

The expression, purification and preliminary X-ray diffraction studies of a novel N-acetyl-l-citrulline deacetylase from X. campestris are reported.
Keywords: argE gene, deacetylase, N-acetyl-l-citrulline, N-acetyl-l-ornithine, arginine biosynthesis
Abstract
A novel N-acetyl-l-citrulline deacetylase that is able to catalyze the hydrolysis of N-acetyl-l-citrulline to acetate and citrulline was identified from Xanthomonas campestris. The protein was overexpressed, purified and crystallized. The crystals belong to the monoclinic space group C2 and diffract to 1.75 Å resolution, with unit-cell parameters a = 94.13, b = 95.23, c = 43.61 Å, β = 93.76°. Since attempts to use homologous structural models to solve the structure via molecular replacement were unsuccessful, the selenomethionine-substituted protein was prepared using an overnight auto-induction overexpression system. Selenomethionine incorporation into the protein was verified by MALDI–TOF/TOF mass-spectroscopic analysis after trypsin digestion. The crystals of the selenomethionine-substituted protein were prepared using crystallization conditions similar to those for the native protein. Multiple anomalous dispersion (MAD) data were collected at Brookhaven National Laboratory. Structure determination is under way using the MAD phasing method.
1. Introduction
In the canonical arginine-biosynthetic pathway, l-ornithine deacetylase catalyzes the conversion of acetylornithine to ornithine, which is then converted into citrulline by ornithine transcarbamylase (OTCase; Cunin et al., 1986 ▶). The gene that encodes OTCase in various organisms is known as argF. In Xanthomonas campestris, however, the encoded protein of the argF gene that was previously annotated as OTCase does not contain the DxxSMG ornithine-binding motif conserved in all known OTCases (Shi et al., 2001 ▶). The gene product of X. campestris argF was structurally and enzymatically confirmed to be a novel N-acetyl-l-ornithine transcarbamylase (AOTCase), which catalyzes the carbamylation of N-acetyl-l-ornithine rather than l-ornithine (Shi et al., 2005 ▶). Therefore, in order to fulfill arginine biosynthesis in X. campestris, a subsequent novel enzyme must exist to catalyze the deacetylation of N-acetyl-l-citrulline or the following acetyl intermediates. A gene whose encoded protein sequence has high sequence similarity to other argE-encoded proteins was cloned from X. campestris (denoted henceforth as argE′). Activity assays indicate that the purified protein is able to catalyze the deacetylation of N-acetyl-l-citrulline to produce l-citrulline. Thus, the arginine-biosynthetic pathway in X. campestris is different from the canonical arginine-biosynthetic pathway found in most bacteria, yeasts and plants.
This novel arginine pathway also seems to be present in Xylella fastidiosa, Xanthomonas axonopodis, Xanthomonas oryzae, Bacteroides fragilis, Bacteroides thetaiotaomicron, Cytophaga hutchinsonii, Tannerella forsythensis, Prevotella ruminicola and Myxococcus xanthus (Shi et al., 2005 ▶). The genera Xanthomonas and Xylella are plant pathogens that attack a wide variety of economically important crops such as citrus, grapes, cotton, tomatoes, peppers and rice (da Silva et al., 2001 ▶, 2002 ▶). In addition, B. fragilis, a Gram-negative anaerobic bacteria most commonly found in the intestine, is considered to be one of the most clinically important anaerobic human pathogens (Miki et al., 2005 ▶). T. forsythensis is found in human gingiva and is thought to be associated with periodontal disease (Sanz et al., 2004 ▶).
We report here the cloning, overexpression, purification, crystallization and preliminary X-ray diffraction data for the novel N-acetyl-l-citrulline deacetylase from X. campestris.
