unit-cell parameters (Å) | a = b = 62.8, c = 160.9 | a = b = 63.2, c = 161.2 | a = b = 63.2, c = 161.8 | a = b = 63.2, c = 160.9 | a = b = 63.2, c = 161.7 |
| X-ray source | SSRL beamline 7.1 | R-AXIS IV | SSRL beamline 7.1 | SSRL beamline 7.1 | R-AXIS IV |
| Wavelength (Å) | 0.98 | 1.54 | 0.98 | 1.08 | 1.54 |
| Resolution (Å) | 20–2.3 | 20–2.7 | 20–2.0 | 20–2.1 | 20–2.2 |
| No. of observations | 42420 | 43512 | 74960 | 65315 | 73589 |
| No. of reflections | 14913 | 9552 | 22859 | 19365 | 17387 |
| Completeness (%) | 99.2 (100) | 97.2 (98.6) | 99.5 (99.7) | 97.8 (89.2) | 99.1 (99.9) |
| Mean I/σ(I) | 4.5 (2.2) | 5.4 (1.9) | 4.4 (1.8) | 8.7 (3.6) | 4.9 (1.8) |
| Rmerge on I† | 0.072 (0.313) | 0.128 (0.382) | 0.079 (0.323) | 0.074 (0.204) | 0.123 (0.372) |
| Cutoff criteria | I < −3σ(I) | I < −3σ(I) | I < −3σ(I) | I < −3σ(I) | I < −3σ(I) |
| Model and refinement statistics | |||||
| Resolution range (Å) | 20–2.3 | 20–2.7 | 20–2.0 | 20–2.1 | 20–2.2 |
| No. of reflections | 14126 | 876 | 21643 | 18317 | 16065 |
| No. of reflections in test set | 754 | 437 | 1167 | 988 | 856 |
| Completeness (%) | 98.96 | 96.34 | 99.36 | 97.46 | 97.32 |
| Cutoff criterion | |F| > 0.0 | |F| > 0.0 | |F| > 0.0 | |F| > 0.0 | |F| > 0.0 |
| No. of residues | 261 | 261 | 261 | 261 | 261 |
| No. of water molecules | 161 | 69 | 280 | 228 | 167 |
| R.m.s.d. bond lengths (Å) | 0.006 | 0.006 | 0.006 | 0.006 | 0.006 |
| R.m.s.d. bond angles (°) | 0.895 | 1.109 | 0.896 | 0.899 | 0.902 |
| Luzzati error (Å) | 0.277 | 0.323 | 0.244 | 0.246 | 0.289 |
| Correlation factor‡ | 0.913 | 0.886 | 0.922 | 0.920 | 0.911 |
| Rcryst§ (%) | 20.40 | 19.93 | 20.10 | 19.75 | 21.18 |
| Rfree (%) | 25.82 | 25.44 | 23.57 | 24.18 | 24.49 |
| Ramachandran plot statistics¶ | |||||
| Most favored | 207 [89.6%] | 206 [89.2%] | 206 [89.2%] | 205 [88.7%] | 207 [89.6%] |
| Additional allowed | 22 [9.5%] | 23 [10.0%] | 23 [10.0%] | 24 [(10.4%] | 21 [9.1%] |
| Generously allowed | 2 [0.9%] | 2 [0.9%] | 2 [0.9%] | 2 [0.9%] | 3 [1.3%] |
| Disallowed | 0 [0%] | 0 [0%] | 0 [0%] | 0 [0%] | 0 [0%] |
| Overall G factor†† | 0.1 | 0.0 | 0.1 | 0.2 | 0.1 |
R
merge = 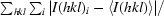
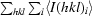 .
.
Correlation factor between the structure factors and the model as calculated by SFCHECK (Vaguine et al., 1999 ▶).
R
cryst = 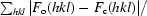
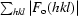 , where F
o and F
c are the observed and calculated structure factors, respectively.
, where F
o and F
c are the observed and calculated structure factors, respectively.
Computed with PROCHECK (Laskowski et al., 1993 ▶).
