

Crystals of the lipase of B. glumae in complex with its specific foldase were obtained in two forms. Crystallization, crystal manipulation and preliminary X-ray diffraction analysis are described.
Keywords: lipase, steric chaperone, dehydration
Abstract
Bacterial lipases that are secreted via the type II secretion pathway require a lipase-specific foldase in order to obtain their native and biologically active conformation in the periplasmic space. The lipase–foldase complex from Burkholderia glumae (319 and 333 residues, respectively) was crystallized in two crystal forms. One crystal form belongs to space group P3121 (P3221), with unit-cell parameters a = b = 122.3, c = 98.2 Å. A procedure is presented which improved the diffraction of these crystals from ∼5 to 2.95 Å. For the second crystal form, which belonged to space group C2 with unit-cell parameters a = 183.0, b = 75.7, c = 116.6 Å, X-ray data were collected to 1.85 Å.
1. Introduction
Lipases (triacylglycerol hydrolases; EC 3.1.1.3) have evolved to be efficient catalysts for lipolytic reactions. In addition to their capacity to hydrolyze an extensive diversity of triacylglycerols, they can reverse this reaction in non-aqueous media in a process which is called transesterification. Therefore, lipases comprise an important group of industrially exploited biocatalysts (Jaeger & Reetz, 1998 ▶; Jaeger & Eggert, 2002 ▶). Many bacterial lipases, such as those produced by Pseudomonas and Burkholderia species, have been exceptionally useful because of their ability to function under extreme conditions and because of their chemo-, regio- and enantiospecific behaviour.
The lipase of B. glumae (previously known as P. glumae) was the first bacterial lipase to be structurally characterized by X-ray crystallography (Urakami et al., 1994 ▶; Cleasby et al., 1992 ▶; Noble et al., 1993 ▶). It is secreted to the extracellular medium via the type II secretion pathway, which involves two separate translocation events (Filloux, 2004 ▶). Firstly, the lipase crosses the inner membrane in an unfolded conformation using the Sec system and is then released into the periplasmic space. The second step comprises the translocation of the fully folded active lipase across the outer membrane by means of the Xcp secreton. One indispensable intermediate step in this secretion process is the transient folding of the lipase in the periplasm, which strictly depends on a highly dedicated chaperone called lipase-specific foldase (Lif; Rosenau et al., 2004 ▶).
Lipase molecules that fail to fold in the periplasm are rapidly degraded and do not reach their final destination. In vitro studies have shown that only Lif is required for proper folding of the lipase and that in the absence of Lif the lipase acquires an enzymatically inactive intermediate conformation. This inactive-folding intermediate is virtually indistinguishable from the native biologically active lipase by circular dichroism and fluorescence spectroscopy (El Khattabi et al., 2000 ▶). Addition of Lif to this folding intermediate leads to rapid activation and the formation of a tight complex between the two proteins. Thus, Lif is considered to play a fundamental role in overcoming a high-energy barrier in the folding landscape of the lipase. Lif is anchored to the inner membrane via an N-terminal hydrophobic α-helix, with the large C-terminal domain of the protein being located in the periplasma (Frenken et al., 1993 ▶). Owing to its specific action, Lif is classified as a steric chaperone (Ellis, 1998 ▶). Because the membrane anchor is not essential for the activation function (El Khattabi et al., 1999 ▶), a truncated Lif in which the transmembrane helix is replaced by a His tag was used in our experiments.
More than a decade after the publication of the first crystal structure of a bacterial lipase and the finding that the folding of such bacterial lipases strictly depends on a specific steric chaperone, the enigmatic question of how the folding of the lipase is catalysed by the foldase remains unanswered. Here, we report the crystallization of the B. glumae lipase (33.1 kDa) in complex with its specific foldase (35.4 kDa). The crystals obtained are suitable for detailed structural studies.
