

The product of the human leukocyte antigen (HLA) gene HLA-B*2703 differs from that of the prototypical subtype HLA-B*2705 by a single amino acid at heavy-chain residue 59 that is involved in anchoring the peptide N-terminus within the A pocket of the molecule. Two B*2703–peptide complexes were crystallized using the hanging-drop vapour-diffusion method using PEG 8000 as a precipitant. A pocket of the molecule, two HLA-B*2703–peptide complexes were crystallized and data sets were collected to high resolution using synchrotron radiation.
Keywords: HLA-B*2703, HLA-B27 subtypes, subtype-dependent peptide-binding modes, ankylosing spondylitis, residue 59 polymorphism
Abstract
The product of the human leukocyte antigen (HLA) gene HLA-B*2703 differs from that of the prototypical subtype HLA-B*2705 by a single amino acid at heavy-chain residue 59 that is involved in anchoring the peptide N-terminus within the A pocket of the molecule. Two B*2703–peptide complexes were crystallized using the hanging-drop vapour-diffusion method using PEG 8000 as a precipitant. The crystals belong to space group P21 (pVIPR peptide) or P212121 (pLMP2 peptide). Data sets were collected to 1.55 Å (B*2703–pVIPR) or 2.0 Å (B*2703–pLMP2) resolution using synchrotron radiation. With B*2705–pVIPR as a search model, a clear molecular-replacement solution was found for both B*2703 complexes.
1. Introduction
Major histocompatibility complex (MHC; in humans, HLA) class I molecules consist of a highly polymorphic heavy chain (HC) that is non-covalently associated with β2-microglobulin (β2m). The HC forms a groove carrying peptides derived from self- or nonself-proteins within the cell. A large number of MHC class I molecules have already been investigated by X-ray crystallography, but the pairwise comparison of very closely related alleles, which additionally may differ in their association with diseases, has only recently been accomplished (Hülsmeyer et al., 2002 ▶, 2004 ▶, 2005 ▶; Macdonald et al., 2003 ▶; Webb et al., 2004 ▶; Zernich et al., 2004 ▶; Fiorillo et al., 2005 ▶).
In case of the human MHC class I allele HLA-B27, which is very strongly associated with a variety of autoimmune diseases, among them ankylosing spondylitis (AS; Ramos & López de Castro, 2002 ▶; Khan & Ball, 2002 ▶), peptide presentation has been suspected to play a role in pathogenesis (Benjamin & Parham, 1990 ▶; Ramos & López de Castro, 2002 ▶). Increased numbers of cytotoxic T lymphocytes (CTL) directed against the self-antigen pVIPR [RRKWRRWHL, derived from vasoactive intestinal peptide type 1 receptor (residues 400–408)] have been found in disease-affected individuals with the HLA-B*2705 subtype (Fiorillo et al., 2000 ▶). About one-sixth of these T cells cross-react with the viral pLMP2 peptide [RRRWRRLTV, derived from latent membrane protein 2 (residues 236–244) of Epstein–Barr virus (EBV); Fiorillo et al., 2000 ▶, 2005 ▶]. Individuals with another subtype, HLA-B*2709, which is not associated with AS and differs from the former only by a single amino acid (His116 instead of Asp116), do not develop CTL responses against the self-peptide pVIPR (Fiorillo et al., 2000 ▶), suggesting a HLA-B27 subtype-dependent connection with AS pathogenesis.
