

The expression, purification, crystallization, and structure determination of NAD-kinase from T. maritima are reported. Similarity to other NAD-kinases as well as homo-oligomrization state of the enzyme from T. maritima are discussed.
Keywords: gi|1564447, NAD, NADP, phosphorylation, kinases
Abstract
NAD kinase is the only known enzyme that catalyzes the formation of NADP, a coenzyme involved in most anabolic reactions and in the antioxidant defense system. Despite its importance, very little is known regarding the mechanism of catalysis and only recently have several NAD kinase structures been deposited in the PDB. Here, an independent investigation of the crystal structure of inorganic polyphosphate/ATP-NAD kinase, PPNK_THEMA, a protein from Thermotoga maritima, is reported at a resolution of 2.3 Å. The crystal structure was solved using single-wavelength anomalous diffraction (SAD) data collected at the Se absorption-peak wavelength in a state in which no cofactors or substrates were bound. It revealed that the 258-amino-acid protein is folded into two distinct domains, similar to recently reported NAD kinases. The N-terminal α/β-domain spans the first 100 amino acids and the last 30 amino acids of the polypeptide and has several topological matches in the PDB, whereas the other domain, which spans the middle 130 residues, adopts a unique β-sandwich architecture and only appreciably matches the recently deposited PDB structures of NAD kinases.
1. Introduction
The designation nicotinamide adenine dinucleotide (NAD) kinase (NAD 2′-phosphotransferase; EC 2.7.1.23) refers to the capacity of an enzyme to catalyze the synthesis of NADP by the phosphorylation of NAD using either inorganic polyphosphates or nucleosidetriphosphates (NTP) as phosphoryl donors (McGuinness & Butler, 1985 ▶). A schematic diagram of this reaction is shown in Fig. 1 ▶.
Figure 1.

Schematic diagram of the reaction catalyzed by NAD kinases.
According to Pfam 16.0 (Bateman et al., 2004 ▶), there are approximately 250 members of this family of enzymes (accession No. PF01513). While some of the enzymes catalyze the phosphorylation of NAD, others belong to a subfamily of kinases that catalyze the phosphorylation of NADH (EC 2.7.1.86). Although not divided into different subfamilies, some of the members utilize only NTP as a phosphoryl group source, whereas others are also able to use polyphosphates. At our present level of understanding, the only source of NADP and/or NADPH in all living organisms is the abovementioned phosphorylation reaction catalyzed by NAD/NADH kinases. Unless otherwise stated, we will refer to the enzymes of this family as NAD kinases.
The enzyme activity associated with NAD kinase was first identified by observing NADP formation in yeast homogenates (Vestin, 1937 ▶; Von Euler & Adler, 1938 ▶). Using the genetic footprinting technique, it has been shown that the NAD kinase in Escherichia coli is required for viability and is thus considered essential (Gerdes et al., 2002 ▶). The indispensability of this enzyme makes it an attractive drug target for the design of new defenses against antibiotic-resistant pathogens if the critical differences between the human and bacterial enzymes can be capitalized upon.
In prokaryotic cells, NAD kinase activity has only been detected in the cytoplasm, while in eukaryotic cells the subcellular distribution varies (McGuinness & Butler, 1985 ▶). There is general agreement that the optimum pH for the NAD kinase reaction occurs in the range 7.0–7.5, although noticeably different values have been reported for the cytoplasmic (pH 9.4) and mitochondrial (pH 8.3) NAD kinases of Saccaromyces cerevisiae. The enzyme is remarkably stabilized by the addition of NAD (K m ≃ 1.9 nM) and this effect is enhanced when 2-mercaptoethanol is added together with NAD (Chung, 1967 ▶, 1968 ▶, 1971 ▶). Although activities measured on NAD kinases from different organisms and/or different subcellular distribution show different NTP specificity, in general the NAD kinases appear to react with all NTPs with different rates (K m ≃ 2.0 mM for ATP). Imidazole-, sulfate- and phosphate-containing buffers are known to inhibit all types of NAD kinases (McGuinness & Butler, 1985 ▶). Irrespective of its origin, the enzyme has an absolute requirement for Mg2+ (10−2 M) as stated in the review by McGuinness & Butler (1985 ▶). The most recent extensive study of divalent cation interactions in NAD kinase was performed on an enzyme from Sphingomonas sp. A1 (Ochiai et al., 2004 ▶), in which it was shown that this specific enzyme exhibited approximately a threefold higher activity in the presence of Mn2+ ions compared with the same concentration of Mg2+ ions and a twofold higher activity with Zn2+ ions. The activities in the presence of Ca2+ and Co2+ ions were 46 and 31%, respectively, compared with Mg2+ ions (100%). Significant thermostability is another property of all NAD kinases (Apps, 1968 ▶; Nemchinskaia et al., 1966 ▶). The partially purified enzymes even from non-hyperthermophilic organisms were able to maintain activity after 30 min exposure to 338–341 K, although the purified enzyme is somewhat less thermostable.