2. Materials and methods
2.1. Expression and purification
The B. glumae PG1 strain carrying plasmid pUR6524 (Frenken et al., 1993 ▶) was kindly provided by Dr Jan Tommassen and used for the production of lipase. An overnight culture of B. glumae pUR6524 grown at 301 K in Luria–Bertani (LB) medium supplemented with 50 µg ml−1 kanamycin was diluted 50 times in PG medium (Frenken et al., 1992 ▶) supplemented with 1% olive oil as a sole carbon source and 50 µg ml−1 kanamycin. After 4 h incubation at 301 K, 1 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG) was added to the culture and cells were grown for 48 h at 301 K. The crude lipase solution obtained by removal of the cells via centrifugation (20 min, 5000 rev min−1; Beckman JLA-8100 rotor) was adjusted to pH 8.0 with Tris (Sigma). Insoluble material was removed by centrifugation (45 min, 8000 rev min−1; Beckman JLA-8100 rotor). Lipase activity was measured by the hydrolysis of p-nitrophenyl caprylate as a substrate by monitoring p-nitrophenol formation for 3 min at OD405nm (El Khattabi et al., 1999 ▶).
Escherichia coli DH5α containing the plasmids pMEK52 and pT7pol23 (a generous gift from Dr Jan Tommassen) was used to express the His-tagged Lif as described previously (El Khattabi et al., 1999 ▶). Cells were harvested 3 h after induction by centrifugation (10 min, 5000 rev min−1; Beckman JA-10 rotor), resuspended in 50 mM Tris–HCl pH 8.0 containing 1 M NaCl and lysed by two passes through a French Press (SLM-Aminco Spectronic Instruments) at 6.9 MPa. Cell debris was removed by centrifugation (60 min at 18 000 rev min−1; Beckman JA-20 rotor). The supernatant solution was subjected to an Ni2+–NTA affinity chromatography (Qiagen) and bound proteins were eluted with column buffer containing 500 mM imidazole. The elution fractions containing Lif were pooled and subsequently concentrated with Vivaspin 10 kDa cutoff PES concentrators (Vivascience). Final purification was achieved by gel-filtration chromatography on a Superdex 75 1690 column (Amersham Biosciences) equilibrated with 20 mM Tris–HCl pH 8.0.
For purification of the lipase–foldase complex, an Ni2+–NTA column equilibrated with 20 mM Tris–HCl pH 8.0, 150 mM NaCl was loaded with His-tagged Lif. Subsequently, a crude lipase solution (pH 8.0) was applied and saturation of the column with lipase was monitored by measuring the lipase activity of the flowthrough. Unbound lipase was removed by washing with 20 mM Tris–HCl pH 8.0, 150 mM NaCl and the complex was eluted with 500 mM imidazole. The elution fractions containing the lipase–foldase complex were pooled, concentrated and desalted by gel filtration using a Superdex 75 1690 column (Amersham Biosciences) in 20 mM Tris–HCl pH 8.0. The purified complex was analyzed via SDS–PAGE with Coomassie blue staining.
In anticipation of determining the structure, we introduced seleno-l-methionine (SeMet) into the amino-acid sequence of Lif, which naturally contains five methionine residues. The expression was similar to that of native Lif, with the concerted feedback inhibition of aspartokinase being exploited and hence inhibiting methionine biosynthesis (Van Duyne et al., 1993 ▶).
An overnight culture of E. coli DH5α pMEK52 pT7pol23 grown in LB medium supplemented with 100 µg ml−1 ampicillin and 25 µg ml−1 kanamycin was diluted 1000 times in 9 × 330 ml M9 minimal medium supplemented with 40 µg ml−1 of all amino acids, 2 mM MgSO4, 0.05%(w/v) NH4Cl, 0.4%(w/v) glucose, 2 µg ml−1 thiamine, 40 µl Q-solution (5 g FeCl2·4H2O, 184 mg CaCl2·2H2O, 64 mg H3BO4, 18 mg CoCl2·6H2O, 4 mg CuCl2·H2O, 340 mg ZnCl2, 605 mg Na2MO4·2H2O and 8 ml concentrated HCl per litre), 100 µg ml−1 ampicillin and 25 µg ml−1 kanamycin and incubated for 24 h at 301 K while shaking. In each culture, the final concentrations of lysine, phenylalanine and threonine and of isoleucine, leucine, valine and methionine were increased to 100 and 50 µg ml−1, respectively. Growth continued for an additional 60 min. Cells were harvested by centrifugation (5 min, 5000 rev min−1; Beckman JA-10 rotor). The pellet was washed twice with 100 ml 10 mM MgSO4 and subsequently divided over 9 × 330 ml M9 minimal media preheated at 301 K with the same supplements as described above except for methionine, which was replaced by 50 µg ml−1 l-(+)-selenomethionine (Acros Organics), and with valine and isoleucine at 50 µg ml−1 and lysine, threonine and phenylalanine at 100 µg ml−1. After 15 min at 310 K the expression of the His-tagged Lif was induced by incubating the cultures for 60 min at 315 K and cell growth continued for an additional 3 h at 310 K.
