

The crystal structure of the 40.8 kDa At2g17340 protein from A. thaliana was determined at 1.7 Å resolution. The structure provides the first insight into the structural organization of the Pfam01937.11 family and establishes that the proteins of this family coordinate a metal in its putative active site.
Keywords: At2g17340 protein, Pfam01937.11 family
Abstract
The crystal structure of the At2g17340 protein from A. thaliana was determined by the multiple-wavelength anomalous diffraction method and was refined to an R factor of 16.9% (R free = 22.1%) at 1.7 Å resolution. At2g17340 is a member of the Pfam01937.11 protein family and its structure provides the first insight into the structural organization of this family. A number of fully and highly conserved residues defined by multiple sequence alignment of members of the Pfam01937.11 family were mapped onto the structure of At2g17340. The fully conserved residues are involved in the coordination of a metal ion and in the stabilization of loops surrounding the metal site. Several additional highly conserved residues also map into the vicinity of the metal-binding site, while others are clearly involved in stabilizing the hydrophobic core of the protein. The structure of At2g17340 represents a new fold in protein conformational space.
1. Introduction
The gene At2g17340 of Arabidopsis thaliana encodes a protein with molecular weight 40.8 kDa (residues 1–367). The function of this protein in A. thaliana is not yet established. At2g17340 is a member of the conserved domain family Pfam01937.11, represented by DUF89 protein, without an experimentally established function (Bateman et al., 2004 ▶). Several established eukaryotic pantothenate kinases contain this conserved domain. At2g17340, as well as other members of the Pfam01937.11 family, shows homology to the carboxy-terminal portion of eukaryotic pantothenate kinases. Pantothenate kinases (EC 2.7.1.33) catalyze the first step in the biosynthetic pathway leading to coenzyme A, an essential prosthetic group of a number of enzymes including the acyl-carrier protein components, citrate lyases and others (Begley et al., 2001 ▶). Interestingly, bacterial and eukaryotic pantothenate kinases share neither a sequence nor a fold-family relationship. Therefore, the bacterial pantothenate kinases have recently become interesting drug targets (Daugherty et al., 2002 ▶). The first inhibitors have already been identified for staphylococcal pantothenate kinase (Gerdes et al., 2002 ▶). Recently, a number of mutations in human pantothenate kinase 2 were linked to the onset of Hallervorden–Spatz syndrome, a neurodegenerative disorder associated with iron accumulation in the basal ganglia (Zhou et al., 2001 ▶). Here, we report the three-dimensional structure of At2g17340 protein at 1.7 Å determined by the multiple-wavelength anomalous dispersion (MAD) method. The structure was determined under the National Institutes of Health NIGMS Protein Structure Initiative.
2. Materials and methods
The A. thaliana gene At2g17340 was cloned and native and selenomethionine (SeMet) labeled At2g17340 proteins were expressed and purified following the standard CESG pipeline protocol for cloning (Thao et al., 2004 ▶), protein expression (Sreenath et al., 2005 ▶), protein purification (Jeon et al., 2005 ▶) and overall information management (Zolnai et al., 2003 ▶). Crystals of At2g17340 were grown by the hanging-drop method from 10 mg ml−1 protein solution in a buffer (50 mM NaCl, 3 mM NaN3, 0.3 mM TCEP, 5 mM Bis-Tris pH 6.0) mixed with an equal amount of well solution containing 22% PEG 8000, 20 mM MgCl2, 100 mM N-(2-hydroxyethyl)piperazine-N′-3-propanesulfonic acid (HEPPS) pH 8.0 at 293 K. Typically, crystals grew as clusters of plates or needles emanating from a nucleation center. Occasionally, crystals with minor morphological defects grew to dimensions of about 200 × 30 × 10 µm. The selenomethionyl crystals of At2g17340 belong to space group P1, with unit-cell parameters a = 40.2, b = 44.0, c = 52.8 Å, α = 74.4, β = 74.7, γ = 84.4°. The native crystals of At2g17340 belong to the