All known NAD kinases, regardless of size, share motifs that are particularly rich in glycine residues. Residues 49–52 (the numbering corresponds to that of PPNK_THEMA) constitute the first invariant motif GGDG. The region 69–76 is conserved as GXXXGXXG. Residue number 119 is a conserved asparagine. This is followed by the motif GXXXXXXXGST in the region 150–160. Position 168 again is occupied by a conserved glycine residue, position 173 is a conserved proline and position 214 is a conserved aspartic acid. Sequence alignment of seven members of the NAD kinase family, including enzymes from Mycobacterium tuberculosis and Archaeoglobus fulgidus, is shown in Fig. 2 ▶.
Figure 2.

Sequence alignment of seven bacterial NAD kinases with the secondary-structure elements of PPNK_THEMA indicated above the sequences. The residues with red background are characteristic of this family of proteins and are conserved throughout the family. Secondary-structure assignment C stands for coil and T stands for turn. Residue numbers above the secondary-structure elements correspond to PPNK_THEMA. The alignment was prepared using CLUSTALW (Thompson et al., 1994 ▶).
Furthermore, it has been shown that the invariant motif GGDG is a common signature in diacylglyceride (EC 2.7.1.107), sphingosine (EC 2.7.1.91), NAD and 6-phosphofructose kinases (EC 2.7.1.11; Labesse et al., 2002 ▶). As a result, it was suggested (Labesse et al., 2002 ▶) that they all possess a similar phosphate donor-binding site and probably a similar phosphorylation mechanism.
As a step towards understanding the mechanism of catalysis by NAD kinases, we have overexpressed in E. coli, crystallized and solved the crystal structure of PPNK_THEMA, an NAD kinase from Thermotoga maritima. The crystal structure was solved by single-wavelength anomalous diffraction (SAD) method using data collected at the Se absorption-peak wavelength and refined to a resolution of 2.3 Å with no cofactors or substrate bound to the enzyme. The results of this structural analysis are reported here.
2. Materials and methods
2.1. Cloning, expression and purification
The DNA encoding PPNK_THEMA was amplified by PCR from T. maritima genomic DNA (American Type Culture Collection) using Deep Vent DNA Polymerase (New England Biolabs, Beverly, MA, USA). The resulting PCR product was purified and prepared for ligation-independent cloning (LIC; Aslanidis & de Jong, 1990 ▶) by treatment with T4 DNA polymerase in the presence of 1 mM dTTP for 30 min at 310 K. The prepared DNA was then mixed with a pB4 vector for 5 min at room temperature and transformed into DH5α. The LIC pB4 vector was designed in our laboratory to express the target protein together with an N-terminal His6-tagged maltose-binding protein fusion containing a tobacco etch virus (TEV) protease cleavage site. The TEV cleavage produces target protein with six glycine residues at the N-terminus. The resulting plasmid was transformed into BL21 (DE3)/pSJS1244 for protein expression (Kim et al., 1998 ▶).