The HLA-B*2703 subtype is nearly exclusively restricted to black individuals (Rojo et al., 1987 ▶; Choo et al., 1988 ▶; Gonzalez et al., 2002 ▶). Unlike B*2705, it exhibits a questionable association with AS (Ramos & López de Castro, 2002 ▶; Khan & Ball, 2002 ▶) and its product differs from that of the former subtype by a single amino acid (His59 instead of Tyr59). The consequences of this exchange for peptide binding (Colbert et al., 1994 ▶; Boisgérault et al., 1996 ▶) or T-cell responses (Villadangos et al., 1994 ▶) are entirely distinct from those found for the B*2705/B*2709 pair (Ramos et al., 2002 ▶; Fiorillo et al., 2000 ▶). HC residue 116 polymorphisms are among the most frequent HLA-B allele exchanges (Reche & Reinherz, 2003 ▶) and lead to altered binding of the C-terminal residue of the peptide. In contrast, of the six HLA class I loci with a current total of 1180 alleles, replacement of Tyr59 occurs only in B*2703 (His59) and B*2717 (Phe59). This exchange is expected to primarily affect the A pocket of the molecule, which binds the peptide N-terminus through hydrogen bonds that are arranged in a highly conserved characteristic pentagonal network (Madden, 1995 ▶; Hülsmeyer et al., 2002 ▶). The influence of another naturally occurring A-pocket amino-acid exchange on the binding of the peptide N-terminus has already been investigated for B*5101, one of the few subtypes where the common Tyr171 is replaced by His171 (Maenaka et al., 2000 ▶). Contrary to residue 59, which is located at the beginning of the α1-helix, residue 171 is part of the end of the α2-helix, opposite residue 59. The His171Tyr exchange results in the complete rearrangement of the above-mentioned pentagonal hydrogen-bonding network within the A pocket (Maenaka et al., 2000 ▶).
Determination of the influence of selected polymorphisms on peptide presentation by HLA-B27 subtypes will aid in understanding subtype-dependent differential disease associations (Ramos & López de Castro, 2002 ▶; López de Castro et al., 2004 ▶). In particular, we addressed the following questions. How are peptides such as pVIPR and pLMP2, whose binding modes have already been determined in the B*2705 and B*2709 subtypes (Hülsmeyer et al., 2004 ▶; Fiorillo et al., 2005 ▶), bound to B*2703? How does the His59Tyr replacement affect the binding mode of the N-terminal amino acid of the peptide and is its effect comparable to that found for His171 in case of B*5101? Furthermore, can the peptide conformation which characterizes pVIPR and pLMP2 binding in the B*2705 subtype (i.e. main-chain ϕ/ψ torsion angles in α-helical conformation at peptide position p6 instead of the common p4; Hülsmeyer et al., 2004 ▶; Fiorillo et al., 2005 ▶) also be observed in B*2703? Our study is the first to determine the structural properties of the B*2703 subtype.
2. Materials and methods
2.1. Protein preparation
The peptides pVIPR (RRKWRRWHL) and pLMP2 (RRRWRRLTV) were synthesized by the solid-phase method and purified by Alta Bioscience (Birmingham, England). The extracellular region of the B*2703 heavy chain (the clone was generated by in vitro mutagenesis from a B*2705 clone) and β2m were expressed separately as inclusion bodies in Escherichia coli, dissolved in 50%(w/v) urea and the HLA-B27–peptide complexes were reconstituted for 14 d at 277 K as described previously (Garboczi et al., 1992 ▶) with slight modifications. Briefly, unfolded HC (12 mg), β2m (10 mg) and 4 mg of either pVIPR or pLMP2 were rapidly injected into 400 ml of refolding buffer (400 mM arginine–HCl, 2 mM EDTA, 5 mM reduced glutathione, 0.5 mM oxidized glutathione and 100 mM Tris–HCl pH 7.5). The mixture was concentrated using Amicon Ultra-15 devices and the complexes were isolated by size-exclusion chromatography and used for crystallization at concentrations of 13–15 mg ml−1 in 20 mM Tris–HCl, 150 mM NaCl, 0.01% sodium azide pH 7.5.