The purification of the of SeMet-labelled Lif was identical to that of native Lif except that all buffers were degassed by argon flushing and 0.05 mM EDTA was added to each step of the purification.
2.2. Mass-spectrometry analysis
To confirm the substitution of methionine with selenomethionine by mass spectrometry, the proteins were desalted on ZipTip C18 (Millipore, Billerica, USA) and eluted in 50% acetonitrile/1%(v/v) formic acid. The samples were loaded into a nanoflow capillary (Proxeon, Odense, Denmark) and ESI mass spectra were acquired using a quadrupole time-of-flight instrument (Q-Tof Ultima, Micromass/Waters, Manchester, UK) operating in the positive-ion mode and equipped with a Z-spray nanoelectrospray source. Data acquisition was performed using a MassLynx 4.0 system. The exact mass of the proteins was determined after processing the spectra with the Transform software (Micromass/Waters, Manchester, UK).
The SeMet-labelled Lif and native Lif were digested overnight at 310 K with sequencing-grade trypsin (Promega, Madison, USA) in 25 mM ammonium bicarbonate. The resulting peptides were analyzed by tandem mass spectrometry (MS/MS). After processing the MS/MS data using the maximum-entropy data-enhancement program MaxEnt3, the amino-acid sequences were semi-automatically deduced using the peptide-sequencing program PepSeq (Micromass/Waters, Manchester, UK).
2.3. Crystallization
The purified B. glumae lipase–Lif complex was concentrated to a final concentration of 45 mg ml−1 in 20 mM Tris–HCl pH 8.0 prior to crystallization experiments. Initial screening was performed using the hanging-drop vapour-diffusion technique at 293 K with the commercially available Crystal Screens I and II and MembFac (Hampton Research). Drops containing 1 µl protein solution and 2 µl precipitant solution were equilibrated against 500 µl reservoir solution in 24-well plates. Two conditions yielded small needles and were further optimized using additive and detergent screens (Hampton Research) and by testing grid screens with varying precipitant concentrations, pH and protein concentrations. Since crystals were obtained in conditions with PEG as precipitant, the PEG/Ion Screen (Hampton Research) was also used.
2.4. X-ray diffraction and data processing
Diffraction data were collected on ESRF beamline ID14-2 (Grenoble, France) and DESY beamline X13 (Hamburg, Germany). For data collection at room temperature, crystals were mounted in 0.7 mm glass capillaries. Alternatively, data collection was performed under cryogenic conditions, in which the crystals were transferred to cryoprotectant solutions, fished out with a nylon loop and flash-cooled directly in a nitrogen stream at 100 K. The exact cryoprotection protocol used is described in §3. All diffraction data were indexed, integrated and scaled with DENZO and SCALEPACK from the HKL suite (Otwinowski & Minor, 1997 ▶). Structure-factor amplitudes were calculated from the intensities using TRUNCATE and the solvent content was estimated using MATTHEWS_COEFF from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
The lipase–foldase complex was purified to homogeneity and eluted as a single peak from the gel-filtration column. Two bands of 33 and 35 kDa appeared on Coomassie blue-stained 12.5% SDS–PAGE gels, corresponding to the lipase and the His-tagged Lif, respectively (Fig. 1 ▶ a). The incorporation of selenomethionine in Lif without making use of a methionine-auxotrophic strain was successful, as demonstrated by the shift of 234.6 Da between the native Lif and LifSeMet. This shift in mass agrees with the theoretical value of 234.47 Da corresponding to the substitution of five methionines for SeMet. The replacement of each methionine by SeMet was further confirmed after microsequencing by tandem mass spectrometry of the corresponding tryptic peptides.
Figure 1.