2. Experimental
2.1. Cloning and protein expression and purification
The argE′ gene was PCR amplified from X. campestris genomic DNA (ATCC 33913D) using hot-started Turbo Pfu DNA polymerase and the primers 5′-GACATATGACCGATCTACTCG-3′ and 5′-GCGAATTCCAGTGCGAGCCGTTGATG-3′. The PCR products were cloned into a Topo vector using a Zero-blunt Topo cloning kit (Invitrogen) with subsequent insertion into a pET28a vector (Novagen) using T4 DNA ligase (New England BioLabs) and transformation into Escherichia coli DH5α cells (Invitrogen). The protein was prepared using the overnight express auto-induction system 1 (Novagen) according to the manufacturer’s protocol. Briefly, 1 l LB media was supplemented with chemicals provided in the kit and cultures were grown overnight at 310 K. The cells were harvested by centrifugation and suspended in 40 ml Ni-affinity lysis buffer [300 mM NaCl, 50 mM NaH2PO4 pH 7.4, 10%(v/v) glycerol, 10 mM β-mercaptoethanol] and disrupted by sonication. The protein was purified first using a HisTrap Ni-affinity column (GE Healthcare) and then with a HisTrap DEAE column (GE Healthcare) using an ÄKTA FPLC system. In the first step, the soluble fraction was loaded onto a 5 ml HisTrap Ni-affinity column previously equilibrated with lysis buffer and the column was then washed with the buffer containing 50 mM imidazole. The protein was eluted with buffer containing 250 mM imidazole. In the second step, the protein was dialyzed into buffer containing 50 mM NaCl, 20 mM Tris–HCl pH 8.0, 1 mM EDTA and 5 mM β-mercaptoethanol. The protein was then loaded onto a DEAE HisTrap column and eluted using the linear-gradient method applied against a buffer containing 500 mM NaCl, 20 mM Tris–HCl pH 8.0, 1 mM EDTA and 5 mM β-mercaptoethanol. The protein was eluted out at 40%(v/v) target buffer. After concentrating the protein to about 12 mg ml−1, 50 units of thrombin were added to a solution containing about 10 mg protein. The solution was incubated at 277 K overnight to remove the six-histidine tag. The protein was used for crystallization screening without further purification. The protein purity was verified by SDS–PAGE [12%(v/v) polyacrylamide gel] followed by Coomassie staining. Protein concentration was determined by the Bradford method using the BioRad protein-assay dye reagent with bovine serum albumin as a standard (Bradford, 1976 ▶).
2.2. Preparation and verification of selenomethionine-substituted protein
The selenomethionine-substituted protein was prepared using the overnight express auto-induction system 2 (Novagen). The plasmid was transformed into a metE − host strain B834(DE3) (Novagen). The clone was inoculated into 1 l sterile deionized water supplemented with the chemicals provided in the kit and 125 mg l-selenomethionine. Vitamin B12 (cyanocobalamin) was added to a final concentration of 100 nM and the culture was incubated at 310 K for 16 h. After reaching stationary phase, the cells were harvested and disrupted and the protein was purified as described above for the native protein. The yield of selenomethionine-substituted protein was similar to that of the native protein.
10 µg aliquots of native protein and selenomethionine-substituted protein were digested overnight at 310 K using Promega sequencing-grade trypsin [enzyme:protein ratio, 1:50(w:w)] in 50 mM ammonium bicarbonate pH 7.4. The resulting peptides were desalted using C18 ZipTip micropipette tips (Millipore) following the manufacturer’s User Guide. The peptides were eluted from the ZipTips in 10 µl acetonitrile/0.1% TFA [70:30(v:v)]. 0.3 µl peptide solution was mixed with 0.3 µl matrix solution (50 mM α-cyano-4-hydroxycinnamic acid in acetonitrile/0.1% TFA [70:30(v:v)]) and spotted on the MALDI plate. Mass-spectrometry (MS) analyses were performed on a 4700 ABI TOF/TOF mass spectrometer (Applied Biosystems) operated in reflection positive-ion mode. A mixture of standard peptides was used to externally calibrate the instrument. Peptide mapping was verified against the theoretical digest of the protein.