The overall G factor is a measure of the overall normality of the structure and is obtained from an average of all the different G factors for each residue in the structure. The factor is computed for torsion angles as well as main-chain bond lengths and angles using the Engh and Huber small-molecule means and standard deviations (Engh & Huber, 1991 ▶). It is essentially a log-odds score based on the observed distributions of these stereochemical parameters (Laskowski et al., 1993 ▶).
3. Results and discussion
3.1. Identification of small fragments that bind to the active site of caspase-1
In this study, our extender (compound 1) is a small aspartic acid-containing molecule that binds irreversibly to the active-site cysteine Cys285 of caspase-1 and is the same extender as that previously used to identify small-molecule inhibitors of caspase-3 (Erlanson et al., 2003 ▶; Choong et al., 2002 ▶). To verify that the extender labelled exclusively the active-site cysteine of caspase-1 and not one of the other four cysteine residues contained within the large subunit, we obtained a cocrystal structure of caspase-1 bound with the extender (Figs. 1 ▶ c and 1 ▶ d). As expected, the extender is covalently attached to the active-site cysteine residue (Fig. 1 ▶ d) and the aspartic acid residue of the extender, buried in the oxyanion hole, hydrogen bonds to Arg179 and Arg341 (Rano et al., 1997 ▶). This observation further confirms our earlier discovery that the obligatory aspartic acid element in caspase substrates, similar to its mimic a malonate ion, may be responsible for the dramatic conformational changes accompanying the transition from the ligand-free to ligand-bound form of the enzyme (Romanowski et al., 2004 ▶). We also observed that the free thiol of the extender is pointed toward the S3–S4 pockets and is likely to capture small fragments that interact with this region of the active site.
The caspase-1–extender complex was screened against our in-house library of approximately 16 000 disulfide-containing fragments. This library was designed so that the average molecular mass of each fragment was less than 250 Da and appropriate attention was paid to the drug-like properties of the fragments and the chemical diversity of the pools. More than ten different disulfide-containing fragments were identified in this screen and two of these fragments are shown in Fig. 1 ▶(b), disulfide-linked to the extender. Fragment 1 is a tricyclic quinoxaline, while fragment 2 is a hydroxyquinoline. Fragment 1 was found to have a β-Me50 of 2.9 mM [β-Me50 is the concentration of β-mercaptoethanol (β-Me) that produces 50% modification of the caspase-1–extender complex with each fragment; Hyde et al., 2003 ▶], whereas fragment 2 had a β-Me50 of 0.9 mM, suggesting that fragment 1 binds with greater affinity than fragment 2.
3.2. Conversion of newly identified fragments into reversible inhibitors
We first determined whether compound 2 (fragment 1 lacking the free thiol and capped as an N-methyl amide) inhibited caspase-1 activity in an in vitro assay. This compound was found to have no discernible inhibitory activity even at concentrations as high as 100 µM (compound 2; Fig. 2 ▶
a) and, as previously published, a reversible aspartyl aldehyde that binds in the S1/S2 region (z-D-CHO) inhibited caspase-1 activity with a of approximately 110 µM (Graybill et al., 1994 ▶). Thus, given its weak affinity for the enzyme, compound 2 would not have been discovered by a traditional functional screen.
Figure 2.

Conversion of fragments into reversible caspase-1 inhibitors. The structure of each compound is shown along with the for both caspase-1 and caspase-5. The number of times each compound was tested is indicated (n) and both the average and the associated standard deviation values are shown. (a) Conversion of fragment 1 and compound 1 (the extender) into compounds 2–6. (b) Conversion of fragment 2 and compound 1 (the extender) into compounds 7–8.