Selenomethionine-labeled protein was expressed in a methionine auxotroph, E. coli strain B834 (DE3)/pSJS1244 (Leahy et al., 1992 ▶), using an auto-inducible selenomethionyl-containing medium (W. Studier, Brookhaven National Laboratory, personal communication). Cells were disrupted using a microfluidizer (Microfluidics, Newton, MA, USA) in 50 mM HEPES pH 7.0, 500 mM NaCl, 5% glycerol, 1 mM PMSF, 10 µg ml−1 DNAse, 0.1 µg ml−1 antipain, 1 µg ml−1 chymostatin, 0.5 µg ml−1 leupeptin and 0.7 µg ml−1 pepstatin A. The supernatant was then spun in a Beckman ultracentrifuge in a Ti-45 rotor at 35 000 rev min−1 for 30 min at 277 K and applied onto a HiTrap Ni2+-chelating column (GE Healthcare, Piscataway, NJ, USA). His-tagged fusion protein was bound to the column in 50 mM HEPES pH 7.0, 500 mM NaCl and 5% glycerol and was eluted with the same buffer supplemented with 300 mM imidazole. Fractions containing the protein were pooled, mixed with TEV and dialyzed overnight at 277 K against 50 mM HEPES pH 7.0, 500 mM NaCl and 5% glycerol. After centrifugation, the supernatant was applied onto a 5 ml HiTrap Ni2+ chelating column. The cleaved target protein was found in the flowthrough. To assess the homo-oligomerization state of the protein, analytical size-exclusion chromatography was performed using a G4000 SWXL (Tosoh Biosciences, Japan) column. The column was equilibrated with 20 mM Tris–HCl pH 7.5, 1 mM EDTA, 100 mM NaCl. The purity and identity of the target protein was confirmed by SDS–PAGE and MALDI–TOF mass spectrometry. Dynamic light-scattering (DynaPro 99, Wyatt Technology, Santa Barbara, CA, USA) experiments were performed in the concentration range 0.1–1.8 mg ml−1. While the values for the hydrodynamic radius were in the range 30–40 Å with no correlation with sample concentration, the values for polydispersity change with respect to protein concentration and were 9.3% at low concentration and 26.5% at high concentration. At low protein concentration a single monodisperse peak was detected, indicating homogeneity of the protein.
The protein was concentrated to 10 mg ml−1 in 20 mM Tris pH 7.5, 300 mM NaCl and 1 mM DTT buffer for crystallization.
2.2. Crystallization and structure determination
Screening for crystallization conditions was performed using the sparse-matrix method (Jancarik & Kim, 1991 ▶) with several screens from Hampton Research (Hampton Research, Aliso Viejo, CA, USA). Crystals of SeMet-containing PPNK_THEMA were obtained at room temperature in hanging drops. Crystals first appeared in 0.1 M MES pH 6.5, 1.8 M (NH4)2SO4. Optimized diffraction-quality crystals were grown in 0.1 M MES pH 6.5, 1.8 M (NH4)2SO4, 10 mM cobalt(II) chloride and 0.3 M non-detergent sulfobetaine 195 to dimensions of 0.30 × 0.20 × 0.15 mm. They belong to space group P21212, with unit-cell parameters a = 131.45, b = 137.15, c = 58.25 Å. The asymmetric unit contains four monomers. X-ray diffraction data were collected from single crystals at 100 K at Se peak wavelength on Berkeley Center for Structural Biology beamline 8.2.2 at the Advanced Light Source (Lawrence Berkeley National Laboratory, Berkeley, CA, USA) equipped with a Quantum 315 CCD detector (Area Detector System Corporation, Poway, CA, USA). All data were processed with HKL2000 (Otwinowski & Minor, 1997 ▶). A total of 520 images were collected (260 images each for direct and inverse beam). To collect Friedel pairs close in time, the crystal was rotated by 180° after every 10° of data collection. Each image was recorded for 0.5° oscillation range and 3 s exposure time.