same space group, with slightly different unit-cell parameters of a = 40.2, b = 43.3, c = 52.6 Å, α = 74.6, β = 74.6, γ = 84.0°. Both native and selenomethionyl crystals were cryoprotected by soaking in a solution containing 25% PEG 8000, 20 mM MgCl2, 100 mM HEPPS pH 8.0 supplemented with increasing concentrations of glycerol up to a final concentration of 20%. X-ray diffraction data for native and selenomethionyl crystals were collected at SBC 19-BM and BioCARS 14-ID beamlines at Argonne National Laboratory Advanced Photon Source, respectively. The data sets of diffraction images were integrated and scaled using the HKL2000 suite (Otwinowski & Minor, 1997 ▶). The selenium substructure of SeMet-labeled At2g17340 crystals was determined using HySS (Grosse-Kunstleve & Adams, 2003 ▶). The protein structure was phased using three-wavelength MAD data in CNS (Brünger et al., 1998 ▶). Initial phase information obtained from CNS to 2.5 Å was further improved and extended to the 1.7 Å resolution of the native data set by electron-density modification. The automatic tracing procedure of ARP/wARP (Perrakis et al., 1999 ▶) produced an inital model with approximately 82% of residues placed, of which 68% had side chains assigned. The structure was completed using alternate cycles of manual building in Xfit (McRee, 1999 ▶) and refinement in REFMAC5 (Murshudov et al., 1997 ▶). All refinement steps were monitored using an R free value based on 5.0% of the independent reflections. The stereochemical quality of the final model was assessed using PROCHECK (Laskowski et al., 1993 ▶) and MolProbity (Lovell et al., 2003 ▶).
3. Results and discussion
The structure of At2g17340 has been solved to a resolution of 1.7 Å. Data collection, refinement and model statistics are summarized in Tables 1 ▶ and 2 ▶. The final model describes a monomer containing residues 6–165, 181–270 and 273–365. In addition, a magnesium ion and 358 water molecules were built into the final structure.
Table 1. Summary of crystal parameters and data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Native | Se peak | Se edge | Se remote | |
|---|---|---|---|---|
| Space group | P1 | |||
| Unit-cell parameters (Å, °) | a = 40.2, b = 43.3, c = 52.6, α = 74.6, β = 74.6, γ = 84.0 | |||
| Data-collection and phasing statistics | ||||
| Energy (keV) | 12.860 | 12.665 | 12.660 | 12.865 |
| Wavelength (Å) | 0.96411 | 0.97896 | 0.97931 | 0.96374 |
| Resolution range (Å) | 34.91–1.70 (1.73–1.70) | 23.60–2.05 (2.10–2.05) | 23.62–2.05 (2.10–2.05) | 23.63–2.00 (2.10–2.00) |
| No. of reflections (measured/unique) | 152307/35346 | 80026/20249 | 80101/20309 | 84786/21810 |
| Completeness (%) | 97.0 (89.5) | 97.4 (95.2) | 97.4 (94.4) | 97.2 (91.5) |
| Rmerge† | 0.039 (0.277) | 0.044 (0.113) | 0.023 (0.100) | 0.031 (0.138) |
| Redundancy | 4.3 (3.9) | 4.0 (3.8) | 3.9 (3.7) | 3.9 (3.2) |
| Mean I/σ(I) | 23.30 (5.24) | 14.30 (6.41) | 24.45 (7.77) | 17.51 (5.23) |
| Mean FOM of phasing | 0.7084 | |||
R
merge = 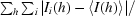
 , where Ii(h) is the intensity of an individual measurement of the reflection and 〈I(h)〉 is the mean intensity of the reflection.
, where Ii(h) is the intensity of an individual measurement of the reflection and 〈I(h)〉 is the mean intensity of the reflection.
Table 2. Summary of refinement statistics.
| Resolution range (Å) | 49.39–1.70 |
| Data set used in refinement | Native |
| No. of reflections (total/test) | 35346/1773 |
| Rcryst† | 0.169 |
| Rfree‡ | 0.221 |
| R.m.s.d. bonds (Å) | 0.017 |
| R.m.s.d. angles (°) | 1.642 |
| Average B factor (Å2) | 21.98 |
| No. of water molecules | 357 |
| No. of ions | 1 Mg2+ |
| Ramachandran plot, residues in | |
| Most favorable region (%) | 90.9 |
| Additional allowed region (%) | 8.8 |
| Generously allowed region(%) | 0.3 |
| Disallowed region (%) | 0.0 |
R
cryst = 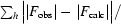
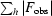 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
R free was calculated as R cryst using 5.0% of the randomly selected unique reflections that were omitted from structure refinement.