2.2. Crystallization and data collection
All crystallization trials were performed in a hanging-drop vapour-diffusion setup at 291 K (1.5 µl protein solution and 1.5 µl precipitant solution), employing the previously described conditions (Hülsmeyer et al., 2002 ▶, 2005 ▶). Crystal formation for both complexes was optimized by varying the PEG concentration in the precipitant solution [18–28%(w/v) PEG 8000, 100 mM Tris–HCl pH 7.0]. To increase the crystal size, streak-seeding was applied by passing a cat whisker through each crystallization drop in the screens. After 4 d, crystals of B*2703–pVIPR grew as plates and maximum dimensions of 200 × 100 × 10 µm were obtained at 18%(w/v) PEG 8000. The crystals of B*2703–pLMP2 had identical morphology, but were always smaller than those from B*2703–pVIPR, with approximate dimensions of 80 × 80 × 5 µm obtained at a PEG 8000 concentration of 22%(w/v) (Fig. 1 ▶).
Figure 1.

Crystals of B*2703–pLMP2. The black bar indicates a length of 80 µm. Crystals of B*2703–pVIPR exhibited the same morphology but were slightly larger (not shown). (a) Crystals within the crystallization drop. (b) Cooled crystal mounted in a cryoloop at beamline ID14-2 (ESRF) equipped with a mini-diffractometer. The rectangle in cyan represents the size of the X-ray beam.
Prior to data collection, the crystals in the crystallization drops were cryoprotected by stepwise increase of glycerol and PEG 8000 to final concentrations of 10 and 21%, respectively. An initial data set for B*2703–pVIPR was collected at the Protein Structure Factory beamline BL14.2 of the Free University Berlin at Berliner Elektronenspeicherring–Gesellschaft für Synchrotronstrahlung mbH (BESSY, Berlin, Germany). Data sets with the highest diffraction limit were collected at the European Synchrotron Radiation Facility (ESRF, Grenoble, France), beamline ID 14-2, at a wavelength of 0.933 Å at 100 K. This beamline is equipped with a novel MD2M mini-diffractometer and an ADSC-Q4 (Area Detector Systems Corperation) CCD detector. The mini-diffractometer simplified the precise centring of the small crystals of B*2703–pLMP2. Visual inspection of the diffraction pattern from crystals of B*2703–pLMP2 clearly showed elongated spots. Flash annealing extended the diffraction limit from the initial 2.4 to 2.0 Å and improved the spot shape. For annealing, the cryostream was blocked with a canteen card for approximately 5 s until melting became visible; the card was then quickly removed to allow re-cooling.
Data were processed with DENZO and scaled with SCALEPACK (Otwinowski & Minor, 1997 ▶). The results are summarized in Table 1 ▶. The B*2703–pVIPR crystals belong to the monoclinic space group P21, whereas the crystals of B*2703–pLMP2 belong to an orthorhombic space group. Molecular replacement was performed using coordinates of the high-resolution crystal structure of B*2705–pVIPR (Hülsmeyer et al., 2004 ▶; PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1ogt; water molecules and peptide were omitted) as a search model. Unambiguous solutions of both structures were found using the program PHASER (Storoni et al., 2004 ▶) with diffraction data in the resolution range 20–3 Å. Initial F o − F c difference maps revealed the undoubted presence of the peptides when inspected with the program O (Jones et al., 1991 ▶).
Table 1. Data-collection statistics of HLA-B*2703–pVIPR and HLA-B*2703–pLMP2.
Values in parentheses refer to the highest resolution shell.