The structure was solved by the SAD method. The heavy-atom substructure was determined using the HySS program (Grosse-Kunstleve & Adams, 2003 ▶) from the PHENIX package (Adams et al., 2002 ▶) at a resolution of 3.0 Å. All 20 Se atoms expected in the asymmetric unit were found. The initial phases were calculated with SOLVE (Terwilliger, 2004 ▶) at 2.3 Å resolution. Statistical density modification was applied using RESOLVE (Terwilliger, 2004 ▶). The resulting electron density was interpretable for 100% of the polypeptide chain in two monomers of four (Fig. 3 ▶). For the other two monomers 75% of the polypeptide chain had interpretable electron density. The model was built for one of the four monomers using O (Jones et al., 1991 ▶). The other three monomers were placed into corresponding densities using MOLREP (Vagin & Teplyakov, 1997 ▶) in ‘slow’ mode (advanced rotation and translation function with rigid-body refinement) from the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶). At this point in structure determination approximately 70 amino acids were excluded from each of those two monomers that did not have fully traceable electron density. They were added into the model upon phase improvement during refinement when non-crystallographic symmetry restraints were released. The refinement was carried out using REFMAC (Murshudov et al., 1997 ▶) to an R factor of 0.21 (R free = 0.28 for 5% of data) for all data in the resolution range 15–2.3 Å. The final electron density featured all the protein residues except for the last two for monomers A and B. For monomers C and D the first 50 residues were placed in a somewhat less defined and noisy electron density. This may partially account for the slightly high R factors. Of 138 ‘solvent’ molecules, 18 were modeled as sulfate ions (probably from ammonium sulfate used as precipitant) based on the tetrahedral shape of the electron density and the basic properties of the protein counterpart they are bound to. The resulting model had 92.4% of the residues in the most favored regions and 7.6% in additionally allowed regions of the ϕ–ψ plot as evaluated with PROCHECK (Laskowski et al., 1993 ▶). The X-ray data and refinement statistics are given in Table 1 ▶.
Figure 3.

The experimental electron-density map of a portion of the refined model of the CTD is shown. The entrance to the open end of the β-sandwich with some of the hydrophobic residues facing inside is depicted. The contour level corresponds to 2σ. Illustration prepared using BOBSCRIPT (Esnouf, 1997 ▶, 1999 ▶).
Table 1. X-ray diffraction data and refinement statistics.
Values in parentheses are for the outermost resolution shell.
| Wavelength (Å) | 0.9794 |
| Resolution (Å) | 15–2.3 (2.37–2.30) |
| Space group | P21212 |
| Unit-cell parameters (Å) | a = 131.45, b = 137.15, c = 58.25 |
| Redundancy | 9.1 (6.7) |
| Unique reflections | 48553 (4872) |
| Completeness (%) | 96.7 (90.1) |
| 〈I/σ(I)〉 | 20.3 (4.7) |
| Rsym† | 0.091 (0.330) |
| Rcryst‡ | 0.21 (0.23) |
| Rfree (5% data) | 0.28 (0.30) |
| No. of protein atoms per AU | 8,147 |
| R.m.s.d. bonds (Å) | 0.016 |
| R.m.s.d. angles (°) | 1.459 |
| Average B (all atoms) (Å2) | 27.6 |
| Solvent molecules (sulfate/water) | 17/120 |
| Ramachandran plot statistics | |
| Favored regions (%) | 92.4 |
| Additional allowed regions (%) | 7.6 |
R
sym = 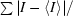
 .
.
R
cryst = 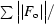
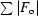 .
.
3. Results and discussion
3.1. Structure overview
The crystal structure shows that the monomer of PPNK_THEMA has overall dimensions of approximately 60 × 45 × 35 Å. It can be divided into two similar-sized structural domains with a total of 14 β-strands and five α-helices (Fig. 4 ▶ a).
Figure 4.

A stereoview of the ribbon diagram of PPNK_THEMA monomer is presented in (a). The secondary-structure elements are numbered according to the text. The NTD (b) and CTD (c) are shown separately. Illustration prepared using MOLSCRIPT (Kraulis, 1991 ▶) and RASTER3D (Merritt & Bacon, 1997 ▶).
The N-terminal domain (NTD) has the structure of a nucleotide-binding domain (also called α/β-hydrolase or a Rossmann fold) and spans residues 1–102 and from residue 233 to the end of polypeptide chain. The core of this domain consists of a twisted six-stranded β-sheet flanked by four α-helices and two long loops (residues 34–43 and 71–82). Strands β1 (residues 1–8), β2 (residues 26–34), β3 (residues 44–50), β4 (residues 65–71) and β14 (residues 222–239) are parallel, while the outermost strand β5 (first half, residues 98–105) is antiparallel (Fig. 4 ▶ b).