The three-dimensional structure of At2g17340 is formed from two domains doubly connected by flexible linkers. The smaller, amino-terminal domain spans residues 37–116 and folds into a three-helix bundle formed by long helices 1, 2 and 4 (see Fig. 1 ▶ b). A short auxiliary helix 3 connects helices 2 and 4 of the bundle. The larger, carboxy-terminal domain spans residues 119–365. The central feature of this domain is a mixed seven-stranded β-sheet formed by five parallel β-strands (H, G, B, C and F) and two antiparallel strands (A and I; see Fig. 1 ▶ a). All of the strands span 5–7 residues, except for strand A, which is shorter. On one side of the central β-sheet is a cluster of six helices (5, 6, 7, 8, 9 and 10) of various sizes that range from two to seven turns. The longest, seven-turn helix 5 is bent about 45° after four turns. A hydrogen-bonding pattern typical of an α-helix is disrupted by Pro136 at that position. A two-stranded β-sheet formed by β-strands D and E connects helix 10 with β-strand F. On the other side of the central β-sheet are three short helices (11, 12 and 13) and a substantial portion of the At2g17340 chain that does not adopt a regular secondary structure. The amino-terminal domain of At2g17340 is anchored to the carboxy-terminal domain by residues 6–25, which do not adopt regular secondary structure. The most important interaction involves the insertion of side chain of Phe10 into a hydrophobic pocket formed by residues Val216, Val315, Phe218 and Phe332.
Figure 1.

(a) A topology diagram of the At2g17340 structure. The central seven-stranded β-sheet (red) is surrounded by an intricate arrangement of helices (cyan), a short two-stranded β-sheet (green) on one side and a polypeptide chain adopting only small segments of regular secondary structure (cyan, bottom) on the other side. A three-helix bundle at the amino-terminal end of the protein forms a separate domain (pink). The figure was generated manually using TopDraw based on a topology analysis of the At2g17340 structure by the TOPS server (Bond, 2003 ▶; Westhead et al., 1999 ▶). (b) A ribbon diagram of the At2g17340 structure. The structure is labeled and colored to match the topology diagram. The yellow sphere represents a putative Mg2+ ion coordinated within the protein. The figure was generated using PyMol (DeLano, 2002 ▶).
To classify the fold of At2g17340, a structural homology search was conducted using the DALI and VAST servers (Holm & Sander, 1993 ▶; Madej et al., 1995 ▶). Both the DALI and VAST servers found a range of weak structural homologs of At2g17340. VAST identified 1181 structural neighbours of At2g17340, while DALI returned 298. The top homolog found by DALI is an aldehyde dehydrogenase from Vibrio harveyi, with Z = 6.2, r.m.s.d. 4.4 Å and 6% sequence identity over 153 aligned Cα residues (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1eyy; Ahvazi et al., 2000 ▶). VAST identified a higher resolution (2.1 Å compared with 2.5 Å; PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1ez0) structure of this protein with a VAST score of 11.6, r.m.s.d. 3.1 Å and 8.7% identity over 93 aligned residues. The top solution from VAST was a leucine carboxy-methyltransferase with a VAST score of 12.3, r.m.s.d. 3.1 Å and 10.5% identity over 114 aligned residues (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1rjd; Leulliot et al., 2004 ▶). The analysis of several top homologs revealed that both prediction servers identified a series of proteins that contain β-sheets with several parallel β-strands in their three-dimensional structures. The best scores were given to proteins that also had a small number of helices on either side of a central β-sheet in a similar location to the search target At2g17340. The four top homologs identified by VAST share among each other a structural similarity with β-strands B, C, F, G and H as well as segments of helices 9 and 11 of At2g17340. The segments from helices 1, 2 and 4 of At2g17340, which form an autonomous three-helix bundle, were spuriously matched in a range of protein structures containing three-helix or four-helix bundles. VAST identified 47 very weak homologs of this domain with VAST scores of 4.0 and less. Overall, structural similarity analysis suggests that the protein fold of At2g17340 is quite unique and not previously represented in the PDB.