(a) SDS–PAGE analysis of the lipase–foldase complex after gel filtration (lane 1) compared with purified lipase (lane 2, 33 kDa) and Lif (lane 3, 35 kDa). (b) Crystals of the lipase–foldase complex grown in 12%(w/v) PEG 6000, 0.1 M Li2SO4, 0.1 M sodium citrate pH 5.6 and 0.2%(w/v) n-octyl-β-d-glucoside. (c) A single crystal grown in 20%(w/v) PEG 3350 and 0.2 M KI. The black bars indicate 0.1 mm.
Screening for suitable crystallization conditions resulted in small needles that appeared within 24 h in 0.1 M NaCl, 0.1 M sodium citrate pH 5.6, 12%(w/v) PEG 4000 and 0.1 M Li2SO4, 0.1 M sodium citrate pH 5.6, 12%(w/v) PEG 6000. Changing the pH, salt and the concentration or type of PEG did not result in larger crystals and neither did the use of various additives. In contrast, application of a detergent screen was successful as several detergents were found to significantly affect the crystal size [n-dodecyl-β-d-maltoside, cyclohexyl-β-d-maltoside (CYMAL-6), cetyltrimethylammonium bromide (CTAB), n-nonyl-β-d-glucoside, n-octyl-β-d-glucoside]. For further crystallization setups, n-octyl-β-d-glucoside was added to the protein solution at a concentration of 0.2%(w/v). This resulted in larger hexagonal shaped crystals (Fig. 1 ▶ b) of average dimensions 85 × 85 × 1000 µm which appeared within 7 d of incubation at 301 K and continued to grow for three weeks in 12%(w/v) PEG 6000, 0.1 M sodium citrate pH 5.6, 0.1 M Li2SO4.
When crystals were transferred to cryoprotectant solution consisting of mother liquor containing 25%(v/v) glycerol, they disintegrated instantaneously. Therefore, diffraction experiments at room temperature were conducted and poor diffraction was observed to a resolution of ∼5 Å (Fig. 2 ▶ a). Unit-cell parameters were derived as a = b = 124.95, c = 99.98 Å. However, rapid deterioration of the diffraction pattern was observed. Serendipitously, we discovered diffraction to 2.8 Å when one crystal dried out during mounting in the glass capillary. The unit-cell parameters were derived as a = b = 122.3, c = 98.6 Å. Unfortunately, we were unable to collect sufficient data at room temperature owing to rapid deterioration of the crystal. Interestingly, the improvement of the diffraction limit appeared to be accompanied by a change in unit-cell parameters. The number of reports on crystal dehydration as a method for improving the diffraction capacity is steadily increasing (Kawashima et al., 1996 ▶; Haebel et al., 2001 ▶; Heras et al., 2003 ▶). Hence, we decided to establish a controlled dehydration protocol in order to improve the diffraction limit and collect a complete data set under cryogenic conditions.
Figure 2.

Diffraction patterns of the hexagonal crystal form of the lipase–Lif complex (a) collected at room temperature to ∼5 Å from a crystal mounted in a glass capillary. (b) After stepwise transfer of the crystals to 30%(w/v) PEG 8000, 160 mM MgSO4, 80 mM sodium cacodylate pH 6.5, 20%(v/v) glycerol, diffraction to 3.4 Å was observed under cryogenic conditions. (c) Equilibration of the crystal in dehydration solution for 30 min against air improved the diffraction to 2.95 Å.
Crystals were transferred from a drop to a stabilizing solution containing 15%(w/v) PEG 8000, 160 mM MgSO4, 80 mM sodium cacodylate pH 6.5. They were then subjected to successive soaks in different drops of this stabilizing solution with increasing amounts of glycerol. Steps of 5% glycerol were used to a final concentration of 20%. Each soaking step lasted for about 15 min. The crystals were subsequently passed to solutions of the same composition [15%(w/v) PEG 8000, 160 mM MgSO4, 80 mM sodium cacodylate pH 6.5, 20%(v/v) glycerol] containing increasing amounts of PEG 8000 in steps of 2.5% up to 30% with an equilibration time of 30 min for each step. When crystals treated in this way were flash-cooled, diffraction was observed to 3.4 Å (Fig. 2 ▶ b). Temperature annealing of the crystals by blocking the cryostream for a few seconds and then flash-cooling the crystal in the N2 flow again was not successful. Despite the successful application of annealing as described in several cases in the literature (Hanson et al., 2003 ▶; Yang et al., 2002 ▶), deterioration or even complete absence of diffraction was observed with these crystals.