The structure of the C-terminal domain (CTD) of PPNK_THEMA consists predominantly of β-strands that form two antiparallel β-sheets of five and six strands each, which in turn build a β-sandwich (Fig. 4 ▶ c). The six-stranded sheet is composed of strands β5 (second half, residues 106–110), β6 (residues 113–118), β8 (residues 131–139), β9 (first half, 143–148), β12 (first half, 200–207) and β13, while the five-stranded sheet is made up of strands β7 (residues 120–125), the second half of β9 (residues 149–154), β10 (residues 176–185), β11 (residues 192–197) and the second half of β12 (residues 208–214). The only helical region, a three-turn distorted α-helix (residues 155–167), is positioned on the outer side of the sandwich behind the five-stranded β-sheet. The inside of the β-sandwich has the shape of an elliptic cylinder and is composed only of hydrophobic residues. One end of that cylinder is open to solvent, but the other side is capped with His115 and three sulfate ions probably from the ammonium sulfate used as precipitant.
The connecting region between the NTD and CTD is composed of a portion of the β5 strand and a loop between strands β13 and β14. Strand β5 participates in both the twisted β-sheet of the NTD and the six-stranded wall of β-sandwich of the CTD.
Given the examples above, the CTD of PPNK_THEMA would be expected to be the nucleotide-binding domain. A homology search for the CTD resulted in only 12 structurally similar proteins, with Z-score values between 20.9 and 2.1. The two known NAD kinases account for the highest scores of 20.9 and 20.7, with an r.m.s.d. of 1.6 Å over 124 residues. The next highest Z score, 3.5, with an r.m.s.d. of 3.5 Å over 67 residues is found with NADP-dependent isocitrate dehydrogenase (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1itw; Yasutake et al., 2002 ▶). A better r.m.s.d. value of 2.5 Å over 64 residues was obtained for tumor repressor protein Smad4 (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1ygs; Z score 2.2; Shi et al., 1997 ▶). The superimposed Cα traces of CTD, isocitrate dehydrogenase and tumor repressor protein Smad4 are shown in Fig. 6 ▶(b). Although the overall shape of the respective fold analogs is a β-sandwich, the topology is quite different. This is in part reflected by the low Z-score values. None of the 12 found proteins are known to bind NTPs.
Figure 6.

Superimposed structures of PPNK_THEMA in gold, M. tuberculosis NAD kinase in cyan (Garavaglia et al., 2004 ▶; Mori et al., 2005 ▶) and A. fulgidus NAD kinase in green (J. Liu, Y. Lou, H. Yokota, P. D. Adams, R. Kim and S.-H. Kim, unpublished work) show great similarity. In contrast to the structure from M. tuberculosis, where the N- and C-terminal domains of monomers in the asymmetric unit deviate from symmetry differently, the PPNK_THEMA does not exhibit any interdomain motion as calculated using DYNDOM from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
3.2. Homo-oligomerization state
A number of studies on NAD kinases from different organisms, dating from 1970 to 2005, have reported different oligomeric states for active enzymes, ranging from dimeric to octameric states (Apps, 1975 ▶; Nemchinskaia et al., 1970 ▶; Kawai et al., 2000 ▶; Mori et al., 2005 ▶; Raffaelli et al., 2004 ▶).
According to the results of size-exclusion chromatography (results not shown), the majority (about 90%) of PPNK_THEMA in solution at aconcentration of about 0.25 mg ml−1 is in a dimeric state. However, the asymmetric unit of the PPNK_THEMA crystal contains four monomers (Fig. 5 ▶ a). These four monomers create six different contacts, which can be then grouped into three types according to the size of the contact area. The most extensive interaction, as judged by the size of the contact area, is created by pairs AD and BC and is about 925 Å2. Somewhat less extensive contacts are created for AB and CD and are about 480 Å2. The AC as well as BD contacts represent the weakest interaction and account for about 50 Å2 of the surface area. Based on the difference in the contact areas as well as the result of the gel-filtration experiment, we propose that if the biological unit of PPNK_THEMA is indeed a dimer, then it is likely to be represented by the AD dimer (Fig. 5 ▶ b).
Figure 5.

(a) The four monomers in the asymmetric unit can be described using local 222 symmetry. The r.m.s.d. values for the coordinates of all atoms vary from 0.88 Å for pair BD to 1.3 Å for pair AD. Imperfect dyads range from 177.7° for pair AD to 180.1° for pair AB. (b) The result of size-exclusion chromatography and the difference in the size of the contact area allowed us to hypothesize discrimination between pairs AB and AD in favor of AD as a dimer in solution. In addition, the crystal structure of NAD kinase from A. fulgidus shows the presence of phosphorylation reaction product, NADP, bound between monomers A and D.