It is straightforward to convert these newly identified fragments into reversible inhibitors. The disulfide linking the extender with the selected fragment can be replaced with a simple methylene spacer and the irreversible acyloxymethyl ketone of the extender can be replaced with a reversible aldehyde to produce a reversible inhibitor (Fig. 1 ▶
a). Our initial linked compound (compound 3) was a surprisingly potent inhibitor of caspase-1 in vitro, with a of 0.15 µM, and displayed specificity against caspase-5 with a of 8.5 µM (57-fold selectivity; Fig. 2 ▶
a). Thus, by linking two weak binding fragments, we generated a compound with a significantly enhanced activity. Compound 4, which contains a rigid thiophene linker, had reduced activity ( = 0.34 µM) and less selectivity against caspase-5 (15-fold). However, neither of these compounds (3 and 4) contains a functional element that can bind in the hydrophobic S2 pocket. As previously demonstrated for caspase-3, the addition of a functional group that binds in the S2 pocket dramatically increases the affinity of an inhibitor for the enzyme (Erlanson et al., 2003 ▶; Choong et al., 2002 ▶; Allen et al., 2003 ▶). Taking into account the known substrate specificity of caspase-1 and previous analysis of caspase-3 compounds, we reasoned that simple hydrophobic groups in this position would likewise increase affinity. We synthesized two variants substituted with either ethyl or 2-thienyl that extend into the S2 pocket. These compounds (5 and 6) were approximately ninefold and 21-fold more potent, respectively, than compound 3. Therefore, simple modifications to our initial fragment resulted in highly potent and selective caspase-1 inhibitors as measured in in vitro assays.
Using a similar approach, we converted the hydroxyquinoline fragment (fragment 2) into a reversible inhibitor (Fig. 2 ▶ b). Compound 7 is a low-micromolar inhibitor of caspase-1 and is approximately 18-fold selective against caspase-5. The activity of this series was less than the quinoxalines (Figs. 2 ▶ a and 2 ▶ b; compounds 3 and 7), which correlates with the predicted relative affinities based upon the β-Me50 values of our two selected fragments. Addition of a 2-thienyl group in the linker that binds in the S2 pocket of the enzyme increased the activity by roughly 13-fold (compound 8) and the selectivity against caspase-5 from 18- to 43-fold.
3.3. Cocrystal structures reveal different binding modes
We obtained cocrystal structures of compounds 4, 5 and 8 containing an aspartic acid-aldehyde moiety in P1, as well as the irreversible 2,3,5,6-tetrafluorophenoxymethyl ketone-containing analog of compound 6 bound to caspase-1. The irreversible ketone group was used to facilitate crystallization. We do not believe that the extra carbon in the P1 element of the irreversible compound affects the way in which the rest of the inhibitor binds to the active site. We have previously determined structures of the aspartic acid-aldehyde and ketone analogs of one inhibitor and discovered that there were no significant differences in the mode of binding of the reversible and irreversible forms of the molecule to the enzyme (results not shown). The main tricyclic portion of compound 4 extends beyond the S4 pocket and interacts with a region of the protein that has not been previously described as important for small-molecule binding (Figs. 3 ▶ a and 3b ▶). Most of the binding affinity of the tricyclic moiety presumably arises from hydrophobic interactions with surrounding amino-acid side chains, as no hydrogen-bonding interactions are apparent. All the hydrogen bonds between the compound and the protein occur in the S1–S3 region, with the majority occurring in the S1 pocket (comprising the Asp-recognition site).
Figure 3.

Crystal structures of various soluble inhibitors derived from Tethering. C atoms are shown in green, N atoms in blue, O atoms in red and inhibitors in yellow. Key amino-acid residues within 8 Å of each inhibitor and the S1–S4 subsites defining the active site of human caspase-1 are indicated. 2F o − F c electron-density maps within a 2.5 Å distance of each compound are contoured at the 1σ level and displayed as blue mesh. (a, b) Structure of compound 4 bound to caspase-1 (2.7 Å resolution). (c, d) Structure of compound 5 bound to caspase-1 (2.0 Å resolution). (e, f) Structure of compound 6 bound to caspase-1 (2.1 Å resolution). (g, h) Structure of compound 8 bound to caspase-1 (2.2 Å resolution).