Several drops containing a crystal in the dehydration solution [30%(w/v) PEG 8000, 160 mM MgSO4, 80 mM sodium cacodylate pH 6.5, 20%(v/v) glycerol] were equilibrated against air for a period of about 20–30 min until phase separation became visible. The crystals were then transferred to the nitrogen stream at 100 K and vitrified. This extended dehydration protocol allowed us to collect data from a single crystal to 2.95 Å (Fig. 2 ▶ c) on EMBL beamline X13 (DESY, Hamburg) which were 99.6% complete. Unit-cell parameters and data-collection statistics are summarized in Table 1 ▶. Interestingly, the unit-cell parameters are highly similar to the unit cell of the unintentionally dehydrated crystal in the glass capillary at room temperature. The asymmetric unit contains a single complex of lipase and Lif, with a corresponding calculated Matthews coefficient of 3.1 Å3 Da−1 and a solvent content of 60.3% (Matthews, 1968 ▶). Untreated crystals diffracting to ∼5 Å at room temperature were calculated to have a solvent content of 62.5%. The diffraction limit could apparently be improved by dehydration of the crystals, with a concomitant contraction of the unit cell and hence a lower solvent content. Kantardjieff & Rupp (2003 ▶) substantiated that a higher packing density (lower solvent content) correlates significantly with increasing resolution. It is probable that application of the post-crystallization treatment influences the crystal packing in such a way that diffraction is enhanced.
Table 1. Crystal parameters and data-collection statistics for the two different crystal forms of the B. glumae lipase–foldase complex.
Values in parentheses indicate statistics for the highest resolution shell.
| Crystal form 1 | Crystal form 2 | |
|---|---|---|
| Space group | P3121 [P3221] | C2 |
| Unit-cell parameters | ||
| a (Å) | 122.3 | 183.0 |
| b (Å) | 122.3 | 75.7 |
| c (Å) | 98.2 | 116.6 |
| Beamline | X13 (DESY) | ID14-2 (ESRF) |
| Wavelength (Å) | 0.8047 | 0.933 |
| Total/unique reflections | 131953/18266 | 875222/119154 |
| Resolution range (Å) | 20–2.95 (3.06–2.95) | 40–1.85 (1.92–1.85) |
| Completeness (%) | 99.6 (99.2) | 99.0 (91.3) |
| Rmerge† (%) | 10.2 (31.5) | 7.2 (52.1) |
| 〈I/σ(I)〉 | 15.96 (4.74) | 15.43 (3.85) |
| Solvent content (%) | 60.3 | 52.9 |
| Lipase–Lif complexes per asymmetric unit | 1 | 2 |
R
merge = 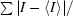
 for all reflections, where I is the reflection intensity and 〈I〉 is the average intensity for multiple measurements of that reflection.
for all reflections, where I is the reflection intensity and 〈I〉 is the average intensity for multiple measurements of that reflection.
After more than four months, a single crystal with a different morphology to the previously obtained hexagonal shaped crystals was observed in 20%(w/v) PEG 3350, 0.2 M KI (Fig. 1 ▶ c). This crystal was transferred to a cryoprotectant solution [mother liquor plus 20%(v/v) PEG 400] by increasing the PEG 400 concentration in steps of 2.5% with a few seconds of equilibration. No crystal cracking was observed during this transfer procedure. A 99% complete data set for this crystal form 2 was collected at the ID14-2 beamline (ESRF, Grenoble). The diffraction data were collected at 100 K after flash-freezing the crystals and the data-collection statistics are summarized in Table 1 ▶. Calculation of the Matthews coefficient (2.6 Å3 Da−1) shows that if two lipase–foldase complexes are assigned to the asymmetric unit, the solvent content of the crystal will be 52.9%. Attempts to reproduce this crystal form have not yet been successful. Therefore, we intend to produce SeMet-labelled crystals of the hexagonal form for initial phasing by MAD and subsequently solve the monoclinic form by molecular replacement.