As expected, the lack of a family relationship between At2g17340 and Escherichia coli pantothenate kinase was also confirmed by a comparison of the available crystal structure of E. coli pantothenate kinase (PDB codes http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1esm and http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1sq5; Yun et al., 2000 ▶; Ivey et al., 2004 ▶) with that of At2g17340. An analysis of the fold similarity between the two proteins by DALI gave a score of Z = 1.6. Since scores of Z < 2 are not consistent with a significant structural similarity, there is no structural relationship between the two proteins.
Sequence analysis of At2g17340 using the profile–profile fold-recognition tool FFAS03 (Jaroszewski et al., 2000 ▶) confirmed that no previous PDB entry with a family relationship to At2g17340 existed in the PDB at the time of manuscript submission. To identify potential biochemically important structural features of At2g17340, we looked for patterns of evolutionary conservation within its primary structure. Analysis by PSI-BLAST revealed that about 65 hypothetical proteins from a range of organisms share a similarity with the sequence of At2g17340 (with threshold E < 4 × 10−5 after three PSI-BLAST cycles). The identified proteins or some of their conserved domains form a core of the Pfam01937.11 family (Bateman et al., 2004 ▶). A multiple sequence alignment of these proteins reveals that several fully or very highly conserved residues exist in their sequences. These residues are shown in bold blue (fully conserved) and green (highly conserved) letters in the aligned sequences of At2g17340 and At4g32180, the two members of the Pfam01937.11 family from A. thaliana (see Fig. 2 ▶ b).
Figure 2.

(a) Sequence-homology relationship between selected proteins homologous to At2g17340 (green). Three-digit numbers indicate the length of the given protein and the percentage values indicate the sequence identity between aligned length of adjacent pairs of proteins. The profile–profile pairwise sequence alignments were made using FFAS03 (Jaroszewski et al., 2000 ▶). The A. thaliana proteins At2g17320 and At4g35360 (cyan and yellow) show a close homology to At2g17340. Furthermore, At4g32180 (blue) and At2g17340 share 32% identity. At4g32180 is currently annotated as a pantothenate kinase and shares 38% identity with human pantothenate kinase 4 (hPanK4, pink). Also, in its amino-terminal part At4g32180 shows a high level of homology to the A. thaliana protein At1g60440 (red). No family sequence relationship between the amino-terminal and carboxy-terminal parts of At4g32180 was found. All diagrammed proteins except At1g60440 are members of the Pfam01937.11 family, the extent of which is indicated by parentheses. (b) A pairwise profile–profile sequence alignment by FFAS03 of A. thaliana sequences At2g17340 and pantothenate kinase At4g32180. Both At2g17340 and At4g32180 belong to the Pfam01937.11 protein family. The identical residues in both sequences are highlighted in red. The residues in blue and green represent fully or very highly conserved amino acids in the Pfam01937.11 protein family, respectively.
Fig. 3 ▶ shows how fully (blue) and highly conserved (green) residues map onto the structure of At2g17340. All fully conserved residues are located in the vicinity of a metal site that was identified in At2g17340. Because the crystals of At2g17340 grew from a solution containing 20 mM MgCl2, we refined the structure under an assumption that the coordinated metal is Mg2+. (However, we did not pursue further biochemical studies to confirm the identity of the metal.) Residues Asp220, Asn221 and Asp256 act as monodentate ligands of the metal; the coordination sphere is completed by three water molecules (see Fig. 4 ▶). The amide N atom of Asn163 forms a hydrogen bond with the carboxyl oxygen of the metal-coordinating residue Asp220. Interestingly, Gly319, located on the loop adjacent to the metal-binding site, is absolutely conserved. Upon closer inspection of the structure it became clear that any other amino acid at this position would interfere with the positioning of the side chain of the metal-coordinating residue Asp220, thus disrupting coordination and likely resulting in the loss of metal binding. The distance