Comparison of the structure of compound 4 with the tetrapeptide inhibitor Ac-YVAD-CHO (Rano et al., 1997 ▶) shows that the tricyclic portion of compound 4 extends beyond the terminal Tyr of the tetrapeptide inhibitor (Fig. 4 ▶). Surprisingly, there are few differences between the protein backbone either in the presence of compound 4 or the tetrapeptide inhibitor (r.m.s.d. = 0.6 Å for 259 Cα atoms). There are, however, two amino-acid residues, Arg383 and Asp381, that adopt a different rotamer in order to accommodate the longer extended tricyclic inhibitor. The side chain of Arg383 rotates away from the S1 site, resulting in a 2.6 Å separation between the Cζ atoms of the residue observed in the structure of caspase-1 in complex with the tetrapeptide inhibitor (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1ice) and that of the protein complexed with compound 4, and toward the S4 pocket of the enzyme. This movement leads to stacking of the guanidinium group of Arg383 with a phenyl ring of the inhibitor. Asp381 also adopts a slightly different conformation, but it is not clear whether the observed difference arises from a real structural rearrangement or differences in model refinement. The Asp381 residue is found in the same three-dimensional position in the structures of four of our compounds that have very different binding modes, while it adopts a different conformation in the structure of the protein–tetrapeptide complex. Asp381 is the central residue of a surface-exposed three-residue turn between an α-helix and a β-strand, has higher than average B factors and its position in the models may be a reflection more of crystallization conditions and resolution than a role in binding the active-site compounds.
Figure 4.

Comparison of the active site of caspase-1 occupied by small-molecule and tetrapeptide inhibitors. (a) Stereo plot of compound 4 bound to the active site (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1rwk). (b) Stereo plot of the tetrapeptide inhibitor Ac-YVAD-CHO bound to the active site (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1ice).
Cocrystal structures of compounds 5 and 6 (Figs. 3 ▶ c, 3 ▶ d, 3 ▶ e and 3 ▶ f) also show the inhibitor extending beyond the S4 pocket. However, in contrast to compound 4, the tricyclic fragment of compound 5 points down and away from Asp381. Compound 6 (Figs. 3 ▶ e and 3 ▶ f) has a 2-thienyl group extending into the S2 pocket and, like compound 5, the tricyclic portion of the inhibitor also points away from Asp381. When bound to compound 5 or 6 (‘down’ conformation), compared with compound 4 (‘up’ conformation), the only salient structural change affecting residues in the active site involves the side chain of Arg383. The side chain of this residue rotates farther away from the S3 pocket when the protein is complexed with inhibitors in the ‘down’ conformation relative to the caspase-1 complexes in which the compound is in the ‘up’ conformation. The wider rotation away from S3 is made possible by the absence of the obstructing quinoxaline or hydroxyquinoline group of the inhibitor.
The structural analysis of the compounds leads us to conclude that the binding mode of the tricyclic moiety depends on the P2 functional group. Compound 4, which does not extend into the S2 pocket, has an ‘up’ conformation, whereas compounds 5 and 6, which have an ethyl or a 2-thienyl, respectively, extending into the S2 pocket, have a ‘down’ conformation. There also appears to be a correlation between the ‘up’ and ‘down’ conformation of the fragment and enzyme activity. Compound 4 (‘up’), has a significantly weaker of 0.34 µM compared with compounds 5 and 6 (‘down’), which have values of 0.016 and 0.007 µM, respectively. However, it is not clear if the increased affinity of compounds 5 and 6 arises from the fragment being in the ‘down’ conformation and/or from the presence of an extended P2 functionality in both compounds.
A cocrystal structure of compound 8 bound to caspase-1 (Figs. 3 ▶ g and 3 ▶ h) demonstrates that the fragment binds predominantly in the S4 pocket, with only very minor differences between the peptide backbone of this structure and our other structures (Figs. 3 ▶ a, 3 ▶ b, 3 ▶ c, 3 ▶ d, 3 ▶ e and 3 ▶ f). One noticeable difference is Arg383, which for compound 8 sits in the same conformation as compound 4, but is rotated relative to its position in the structures obtained with compounds 5 and 6.
The S2 pocket of caspase-1 and specifically Val338, which forms the uppermost boundary of the pocket, appears to lack flexibility and exists in an ‘open’ conformation, whether or not an appropriate P2 element is present in the inhibitor or substrate. This stands in sharp contrast to caspase-3, in which the S2 pocket appears to be very flexible and is found either in an open or closed (collapsed) conformation (Erlanson et al., 2003 ▶). Tyr204 in caspase-3, which is in a homologous position to Val338 in caspase-1, was found to collapse when the S2 pocket was unoccupied, even when an inhibitor lacking an S2-binding group was bound to the active site. Thus, the S2 pocket of caspase-1 appears to be less flexible than the corresponding pocket of caspase-3 and remains in an ‘open’ conformation even when unoccupied.