| HLA-B*2703–pVIPR | HLA-B*2703–pLMP2 | |
|---|---|---|
| Space group | P21 | P212121 |
| Unit-cell parameters | ||
| a (Å) | 50.9 | 50.7 |
| b (Å) | 81.6 | 82.6 |
| c (Å) | 65.3 | 108.3 |
| α (°) | 90 | 90 |
| β (°) | 107 | 90 |
| γ (°) | 90 | 90 |
| Solvent content (%) | 58 | 52 |
| Matthews coefficient† (Å3 Da−1) | 2.9 | 2.6 |
| Resolution (Å) | 30.0–1.55 (1.58–1.55) | 20.0–2.0 (2.03–2.00) |
| Unique reflections | 70574 (3112) | 30890 (1512) |
| Completeness (%) | 95.3 (85.1) | 98.3 (97.3) |
| Redundancy | 3.1 (2.7) | 4.2 (3.7) |
| 〈I/σ(I)〉 | 22.2 (3.4) | 16.3 (3.9) |
| Rsym‡ | 0.045 (0.281) | 0.074 (0.301) |
| Rmerge§ | 0.045 (0.280) | 0.070 (0.300) |
| Rr.i.m.§ | 0.054 (0.342) | 0.085 (0.346) |
| Rp.i.m.§ | 0.030 (0.193) | 0.039 (0.168) |
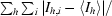
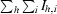
3. Results
The B*2703–pVIPR and B*2703–pLMP2 complexes were crystallized and crystal formation was optimized using streak-seeding techniques (Fig. 1 ▶). X-ray diffraction analysis revealed that the crystals of the two complexes were not isomorphous (Table 1 ▶), contrary to our expectation from the experiments with the B*2705 and B*2709 subtypes, which crystallized in the P21 space group when complexed with pVIPR and pLMP2 (Hülsmeyer et al., 2004 ▶; Fiorillo et al., 2005 ▶). B*2703–pLMP2 crystallized in space group P212121, which has previously been observed in crystals of B*2705 and B*2709 [each in complex with two peptides, m9 (Hülsmeyer et al., 2002 ▶) or TIS (Hülsmeyer et al., 2005 ▶)]. This finding indicates that B*2703–pLMP2 as well as B*2705 and B*2709 complexed with m9 or TIS adopt similar conformations. The crystals diffracted to 1.55 Å (B*2703–pVIPR) and 2.0 Å (B*2703–pLMP2). Therefore, a detailed comparison will be possible not only between the two B*2703–peptide complexes, but also between them and peptides complexed with B*2705, B*2709 and B*5101. Further refinement of both B*2703–peptide complexes is in progress.
Acknowledgments
We thank W. Weihofen for helpful comments on the manuscript. Financial support for this work was provided by the Deutsche Forschungsgemeinschaft (to BL and JB as well as to WS, BU-Z, AV and A. Ziegler through SFB 449/B5,B6) and the Fonds der Chemischen Industrie [to A. Zawacka (Kekulé Fellowship) and WS]. We are also grateful for allocation of beam time and support at BESSY (Berlin) and ESRF (Grenoble).
References
- Benjamin, R. & Parham, P. (1990). Immunol. Today, 11, 137–142. [DOI] [PubMed] [Google Scholar]
- Boisgérault, F., Tieng, V., Stolzenberg, M. C., Dulphy, N., Khalil, I., Tamouza, R., Charron, D. & Toubert, A. (1996). J. Clin. Invest.98, 2764–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo, S. Y., St John, T., Orr, H. T. & Hansen, J. A. (1988). Hum. Immunol.21, 209–219. [DOI] [PubMed] [Google Scholar]
- Colbert, R. A., Rowland-Jones, S. L., McMichael, A. J. & Frelinger, J. A. (1994). Immunity, 1, 121–130. [DOI] [PubMed] [Google Scholar]
- Fiorillo, M. T., Maragno, M., Butler, R., Dupuis, M. L. & Sorrentino, R. (2000). J. Clin. Invest.106, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo, M. T., Rückert, C., Hülsmeyer, M., Sorrentino, R., Saenger, W., Ziegler, A. & Uchanska-Ziegler, B. (2005). J. Biol. Chem.280, 2962–2971. [DOI] [PubMed] [Google Scholar]