between the Cα atom of residue Gly319 and the carboxyl O atom of Asp220 in the structure of At2g17340 is 3.5 Å. Another fully conserved glycine residue is Gly223. This residue is located at the beginning of an α-helix on the loop that harbors the metal-coordinating Asp220 and Asn221. Residues Asp118 and Lys124 are involved in a conserved salt bridge. Residue Lys124 also forms a salt bridge with residue Glu261. This residue is located near the metal-coordinating residue Asp256. It is possible that Glu261 is involved in the stabilization of a loop conformation required for positioning of the side chain of Asp256. The fully conserved Pro252 may play a similar role in stabilizing this loop. A highly conserved aromatic residue at a position equivalent to Phe120 is observed in Pfam01937.11 proteins. This residue is cushioning Ile254 and may also stabilize the loop that harbors the metal-coordinating Asp256. The last fully conserved residue is Asp166. The residues 166–180 were disordered in the structure of At2g17340 and we could not define the position of Asp166. It is possible that it could form a salt bridge with a conserved Lys343. The disordered loop 166–180 is adjacent to the metal-binding site and it is therefore possible that some of the residues within this loop could be involved in substrate recognition. It is often observed that loops involved in substrate recognition remain unstructured until a substrate is bound (Dyson & Wright, 2005 ▶). Several other conserved residues located in the carboxy-terminal domain of At2g17340 are likely to be involved in stabilizing the hydrophobic core of the protein. These include Leu328 and Leu351, which are surrounded by the hydrophobic residues Ile197, Ile324, Tyr329, Phe350 and Val359, and Val224, which is surrounded by the hydrophobic residues Ala215, Ile217, Phe233, Ala234, Leu237, Leu238, Leu246 and Leu284. Also, residue Val315, which was discussed above, is part of a hydrophobic pocket within the carboxy-terminal domain that anchors the amino-terminus of the At2g17340 via hydrophobic contact with Phe10. In summary, the pattern of primary structure conservation within the Pfam01937.11 family suggests the crucial importance of the metal-binding site for the function of proteins in this family.
Figure 3.

Fully (blue) and highly conserved (green) residues of the Pfam01937.11 family mapped onto a Cα trace of the At2g17340 structure. The color coding of the structure is consistent with that introduced in Fig. 1 ▶. The yellow sphere represents a putative Mg2+ ion coordinated within the protein. The clustering of conserved amino acids around the metal atom and in the cleft between the amino-terminal three-helix cluster (pink) and the carboxy-terminal domain (cyan and red) suggests the location of the active site. The figure was generated using PyMol (DeLano, 2002 ▶).
Figure 4.

A stereo representation of the A. thaliana At2g17340 electron-density map and the final refined model (yellow sticks) in the vicinity of the metal-binding site. The centrally positioned residue Asn221 together with Asp220 and Asp256 act as monodentate ligands for the metal (purple cross); the coordination sphere is completed by three water molecules (red crosses). The figure was generated using PyMol (DeLano, 2002 ▶).
A limited number of proteins identified by the PSI-BLAST search show homology to the amino-terminal domain of At2g17340. The multiple sequence alignment of these proteins reveals several additional highly conserved residues (see Fig. 2 ▶ b). Phe76 is most likely to be involved in stabilizing the core of the three-helix bundle. This residue is surrounded by the hydrophobic residues Leu111, Val110, Tyr80 and Ala56. Also, a highly conserved residue Ala60 is in the close vicinity of Phe76. The very highly conserved Trp45 is located in the hinge region between the amino-terminal and the carboxy-terminal domain of At2g17340. The indole N atom of Trp45 forms a hydrogen bond with a water molecule that is also hydrogen bonded to a carbonyl O atom of Glu346, thus bridging the two domains of At2g17340. Residue Arg72 is likely to be involved in the stabilization of the loop connecting helices 1 and 2 of the three-helix bundle. The guanidinum N atom of this residue forms a hydrogen bond with the carbonyl O atom of Pro67.