3.4. Cell activity of soluble inhibitors
All compounds were tested for their ability to block IL-1β secretion from human cells. Human peripheral blood mononuclear cells (PBMCs) were purified from normal healthy donors and treated with LPS to stimulate IL-1β processing and secretion. All our compounds inhibited IL-1β secretion, but did not inhibit IL-6 secretion at concentrations below 200 µM (Table 2 ▶), as expected since the secretion of IL-1β and not IL-6 is dependent upon caspase-1 activity (Li et al., 1995 ▶; Kuida et al., 1995 ▶; Gu et al., 1997 ▶; Ghayur et al., 1997 ▶). Of the compounds in the tricyclic series, compound 3 was the most potent in cells, whereas compound 6, the most potent in the enzyme assay, only inhibited IL-1β secretion with an EC50 of approximately 20 µM. Compound 5, however, was almost as potent as compound 3. Our second series of compounds, compounds 7 and 8, also displayed cell activity and, as expected based upon their respective values (Fig. 2 ▶
b), compound 8 was more active at inhibiting IL-1β secretion than compound 7. In summary, although the activities of the inhibitors measured in enzyme assays varied greatly from compound to compound, the molecules displayed overall similar activities in cell-based assays. The discrepancy implies the need for improvement of cell permeability and possibly further optimization of specificity of the inhibitors for caspase-1.
Table 2. Reduction of IL-1β and IL-6 secretion from human PBMCs by caspase-1 inhibitors.
Compounds were tested for their ability to inhibit IL-1β and IL-6 secretion from human PBMCs. EC50 values were calculated as the average of several measurements (n) and are shown with their associated standard deviations.
| Compound | EC50 IL-1β (µM) | EC50 IL-6 (µM) | n |
|---|---|---|---|
| 3 | 4.1 ± 2.7 | >200 | 4 |
| 4 | 18.4 ± 2.8 | >200 | 4 |
| 5 | 8.9 ± 4.3 | >200 | 3 |
| 6 | 20.5 ± 10.7 | >200 | 5 |
| 7 | 36.6 ± 7.4 | >200 | 3 |
| 8 | 24 ± 7 | >200 | 2 |
4. Conclusions
Here we demonstrate that Tethering with extenders, aided by detailed structural analysis, is a powerful approach for identifying fragments that can be rapidly and productively converted into reversible inhibitors of caspase-1. Moreover, the method allows us to identify fragments that are unlikely to be discovered by more traditional functional screening approaches and the use of an S1-binding extender eliminates the challenges associated with de novo linking of two novel fragments. Several compounds synthesized from one of these fragments were found to reach a distal region of the active site and bound in one of two very different ways to that portion of the enzyme. The differences in the mode of binding did not result in major changes to the conformation of the active site. Although the extender applied here was previously used to capture fragments interacting with a related caspase member, caspase-3 (Erlanson et al., 2003 ▶), the fragments identified in this study are completely unrelated to those discovered for caspase-3. Thus, Tethering with a single extender is an efficient and viable approach to the discovery of unrelated fragments that bind to different members of a related family of proteins and, as demonstrated in this study, it can be used to generate specific inhibitors. Furthermore, this approach is likely to be widely applicable to other classes of targets with two distinct proximal binding sites, such as other caspases, kinases and protein–protein interaction interfaces.
Supplementary Material
PDB reference: caspase-1 complexes with compound 1, 1rwk, r1rwksf
PDB reference: compound 4, 1rwm, r1rwmsf
PDB reference: compound 5, 1rwn, r1rwnsf
PDB reference: compound 6, 1rwo, r1rwosf
PDB reference: compound 8, 1rwp, r1rwpsf
Acknowledgments
This work was supported in part by SBIR grant No. 1 R43 AR 049976-01. We would like to thank the staff at SSRL beamline 7.1 for assistance with data collection, the Sunesis automation group (Stuart Lam, Tom Webb and Alex Hsi) for help in preparatory-scale HPLC purifications and Drs W. Michael Flanagan, Mike Randal, Robert S. McDowell and James A. Wells for critical reading of this manuscript.