This conclusion contradicts that made on the basis of the crystal structure of the NAD kinase from M. tuberculosis (Garavaglia et al., 2004 ▶), where the authors also detected a dimeric form and decided in favor of AB contacts based only on the presence of these two monomers in the asymmetric unit.
However, all crystal structures of this family show homotetrameric organization. It is possible that the molecule may assume more than one oligomeric state during the various stages of its functional process.
3.3. Structural similarity to NAD kinases
Recently, the coordinates of two NAD kinases from M. tuberculosis (PDB codes http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1u0r, http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1u0t, http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1y3h, http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1y3i; Garavaglia et al., 2004 ▶; Mori et al., 2005 ▶) and A. fulgidus (PDB codes http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1suw, http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1z0s; J. Liu, Y. Lou, H. Yokota, P. D. Adams, R. Kim and S.-H. Kim, unpublished work) have been deposited in the PDB. A homology search for full-length PPNK_THEMA using DALI resulted only in these two proteins, with Z-score values as high as 28.2. The enzyme from M. tuberculosis has an extra 20 residues at the N-terminus which are probably partially unfolded and not present in the crystal structure. Also, several additional residues at the C-terminus of the M. tuberculosis enzyme fold in such a way that they interact with one of the sides of the β-sandwich in the neighboring monomer. This may contribute to a difference in oligomerization state between the T. maritima protein and the M. tuberculosis protein, for which a tetrameric state was detected in solution. Otherwise, the rest of the M. tuberculosis NAD kinase structure as well as that from A. fulgidus show a great degree of similarity to PPNK_THEMA (Fig. 6 ▶).
The NAD kinase crystal structures from M. tuberculosis and A. fulgidus have an endogenous NADP and/or NAD bound in the cleft between the NTD and CTD which can also be described as the contact area between monomers A and D. The NADP molecule creates some 124 contacts with monomers A and D, including six contacts with solvent molecules ranging between 2.6 and 3.8 Å. All the residues whose side chains interact with NADP are identical in the A. fulgidus and T. maritima enzyme, except for two conservative substitutions (Arg to Lys and Glu to Asp) and Gln211 of the A. fulgidus enzyme, which does not have an analog in PPNK_THEMA. Furthermore, PPNK_THEMA has a bound sulfate ion which mimics the presence of the newly attached phosphate group of NADP at almost exactly the same position when the structures of these two enzymes are superimposed using DALI-derived matrices. The result of this comparison allows us to hypothesize that the NADP molecule will probably bind to PPNK_THEMA in same way as found in the crystal structure of the enzyme from A. fulgidus. Two possible reasons for NAD or NADP not being found in the PPNK_THEMA may be that (i) PPNK_THEMA was exposed for some period of time to high concentrations of imidazole and (ii) the crystallization condition that yielded crystals contained a high concentration of ammonium sulfate. Both imidazole and sulfate ions are known inhibitors of NAD kinases.
Supplementary Material
PDB reference: NAD kinase, 1yt5, r1yt5sf
Acknowledgments
The authors thank Dr John-Marc Chandonia for the bioinformatics search, Barbara Gold for cloning, Marlene Henriquez and Bruno Martinez for expression and cell-paste preparation, Dr Alexander Iakounine of the University of Toronto for functional assays and staff members for user support at beamline 8.2.2 at the Advanced Light Source. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences and Material Sciences Division of the US Department of Energy under contract No. DE-AC03-76SF00098 at Lawrence Berkeley National Laboratory. The research presented here was supported by a Protein Structure Initiative grant from the National Institutes of Health (grant No. 62412).