The A. thaliana genome codes for several proteins that share sequence similarity with At2g17340 (see Fig. 2 ▶ a). Two of them are highly homologous to At2g17340: At2g17320 is 87% identical and At4g35360 is 86% identical to At2g17340. At2g17340 also shows sequence homology to A. thaliana protein At4g32180. The profile–profile pairwise sequence alignment of these proteins obtained by FFAS03 is presented in Fig. 2 ▶(b). The proteins share 32% identity in the aligned region. At2g17340 is homologous to a carboxy-terminal part of the 901 amino-acid-long At4g32180. At4g32180 is anotated as a pantothenate kinase owing to the protein’s extensive homology to several human pantothenate kinases. For example, At4g32180 shares 38% identity over 862 aligned residues with human pantothenate kinase 4. Based on the similarity to these enzymes, At2g17340 is currently annotated as ‘pantothenate kinase-like’ protein. Interestingly, another protein from A. thaliana, At1g60440, shows 61% identity to an amino-terminal part of At4g32180. Recently, At1g60440 was shown to have pantothenate kinase activity in an in vitro reconstitution of a biosynthetic pathway of coenzyme A in A. thaliana (Kupke et al., 2003 ▶). (The manuscript refers to this protein by the name AtCoaA.) However, no direct family relationship between At2g17340 and At1g60440 was detected by profile–profile sequence alignment using FFAS03. Also, all mutations in human pantothenate kinase 2 that were associated with Hallervorden–Spatz syndrome were located within the amino-terminal portion of the protein (Zhou et al., 2001 ▶). The in vitro reconstitution of coenzyme A biosynthesis from A. thaliana proteins and genetic evidence from humans indicate that it is possible that the pantothenate kinase enzymatic activity resides exclusively in the amino-terminal portion of these proteins. The exact role of the carboxy-terminal portion of longer chain pantothenate kinases and thus members of the Pfam01937.11 family that are homologous to it remains elusive.
In conclusion, the crystal structure of At2g17340 provides the first insight into the Pfam01937.11 family of previously structurally uncharacterized eukaryotic proteins. The pattern of evolutionary conservation within the sequences of the proteins in this family is consistent with the crucial role of the metal-binding site, an area that is likely to be an active site of the protein. Also, the 1.7 Å structure of At2g17340 represents a well defined new fold in the universe of protein conformational space.
Supplementary Material
PDB reference: At2g17340, 1xfi, r1xfisf
Acknowledgments
We acknowledge financial support from NIH National Institute for General Medical Sciences grant P50 GM64598. Use of the Advanced Photon Source and the Argonne National Laboratory Structural Biology Center beamlines at the Advanced Photon Source was supported by the US Department of Energy, Office of Energy Research under contract No. W-31-109-ENG-38. Use of BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources under grant No. RR07707. We would like to thank 19-BM SBC beamline scientists Norma E. C. Duke PhD and Stephan L. Ginell PhD for help we have received during data collection. Special thanks goes to all members of the CESG team including Todd Kimball, John Kunert, Nicholas Dillon, Rachel Schiesher, Juhyung Chin, Megan Riters, Andrew C. Olson, Jason M. Ellefson, Janet E. McCombs, Brendan T. Burns, Blake W. Buchan, Holalkere V. Geetha, Zhaohui Sun, Ip Kei Sam, Eldon L. Ulrich, Nathan S. Rosenberg, Janelle Warrick, Bryan Ramirez, Zsolt Zolnai, Peter T. Lee, Jianhua Zhang, David J. Aceti, Russell L.Wrobel, Ronnie O. Frederick, Hassan Sreenath, Frank C. Vojtik, Won Bae Jeon, Craig S. Newman, John Primm, Michael R. Sussman, Brian G. Fox and John L. Markley.
References
- Ahvazi, B., Coulombe, R., Delarge, M., Vedadi, M., Zhang, L., Meighen, E. & Vrielink, A. (2000). Biochem J.349, 853–861. [PMC free article] [PubMed] [Google Scholar]
- Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E. L., Studholme, D. J., Yeats, C. & Eddy, S. R. (2004). Nucleic Acids Res.32, D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley, T. P., Kinsland, C. & Strauss, E. (2001). Vitam. Horm.61, 157–171. [DOI] [PubMed] [Google Scholar]
- Bond, C. S. (2003). Bioinformatics, 19, 311–312. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Daugherty, M., Polanuyer, B., Farrell, M., Scholle, M., Lykidis, A., de Crecy-Lagard, V. & Osterman, A. (2002). J. Biol. Chem.277, 21431–21439. [DOI] [PubMed] [Google Scholar]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System, http://www.pymol.org. DeLano Scientific, San Carlos, CA, USA.