Footnotes
The term Tethering® is a registered trademark of Sunesis Pharmaceuticals Inc. for its fragment-based drug-discovery methods.
References
- Allen, D., Pham, P., Choong, I. C., Fahr, B., Burdett, M., Lew, W., DeLano, W. L., Gordon, E., Lam, J., O’Brien, T. & Lee, D. (2003). Biol. Med. Chem. Lett.13, 3651–3655. [DOI] [PubMed]
- Cerretti, D., Kozlosky, C. J., Mosley, B., Nelson, N., Ness, K., Greenstreet, T., March, K., Kornhein, S., Druck, T., Cannizzaro, L., Heubner, K. & Black, R. (1992). Science, 256, 97–99. [DOI] [PubMed] [Google Scholar]
- Choong, I. C., Lew, W., Lee, D., Pham, P., Burdett, M., Lam, J., Wiesmann, C., Luong, T., Fahr, B., DeLano, W. L., McDowell, R., Allen, D., Erlanson, D., Gordon, E. & O’Brien, T. (2002). J. Med. Chem.45, 5005–5022. [DOI] [PubMed] [Google Scholar]
- Dinarello, C. A. (1998). Ann. NY Acad. Sci.856, 1–11. [DOI] [PubMed] [Google Scholar]
- DeLano, W. L. (2002). The PyMOL User’s Manual San Carlos, CA, USA: DeLano Scientific.
- Engh, R. & Huber, R. (1991). Acta Cryst. A47, 392–400. [Google Scholar]
- Erlanson, D. A. & Hansen, S. K. (2004). Curr. Opin. Chem. Biol.8, 399–406. [DOI] [PubMed] [Google Scholar]
- Erlanson, D., Lam, J., Wiesmann, C., Luong, T., Simmons, B., DeLano, W. L., Choong, I. C., Burdett, M., Flanagan, M., Lee, D., Gordon, E. & O’Brien, T. (2003). Nature Biotechnol.21, 308–314. [DOI] [PubMed]
- Erlanson, D. A., Wells, J. A. & Braisted, A. C. (2004). Annu. Rev. Biophys. Biomol. Struct.33, 199–223. [DOI] [PubMed] [Google Scholar]
- Ghayur, T., Banerjee, S., Hugunin, M., Butler, D., Herzog, L., Carter, A., Quintal, L., Sekut, L., Talanian, R., Paskind, M., Wong, W., Kamen, R., Tracey, D. & Allen, H. (1997). Nature (London), 386, 619–623. [DOI] [PubMed] [Google Scholar]
- Graybill, T. L., Dolle, R. E., Helaszek, C. T., Miller, R. E. & Ator, M. A. (1994). Int. J. Pept. Protein Res.44, 173–182. [DOI] [PubMed] [Google Scholar]
- Gu, Y., Kuida, K., Tsutsui, H., Ku, G., Hsiao, K., Fleming, M. A., Hayashi, N., Higashino, K., Okamura, H., Nakanishi, K., Kurimoto, M., Tanimoto, T., Flavell, R. A., Sato, V., Harding, M. W., Livingston, D. J. & Su, M. S. (1997). Science, 275, 206–209. [DOI] [PubMed] [Google Scholar]
- Gu, Y., Wu, J., Faucheu, C., Lalanne, J. L., Diu, A., Livingston, D. J. & Su, M. S. (1995). EMBO J.14, 1923–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard, A., Kostura, M., Thornberry, N. A., Ding, G., Limjuco, G., Weidner, J., Salley, J., Hogquist, K., Chaplin, D., Mumford, R., Schmidt, J. & Tocci, M. (1991). J. Immunol.147, 2964–2969. [PubMed] [Google Scholar]
- Hyde, J., Braisted, A., Randal, M. & Arkin, M. (2003). Biochemistry, 42, 6475–6483. [DOI] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kang, S. J., Wang, S., Hara, H., Peterson, E. P., Namura, S., Amin-Hanjani, S., Huang, Z., Srinivasan, A., Tomaselli, K. J., Thornberry, N. A., Moskowitz, M. A. & Yuan, J. (2000). J. Cell Biol.149, 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida, K., Loppke, J., Ku, G., Harding, M., Livingston, D. L., Su, M. S.-S. & Flavell, R. A. (1995). Science, 267, 2000–2003. [DOI] [PubMed] [Google Scholar]