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L. W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Apps, D. K. (1968). Eur. J. Biochem.5, 444–450. [DOI] [PubMed] [Google Scholar]
- Apps, D. K. (1975). Eur. J. Biochem.55, 475–483. [DOI] [PubMed] [Google Scholar]
- Aslanidis, C. & de Jong, P. J. (1990). Nucleic Acids Res.18, 6069–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E. L., Studholme, D. J., Yeats, C. & Eddy, S. R. (2004). Nucleic Acids Res.32, D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, A. E. (1967). J. Biol. Chem.242, 1182–1186. [PubMed] [Google Scholar]
- Chung, A. E. (1968). Biochim. Biophys. Acta, 159, 490–495. [DOI] [PubMed] [Google Scholar]
- Chung, A. E. (1971). Methods Enzymol.17, 149–156.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Esnouf, R. M. (1997). J. Mol. Graph. Model.15, 132–134. [DOI] [PubMed] [Google Scholar]
- Esnouf, R. M. (1999). Acta Cryst. D55, 938–940. [DOI] [PubMed] [Google Scholar]
- Garavaglia, S., Raffaelli, N., Finaurini, L., Magni, G. & Rizzi, M. (2004). J. Biol. Chem.279, 40980–40986. [DOI] [PubMed] [Google Scholar]
- Gerdes, S. Y., Scholle, M. D., D’Souza, M., Bernal, A., Baev, M. V., Farrell, M., Kurnasov, O. V., Daugherty, M. D., Mseeh, F., Polanuyer, B. M., Campbell, J. W., Anantha, S., Shatalin, K. Y., Chowdhury, S. A., Fonstein, M. Y. & Osterman, A. L. (2002). J. Bacteriol.184, 4555–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse-Kunstleve, R. W. & Adams, P. D. (2003). Acta Cryst. D59, 1966–1973. [DOI] [PubMed] [Google Scholar]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kawai, S., Mori, S., Mukai, T., Suzuki, S., Yamada, T., Hashimoto, W. & Murata, K. (2000). Biochem. Biophys. Res. Commun.276, 57–63. [DOI] [PubMed] [Google Scholar]
- Kim, R., Sandler, S. J., Goldman, S., Yokota, H., Clark, A. J. & Kim, S.-H. (1998). Biotechnol. Lett.20, 207–210.
- Kraulis, P. J. (1991). J. Appl. Cryst.24, 946–950. [Google Scholar]
- Labesse, G., Douguet, D., Assairi, L. & Gilles, A. M. (2002). Trends Biochem. Sci.27, 273–275. [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., Macarthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291. [Google Scholar]
- Leahy, D. J., Hendrickson, W. A., Aukhil, I. & Erickson, H. P. (1992). Science, 258, 987–991. [DOI] [PubMed] [Google Scholar]
- McGuinness, E. T. & Butler, J. R. (1985). Int. J. Biochem.17, 1–11. [DOI] [PubMed] [Google Scholar]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol.277, 505–524. [DOI] [PubMed]
- Mori, S., Yamasaki, M., Maruyama, Y., Momma, K., Kawai, S., Hashimoto, W., Mikami, B. & Murata, K. (2005). Biochem. Biophys. Res. Commun.327, 500–508. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Nemchinskaia, V. L., Bozhkov, V. M. & Kushner, V. P. (1970). Biokhimiia, 35, 1067–1072. [PubMed] [Google Scholar]
- Nemchinskaia, V. L., Kushner, V. P., Bozhkov, V. M., Turchenko, E. I. & Tukachinskii, S. E. (1966). Biokhimiia, 31, 306–314. [PubMed] [Google Scholar]
- Ochiai, A., Mori, S., Kawai, S. & Murata, K. (2004). Protein Expr. Purif.36, 124–130. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Raffaelli, N., Finaurini, L., Mazzola, F., Pucci, L., Sorci, L., Amici, A. & Magni, G. (2004). Biochemistry, 43, 7610–7617. [DOI] [PubMed] [Google Scholar]
- Shi, Y., Hata, A., Lo, R. S., Massague, J. & Pavletich, N. P. (1997). Nature (London), 388, 87–93. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T. (2004). J. Synchrotron Rad.11, 49–52. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res.22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025. [Google Scholar]
- Vestin, R. (1937). Naturwissenschaften, 25, 667–668.
- Von Euler, H. & Adler, E. (1938). Z. Physiol. Chem.252, 41–48.
- Yasutake, Y., Watanabe, S., Yao, M., Takada, Y., Fukunaga, N. & Tanaka, I. (2002). Structure, 10, 1637–1648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: NAD kinase, 1yt5, r1yt5sf