- Dyson, H. J. & Wright, P. E. (2005). Nature Rev. Mol. Cell Biol.6, 197–208. [DOI] [PubMed]
- Gerdes, S. Y., Scholle, M. D., D’Souza, M., Bernal, A., Baev, M. V., Farrell, M., Kurnasov, O. V., Daugherty, M. D., Mseeh, F., Polanuyer, B. M., Campbell, J. W., Anantha, S., Shatalin, K. Y., Chowdhury, S. A., Fonstein, M. Y. & Osterman, A. L. (2002). J. Bacteriol.184, 4555–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse-Kunstleve, R. W. & Adams, P. D. (2003). Acta Cryst. D59, 1966–1973. [DOI] [PubMed] [Google Scholar]
- Holm, L. & Sander, C. (1993). J. Mol. Biol.233, 123–138. [DOI] [PubMed] [Google Scholar]
- Ivey, R. A., Zhang, Y. M., Virga, K. G., Hevener, K., Lee, R. E., Rock, C. O., Jackowski, S. & Park, H. W. (2004). J. Biol. Chem.279, 35622–35629. [DOI] [PubMed] [Google Scholar]
- Jaroszewski, L., Rychlewski, L. & Godzik, A. (2000). Protein Sci.9, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon, W., Aceti, D. J., Bingman, C., Vojtik, F., Olson, A., Ellefson, J., McCombs, J., Sreenath, H., Blommel, P., Seder, K., Buchan, B., Burns, B., Geetha, H., Harms, A., Sabat, G., Sussman, M., Fox, B. & Phillips, G. (2005). In the press.
- Kupke, T., Hernandez-Acosta, P. & Culianez-Macia, F. A. (2003). J. Biol. Chem.278, 38229–38237. [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., Macarthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291. [Google Scholar]
- Leulliot, N., Quevillon-Cheruel, S., Sorel, I., de La Sierra-Gallay, I. L., Collinet, B., Graille, M., Blondeau, K., Bettache, N., Poupon, A., Janin, J. & van Tilbeurgh, H. (2004). J. Biol. Chem.279, 8351–8358. [DOI] [PubMed] [Google Scholar]
- Lovell, S. C., Davis, I. W., Arendall, W. B. III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2003). Proteins, 50, 437–450. [DOI] [PubMed] [Google Scholar]
- McRee, D. E. (1999). J. Struct. Biol.125, 156–165. [DOI] [PubMed] [Google Scholar]
- Madej, T., Gibrat, J. F. & Bryant, S. H. (1995). Proteins, 23, 356–369. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Perrakis, A., Morris, R. & Lamzin, V. S. (1999). Nature Struct. Biol.6, 458–463. [DOI] [PubMed] [Google Scholar]
- Sreenath, H. K., Bingman, C. A., Buchan, B. W., Seder, K. D., Burns, B. T., Geetha, H. V., Jeon, W. B., Vojtik, F. C., Aceti, D. J., Frederick, R. O., Phillips, G. N. Jr & Fox, B. G. (2005). Protein Expr. Purif.40, 256–267. [DOI] [PubMed] [Google Scholar]
- Thao, S., Zhao, Q., Kimball, T., Steffen, E., Blommel, P. G., Riters, M., Newman, C., Fox, B. & Wrobel, R. (2004). J. Struct. Funct. Genomics, 5, 267–276. [DOI] [PubMed] [Google Scholar]
- Westhead, D. R., Slidel, T. W., Flores, T. P. & Thornton, J. M. (1999). Protein Sci.8, 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun, M., Park, C. G., Kim, J. Y., Rock, C. O., Jackowski, S. & Park, H. W. (2000). J. Biol. Chem.275, 28093–28099. [DOI] [PubMed] [Google Scholar]
- Zhou, B., Westaway, S. K., Levinson, B., Johnson, M. A., Gitschier, J. & Hayflick, S. J. (2001). Nature Genet.28, 345–349. [DOI] [PubMed] [Google Scholar]
- Zolnai, Z., Lee, P. T., Li, J., Chapman, M. R., Newman, C. S., Phillips, G. N. Jr, Rayment, I., Ulrich, E. L., Volkman, B. F. & Markley, J. L. (2003). J. Struct. Funct. Genomics, 4, 11–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: At2g17340, 1xfi, r1xfisf