2.3. Enzyme activity
The putative substrate N-acetyl-l-citrulline was prepared by reacting l-citrulline with acetic anhydride and purified by recrystallization (Strandholm et al., 1971 ▶). Purity was confirmed to be greater than 98% by proton and carbon NMR spectroscopy. Activity was assayed by determining the production of l-citrulline from N-acetyl-l-citrulline using an LC–MS system. Enzymatic activity was assayed in 70 µl 20 mM HEPES buffer pH 8.0 containing 50 mM NaCl and 200 mM CoCl2, 10 µl 10 mM N-acetyl-l-citrulline and 20 µl purified enzyme (∼0.1 mg ml−1). The reaction was quenched with 100 µl 30%(w/v) trichloroacetic acid solution after 5 min, the precipitated protein was removed by micro-centrifugation for 5 min and 10 µl of the supernatant solution was injected into a reverse-phase HPLC–MS system. The mobile phase consisted of 93% solvent A [1%(v/v) trifluoroacetic acid] and 7% solvent B [1 ml trifluoroacetic acid in 1 l 1:9(v:v) water/acetonitrile] and the flow rate was 0.6 ml min−1. Detection was by mass-spectroscopy using selected ion monitoring in atmospheric pressure chemical ionization mode with positive polarity. The mass ions of the protonated compounds of interest were monitored as follows: N-acetyl-l-citrulline, m/z = 218; l-citrulline, m/z = 176. N-acetyl-l-citrulline was eluted from the HPLC with a retention time of 2.626 min, while l-citrulline had a retention time of 2.039 min.
2.4. Crystallization and data collection
The purified protein was concentrated to ∼12 mg ml−1 with an Amicon-Y30 membrane concentrator (Millipore). The Index screening kit was used for crystallization tests (Hampton Research) at 291 K. Crystals consisting of thin plate clusters formed using conditions Nos. 43, 71, 79 and 83 within a week. Since all conditions contained PEG 3350 as a precipitant at pH 6.5, crystallization conditions were further screened with the PEG/Ion screening kit (Hampton Research). Well separated thin plate crystals formed using condition No. 20. After optimization, diffracting crystals with a maximum dimension of 0.4 mm were grown from a solution containing 0.2 M magnesium formate, 12–15%(w/v) PEG 3350 pH 5.9 (Fig. 1 ▶). The selenomethionine-substituted protein was crystallized in the same way.
Figure 1.

Typical thin plate-shaped crystals of N-acetyl-l-citrulline deacetylase. The largest dimension was approximately 0.4 mm.
Prior to data collection, a crystal was dipped quickly into a solution [0.2 M magnesium formate, 15%(w/v) PEG 3350 pH 5.9 supplemented with 20%(v/v) ethylene glycerol] for cryoprotection (less than 1 min). The cryoprotected crystal was frozen by plunging it directly into liquid nitrogen. Data to 2.0 Å resolution (not shown) were collected on an R-AXIS IV image-plate diffractometer mounted on a Rigaku RU-200 rotating-anode generator with a Cu target (λ = 1.54178 Å). Data sets to 1.75 Å resolution for both native and selenomethionine-substituted crystals were later collected using a synchrotron source (X26C and X12B beamlines, Brookhaven National Laboratory). The data sets for the selenomethionine-substituted crystals were collected using three wavelengths at the selenium adsorption edge, inflection and remote positions. All data were processed using the HKL2000 package (Otwinowski & Minor, 1997 ▶) and are summarized in Table 1 ▶.
Table 1. Summary of crystallographic data for native protein and Se-MAD data.