- Garboczi, D. N., Hung, D. T. & Wiley, D. C. (1992). Proc. Natl Acad. Sci. USA, 89, 3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, S., Garcia-Fernandez, S., Martinez-Borra, J., Blanco-Gelaz, M. A., Rodrigo, L., Sanchez del Rio, J., López-Vazquez, A., Torre-Alonso, J. C. & López-Larrea, C. (2002). Hum. Immunol.63, 673–676. [DOI] [PubMed] [Google Scholar]
- Hülsmeyer, M., Fiorillo, M. T., Bettosini, F., Sorrentino, R., Saenger, W., Ziegler, A. & Uchanska-Ziegler, B. (2004). J. Exp. Med.199, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hülsmeyer, M., Hillig, R. C., Volz, A., Rühl, M., Schröder, W., Saenger, W., Ziegler, A. & Uchanska-Ziegler, B. (2002). J. Biol. Chem.277, 47844–47853. [DOI] [PubMed] [Google Scholar]
- Hülsmeyer, M., Welfle, K., Pöhlmann, T., Misselwitz, R., Alexiev, U., Welfle, H., Saenger, W., Uchanska-Ziegler, B. & Ziegler, A. (2005). J. Mol. Biol.346, 1367–1379. [DOI] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Khan, M. A. & Ball, E. J. (2002). Best Pract. Res. Clin. Rheumatol.16, 675–690. [PubMed] [Google Scholar]
- López de Castro, J. A., Alvarez, I., Marcilla, M., Paradela, A., Ramos, M., Sesma, L. & Vazquez, M. (2004). Tissue Antigens, 63, 424–445. [DOI] [PubMed] [Google Scholar]
- Macdonald, W. A., Purcell, A. W., Mifsud, N. A., Ely, L. K., Williams, D. S., Chang, L., Gorman, J. J., Clements, C. S., Kjer-Nielsen, L., Koelle, D. M., Burrows, S. R., Tait, B. D., Holdsworth, R., Brooks, A. G., Lovrecz, G. O., Lu, L., Rossjohn, J. & McCluskey, J. (2003). J. Exp. Med.198, 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden, D. R. (1995). Annu. Rev. Immunol.13, 587–622. [DOI] [PubMed] [Google Scholar]
- Maenaka, K., Maenaka, T., Tomiyama, H., Takiguchi, M., Stuart, D. I. & Jones, E. Y. (2000). J. Immunol.165, 3260–3267. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Ramos, M. & López de Castro, J. A. (2002). Tissue Antigens, 60, 191–205. [DOI] [PubMed] [Google Scholar]
- Ramos, M., Paradela, A., Vazquez, M., Marina, A., Vazquez, J. & López de Castro, J. A. (2002). J. Biol. Chem.277, 28749–28756. [DOI] [PubMed] [Google Scholar]
- Reche, P. A. & Reinherz, E. L. (2003). J. Mol. Biol.331, 623–641. [DOI] [PubMed] [Google Scholar]
- Rojo, S., Aparicio, P., Hansen, J. A., Choo, S. Y. & López de Castro, J. A. (1987). J. Immunol.139, 3396–3401. [PubMed] [Google Scholar]
- Storoni, L. C., McCoy, A. J. & Read, R. J. (2004). Acta Cryst. D60, 432–438. [DOI] [PubMed] [Google Scholar]
- Villadangos, J. A., Galocha, B., Garcia-Hoyo, R., Lopez, D., Garcia, F. & López de Castro, J. A. (1994). Eur. J. Immunol.24, 2548–2555. [DOI] [PubMed] [Google Scholar]
- Webb, A. I., Borg, N. A., Dunstone, M. A., Kjer-Nielsen, L., Beddoe, T., McCluskey, J., Carbone, F. R., Bottomley, S. P., Aguilar, M. I., Purcell, A. W. & Rossjohn, J. (2004). J. Immunol.173, 402–409. [DOI] [PubMed] [Google Scholar]
- Weiss, M. (2001). J. Appl. Cryst.34, 130–135. [Google Scholar]
- Zernich, D., Purcell, A. W., Macdonald, W. A., Kjer-Nielsen, L., Ely, L.K., Laham, N., Crockford, T., Mifsud, N. A., Bharadwaj, M., Chang, L., Tait, B. D., Holdsworth, R., Brooks, A. G., Bottomley, S. P., Beddoe, T., Peh, C. A., Rossjohn, J. & McCluskey, J. (2004). J. Exp. Med.200, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]