The structure of the Lif in complex with its lipase substrate will provide the first atomic picture of such an intermolecular steric chaperone. In addition, it will be a seminal contribution to our understanding of the mechanism of action of a steric chaperone in the bimolecular folding event and in secretion of the lipase.
Acknowledgments
The authors acknowledge the use of ESRF beamline ID14-2 (Grenoble, France) and the EMBL X13 beamline at DESY (Hamburg, Germany). B. glumae pUR6524 strain and E. coli DH5α pMEK52 pT7pol23 were generously provided by Dr Jan Tommassen. KP is a pre-doctoral fellow supported by the Instituut voor de Aanmoediging van Innovatie door Wetenschap en Technologie. This research was sponsored by grants from the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO-Vlaanderen), the Onderzoeksfonds (OZR) of the Vrije Universiteit Brussel and the Nanofoldex Research Project QLRT-2001-02086 of the European Commission.
References
- Cleasby, A., Garman, E., Egmond, M. R. & Batenburg, M. (1992). J. Mol. Biol.224, 281–282. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- El Khattabi, M., Ockhuijsen, C., Bitter, W., Jaeger, K. E. & Tommassen, J. (1999). Mol. Gen. Genet.261, 770–776. [DOI] [PubMed] [Google Scholar]
- El Khattabi, M., Van Gelder, P., Bitter, W. & Tommassen, J. (2000). J. Biol. Chem.275, 26885–26891. [DOI] [PubMed] [Google Scholar]
- Ellis, R. J. (1998). Trends Biochem. Sci.23, 43–45. [DOI] [PubMed] [Google Scholar]
- Filloux, A. (2004). Biochim. Biophys. Acta, 1694, 163–179. [DOI] [PubMed] [Google Scholar]
- Frenken, L. G. J., de Groot, R., Tommassen, J. & Verrips, C. T. (1993). Mol. Microbiol.9, 591–599. [DOI] [PubMed] [Google Scholar]
- Frenken, L. G. J., Egmond, M. R., Batenburg, A. M., Bos, J. W., Visser, C. & Verrips, C. T. (1992). Appl. Environ. Microbiol.58, 3787–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haebel, P. W., Wichman, S., Goldstone, D. & Metcalf, P. (2001). J. Struct. Biol.136, 162–166. [DOI] [PubMed] [Google Scholar]
- Hanson, B. L., Schall, C. A. & Bunick, G. J. (2003). J. Struct. Biol.142, 77–87. [DOI] [PubMed] [Google Scholar]
- Heras, B., Edeling, M. A., Byriel, K. A., Jones, A., Raina, S. & Martin, J. L. (2003). Structure, 11, 139–145. [DOI] [PubMed] [Google Scholar]
- Jaeger, K. E. & Eggert, T. (2002). Curr. Opin. Biotechnol.13, 390–403. [DOI] [PubMed] [Google Scholar]
- Jaeger, K. E. & Reetz, M. T. (1998). Trends Biotechnol.16, 396–403. [DOI] [PubMed] [Google Scholar]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci.12, 1265–1871. [DOI] [PMC free article] [PubMed]
- Kawashima, T., Berthet-Colominas, C., Cusack, S. & Leberman, R. (1996). Acta Cryst. D52, 799–805. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.28, 491–497. [DOI] [PubMed]
- Noble, M. E. M., Cleasby, A., Johnson, L. N., Egmond, M. R. & Frenken, L. G. J. (1993). FEBS Lett.331, 123–128. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Rosenau, F., Tommassen, J. & Jaeger, K. E. (2004). Chembiochem, 5, 152–161. [DOI] [PubMed] [Google Scholar]
- Urakami, T., Ito-Yoshida, C., Araki, H., Kijima, T., Suzuki, K. I. & Komagata, K. (1994). Int. J. Syst. Bacteriol.44, 235–245.
- Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L. & Clardy, J. (1993). J. Mol. Biol.229, 105–124. [DOI] [PubMed] [Google Scholar]
- Yang, J. K., Yoon, H.-J., Ahn, H. J., Lee, B. I., Cho, S. H., Waldo, G. S., Park, M. S. & Suh, S. W. (2002). Acta Cryst. D58, 303–305. [DOI] [PubMed] [Google Scholar]