- Kuzmic, P., Elrod, K., Cregar, L., Sideris, S., Rai, R. & Janc, J. (2000). Anal. Biochem.286, 45–50. [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291. [Google Scholar]
- Li, P., Allen, H., Banerjee, S., Franklin, S., Herzog, L., Johnston, C., McDowell, J., Paskind, M., Rodman, L., Salfeld, J., Towne, E., Tracey, D., Wardwell, S., Wei, F.-Y., Wong, W., Kamen, R. & Seshardi, T. (1995). Cell, 80, 401–411. [DOI] [PubMed] [Google Scholar]
- Martinon, F., Burns, K. & Tschopp, J. (2002). Mol. Cell, 10, 417–426. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. (1999). Acta Cryst. D55, 247–255. [DOI] [PubMed] [Google Scholar]
- Navaza, J. (2001). Acta Cryst. D57, 1367–1372. [DOI] [PubMed] [Google Scholar]
- O’Brien, T. & Lee, D. (2004). Mini Rev. Med. Chem.4, 153–165. [DOI] [PubMed] [Google Scholar]
- Pannu, N. S., Murshudov, G. N., Dodson, E. J. & Read, R. J. (1998). Acta Cryst. D54, 1285–1294. [DOI] [PubMed] [Google Scholar]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed] [Google Scholar]
- Ramage, P., Cheneval, D., Chvei, M., Graff, P., Hemmig, R., Heng, R., Kocher, H. P., Mackenzie, A., Memmert, K., Revesz, L. & Wishart, W. (1995). J. Biol. Chem.270, 9378–9383. [DOI] [PubMed] [Google Scholar]
- Rano, T. A., Timkey, T., Peterson, E. P., Rotonda, J., Nicholson, D., Becker, J., Chapman, K. & Thornberry, N. A. (1997). Chem. Biol.4, 149–155. [DOI] [PubMed] [Google Scholar]
- Riedl, S. J. & Shi, Y. (2004). Nature Rev. Mol. Cell Biol.5, 897–907. [DOI] [PubMed]
- Romanowski, M. J., Scheer, J. M., O’Brien, T. & McDowell, R. S. (2004). Structure, 12, 1361–1371. [DOI] [PubMed] [Google Scholar]
- Solary, E., Eymin, B., Droin, N. & Haugg, M. (1998). Cell Biol. Toxicol.14, 121–132. [DOI] [PubMed] [Google Scholar]
- Thornberry, N. A. et al. (1992). Nature (London), 356, 768–774. [DOI] [PubMed] [Google Scholar]
- Vaguine, A. A., Richelle, J. & Wodak, S. J. (1999). Acta Cryst. D55, 191–205. [DOI] [PubMed] [Google Scholar]
- Wang, S., Miura, M., Jung, Y., Zhu, H., Li, E. & Yuan, J. (1998). Cell, 92, 501–509. [DOI] [PubMed] [Google Scholar]
- Wilson, K., Black, J., Thompson, J., Kim, E., Griffith, J., Navia, M., Murcko, M., Chambers, S., Aldape, R. & Raybuck, S. A. (1994). Nature (London), 370, 270–275. [DOI] [PubMed] [Google Scholar]
- Yang, X., Chang, H. Y. & Baltimore, D. (1998). Mol. Cell, 1, 319–325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: caspase-1 complexes with compound 1, 1rwk, r1rwksf
PDB reference: compound 4, 1rwm, r1rwmsf
PDB reference: compound 5, 1rwn, r1rwnsf
PDB reference: compound 6, 1rwo, r1rwosf
PDB reference: compound 8, 1rwp, r1rwpsf