Values in parentheses are for the highest resolution shell.
| Native | Se edge | Se inflection | Se remote | |
|---|---|---|---|---|
| Wavelength (Å) | 0.9795 | 0.9780 | 0.9761 | 0.9550 |
| Resolution (Å) | 50–1.75 (1.81–1.75) | 50–1.74 (1.80–1.74) | 50–1.74 (1.80–1.74) | 50–1.90 (1.97–1.90) |
| Unit-cell parameters (Å, °) | a = 94.13, b = 95.23, c = 43.61, β = 93.76 | a = 93.87, b = 95.41, c = 43.61, β = 93.54 | a = 93.89, b = 95.46, c = 43.65, β = 93.49 | a = 93.95, b = 95.57, c = 43.69, β = 93.43 |
| No. of measurements | 286692 | 146669 | 146099 | 113866 |
| No. of unique reflections | 37941 | 76016 | 75596 | 58606 |
| Completeness (%) | 98.1 (96.0) | 98.7 (95.6) | 98.2 (92.3) | 99.0 (98.6) |
| Rmerge† | 0.065 (0.397) | 0.067 (0.259) | 0.070 (0.315) | 0.059 (0.224) |
| 〈I/σ(I)〉 | 22.3 (3.5) | 15.0 (3.8) | 13.9 (3.2) | 17.0 (4.8) |
R
merge = 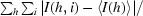
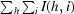 , where I(h, i) is the intensity of the ith observation of reflection h and 〈I(h)〉 is the average intensity of redundant measurements of reflection h.
, where I(h, i) is the intensity of the ith observation of reflection h and 〈I(h)〉 is the average intensity of redundant measurements of reflection h.
3. Results and discussion
3.1. Protein preparation and purification
The enzyme was expressed in large amounts and in soluble form using E. coli BL21(DE3) cells transformed with plasmid containing the target gene and overnight express auto-induction systems. The overexpressed protein was obtained in high yield and to >95% purity using a two-step procedure consisting of Ni-affinity and DEAE columns. The protein was highly soluble and could be concentrated to greater than 12 mg ml−1. The selenomethionine-substituted protein could also be prepared easily using the metE − strain B834(DE3) for overexpression.
3.2. Verification of selenomethionine incorporation
There are seven methionine codons in the argE′ reading frame. MALDI TOF/TOF mass-spectrometric analysis covered 72% of the predicted protein sequence, including five peptides containing methionine residues. The peptide signals with selenomethionine incorporation were identified based on a shift of +47 Da and a typical isotope peak distribution representing the Se atom. The mass spectra of the peptide GSHMTDLLASTLEHLETLVSFDTR detected in the digest of the native and selenomethionine-substituted proteins are shown in Fig. 2 ▶. The yield of selenomethionine incorporation was determined from the spectrum by measuring the ratio of the intensity of the selenomethionine-labelled peptide to that of the remaining unlabelled peptide. This ratio was determined for all the five methionine-containing peptides and indicated that 82% of the total N-acetyl-l-citrulline deacetylase methionine residues were replaced by selenomethionine.
Figure 2.

MALDI–TOF/TOF mass spectroscopy for the N-acetyl-l-citrulline deacetylase-derived peptide GSHMTDLLASTLE-HLETLVSFDTR. (a) Native protein; (b) selenomethionine-substituted protein.
3.3. Enzyme activity
The signals for N-acetyl-l-citrulline and l-citrulline are well separated since they have different retention times and masses. The assay clearly shows that the enzyme is able to catalyze the deacetylation of N-acetyl-lcitrulline, a product of the upstream enzyme N-acetyl-l-ornithine transcarbamylase. This represents the first report of the existence of such an enzyme. Because only a few bacterial species, both plant and human pathogens, have this unique enzyme in the arginine-biosynthesis pathway, this enzyme could be particularly suitable as a target for non-toxic inhibitors.
3.4. Crystallization and structural determination
Thin plate-shaped crystals of maximum dimensions approximately 0.02 × 0.2 × 0.4 mm were obtained for the native and selenomethionine-substituted proteins (Fig. 1 ▶). X-ray data were collected to 1.75 Å resolution using the Brookhaven National Laboratory synchrotron-radiation source (Table 1 ▶). The space group was identified as C2, with unit-cell parameters a = 94.13, b = 95.23, c = 43.61 Å, β = 93.76°. A packing-density calculation (Matthews, 1968 ▶) for the monomer weight of 40 kDa agrees with the presence of one monomer in the asymmetric unit (V M = 2.31 Å3 Da−1, giving a solvent content of 46.3%). If the catalytic unit of the enzyme is a dimer, as are other related proteins such as carboxypeptidase G2 and succinyl diaminopimelate desuccinylase (Rowsell et al., 1997 ▶), the molecular twofold symmetry axis will be coincident with the crystallographic twofold axis.
Molecular replacement was attempted with the program AMoRe (Navaza, 2001 ▶) from the CCP4 crystallographic suite (Collaborative Computational Project, Number 4, 1994 ▶) using structural models of Pseudomonas sp. strain RS-16 carboxypeptidase G2 and Neisseria meningitidis succinyl diaminopimelate desuccinylase (PDB codes http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1cg2 and http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1vgy, respectively) as search models as the sequence of N-acetylcitrulline deacetylase has 33–35% sequence similarity to those of these proteins. No significant solutions were identified.
When the selenomethionine-substituted protein crystals were obtained, diffraction data to 1.75 Å at three wavelengths around the selenium edge, inflection and remote positions were collected. The six selenium positions were easily identified using the program SOLVE (Terwilliger & Berendzen, 1999 ▶). The electron-density map from the programs SOLVE and RESOLVE (Terwilliger, 2000 ▶) was easily interpretable, confirming the presence of one molecule per asymmetric unit. Model building is currently in progress.
Acknowledgments
This work was supported in part by Public Health Service grant DK-47870 (MT) and DK-067935 (DS) from the National Institute of Diabetes, Digestive and Kidney Diseases and HD 32652 from the National Institute of Child Health and Human Development. We thank Dr Michael Doyle and Darren Bykowski for preparing N-acetylcitrulline, Dr David Davies for facilitating the use of the diffraction equipment in the Molecular Structure Section of the National Institutes of Health and Dr Fred Dyda for help in data collection. We also thank Drs Alexei Soares and Stu Myers for their assistance during data collection at beamline X26C and X12B at Brookhaven National Laboratory. This facility is supported by the United States Department of Energy Offices of Health and Environmental Research and of Energy Sciences and by the National Science Foundation.
References
- Bradford, M. (1976). Anal. Biochem.72, 1219–1223. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Cunin, R., Glansdorff, N., Pierard, A. & Stalon, V. (1986). Microbiol. Rev.50, 314–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Miki, T., Kuwahara, T., Nakayama, H., Okada, N., Kataoka, K., Arimochi, H. & Ohnishi, Y. (2005). J. Med. Invest.52, 101–108. [DOI] [PubMed] [Google Scholar]
- Navaza, J. (2001). Acta Cryst. D57, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Rowsell, S., Pauptit, R. A., Tucker, A. D., Melton, R. G., Blow, D. M. & Brick, P. (1997). Structure, 5, 337–347. [DOI] [PubMed] [Google Scholar]
- Sanz, M., Lau, L., Herrera, D., Morillo, J. M. & Silva, A. (2004). J. Clin. Periodontol.31, 1034–1047. [DOI] [PubMed] [Google Scholar]
- Shi, D., Morizono, H., Yu, X., Roth, L., Caldovic, L., Allewell, N. M., Malamy, M. H. & Tuchman, M. (2005). J. Biol. Chem.280, 14366–14369. [DOI] [PubMed] [Google Scholar]
- Shi, D., Morizono, H., Yu, X., Tong, L., Allewell, N. M. & Tuchman, M. (2001). Biochem. J.354, 501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, A. C. da et al. (2002). Nature (London), 417, 459–463. [Google Scholar]
- Silva, F. R. da, Vettore, A. L., Kemper, E. L., Leite, A. & Arruda, P. (2001). FEMS Microbiol. Lett.203, 165–171. [DOI] [PubMed] [Google Scholar]
- Strandholm, J. J., Buist, N. R., Kennaway, N. G. & Curtis, H. T. (1971). Biochem. Biophys. Acta, 244, 214–216. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T. C. (2000). Acta Cryst. D56, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]