

Abstract
Integrins are cell adhesion molecules that mediate cell-cell, cell-extracellular matrix, and cell-pathogen interactions. They play critical roles for the immune system in leukocyte trafficking and migration, immunological synapse formation, costimulation, and phagocytosis. Integrin adhesiveness can be dynamically regulated through a process termed inside-out signaling. In addition, ligand binding transduces signals from the extracellular domain to the cytoplasm in the classical outside-in direction. Recent structural, biochemical, and biophysical studies have greatly advanced our understanding of the mechanisms of integrin bidirectional signaling across the plasma membrane. Large-scale reorientations of the ectodomain of up to 200 Å couple to conformational change in ligand-binding sites and are linked to changes in α and β subunit transmembrane domain association. In this review, we focus on integrin structure as it relates to affinity modulation, ligand binding, outside-in signaling, and cell surface distribution dynamics.
Keywords: cell adhesion, conformational change, ICAM-1, I domain, migration
Introduction
The immune system relies heavily on integrins for (a) adhesion during leukocyte trafficking from the bloodstream, migration within tissues, immune synapse formation, and phagocytosis; and (b) signaling during costimulation and cell polarization. Integrins are so named because they integrate the extracellular and intracellular environments by binding to ligands outside the cell and cytoskeletal components and signaling molecules inside the cell. Integrins are noncovalently associated heterodimeric cell surface adhesion molecules. In vertebrates, 18 α subunits and 8 β subunits form 24 known αβ pairs (Figure 1). This diversity in subunit composition contributes to diversity in ligand recognition, binding to cytoskeletal components and coupling to downstream signaling pathways. Immune cells express at least 10 members of the integrin family belonging to the β2, β7, and β1 subfamilies (Table 1). The β2 and β7 integrins are exclusively expressed on leukocytes, whereas the β1 integrins are expressed on a wide variety of cells throughout the body. Distribution and ligand-binding properties of the integrins on leukocytes are summarized in Table 1. For reviews, see References 1 and 2. Mutations that block expression of the β2 integrin subfamily lead to leukocyte adhesion deficiency, a disease associated with severe immunodeficiency (3).
Figure 1.
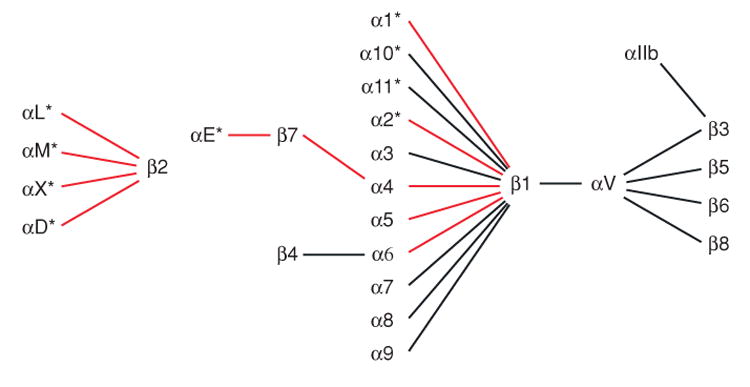
The 24 integrin heterodimers. The α subunits with α I domains are asterisked. Integrin heterodimers on immune cells are shown with red lines.
Table 1.
Integrins on leukocytesa
| Integrin | Distributionb | Ligandc |
|---|---|---|
| αLβ2, LFA-1, CD11a/CD18 | Lymphocytes, NK cells, monocytes, macrophages, dendritic cells, neutrophils | ICAM-1, -2, -3, -5 |
| αMβ2, Mac-1, CR3, CD11b/CD18 | Monocytes, macrophages, neutrophils, NK cells | iC3b, fibrinogen, heparin, many others |
| αXβ2, p150,95, CR4, CD11c/CD18 | Monocytes, macrophages, NK cells, dendritic cells | iC3b, fibrinogen, heparin, many others |
| αDβ2 | Monocytes, macrophages, eosinophils, neutrophils | ICAM-3, VCAM-1 |
| α4β1, VLA-4, CD49d/CD29 | Lymphocytes, monocytes, eosinophils | VCAM-1, fibronectin |
| α4β7, LPAM-1 | Lymphocytes, monocytes, NK cells | MAdCAM-1, fibronectin |
| αEβ7, HML-1 | Intra-epithelial T lymphocytes | E-cadherin |
| α1β1, VLA-1, CD49a/CD29 | Long-term activated T lymphocytes, B lymphocytes, monocytes | Collagen |
| α2β1, VLA-2, GPIa, CD49b/CD29 | long-term activated T lymphocytes, B lymphocytes, monocytes | Collagen |
| α5β1, VLA-5, CD49e/CD29 | T lymphocytes, monocytes | Fibronectin |
| α6β1, VLA-6, GPIc, CD49f/CD29 | T lymphocytes, monocytes | Laminin |
As adhesion molecules, integrins are unique in that their adhesiveness can be dynamically regulated through a process termed inside-out signaling or priming. Thus, stimuli received by cell surface receptors for chemokines, cytokines, and foreign antigens initiate intracellular signals that impinge on integrin cytoplasmic domains and alter adhesiveness for extracellular ligands. In addition, ligand binding transduces signals from the extracellular domain to the cytoplasm in the classical outside-in direction (outside-in signaling). These dynamic properties of integrins are central to their proper function in the immune system. Indeed, mutations or small molecules that stabilize either the inactive state or the active adhesive state—and thereby block the adhesive dynamics of leukocyte integrins—inhibit leukocyte migration and normal immune responses.
Integrin α I Domains
Half of integrin α subunits contain a domain of about 200 amino acids known as an inserted (I) domain, or a von Willebrand factor A domain (Figure 1). In integrins in which it is present, the α I domain is the major or exclusive ligand-binding site. In this review, we begin with the α I domain because it serves as a paradigm for understanding conformational regulation and ligand binding for all integrins. Subsequently, we describe the complex ectodomain architecture shared by all integrins, including 12 different domains, one of which in the β subunit is homologous to the α I domain.
α I Domain Structure
The α I domain can be expressed independently of other integrin domains and was the first domain to be crystallized (4). Several structures of α I domains bound to ligands are now available, including the α2 I domain bound to a triple-helical collagen peptide (5) and αL I domains with mutationally introduced disulfide bonds bound to intercellular adhesion molecule (ICAM)-1 and ICAM-3 (6, 7) (Figure 2). The α I domain adopts the dinucleotide-binding or Rossmann fold, with α-helices surrounding a central β-sheet (Figure 2). β-strands and α-helices tend to alternate in the secondary structure, with the α-helices wrapping around the domain in counterclockwise order when viewed from the “top” face. A divalent cation-binding site, which physiologically binds Mg2+, defines the top face of the domain. The bound Mg2+ is ligated by five side chains located in three different loops (Figure 3). The first of these loops, between β-strand 1 and α-helix 1, i.e., the β1-α1 loop, contains three coordinating residues in a sequence that is a signature of I domains, Asp-Xaa-Ser-Xaa-Ser or DXSXS. The second loop donates a coordinating Thr residue, and the third loop donates an Asp. Divalent cations are universally required for ligand binding by integrins, and in α I domains the metal-coordinating residues, and the residues surrounding the metal-binding site, are important for ligand binding. Therefore, this site has been designated the metal ion-dependent adhesion site (MIDAS).
Figure 2.
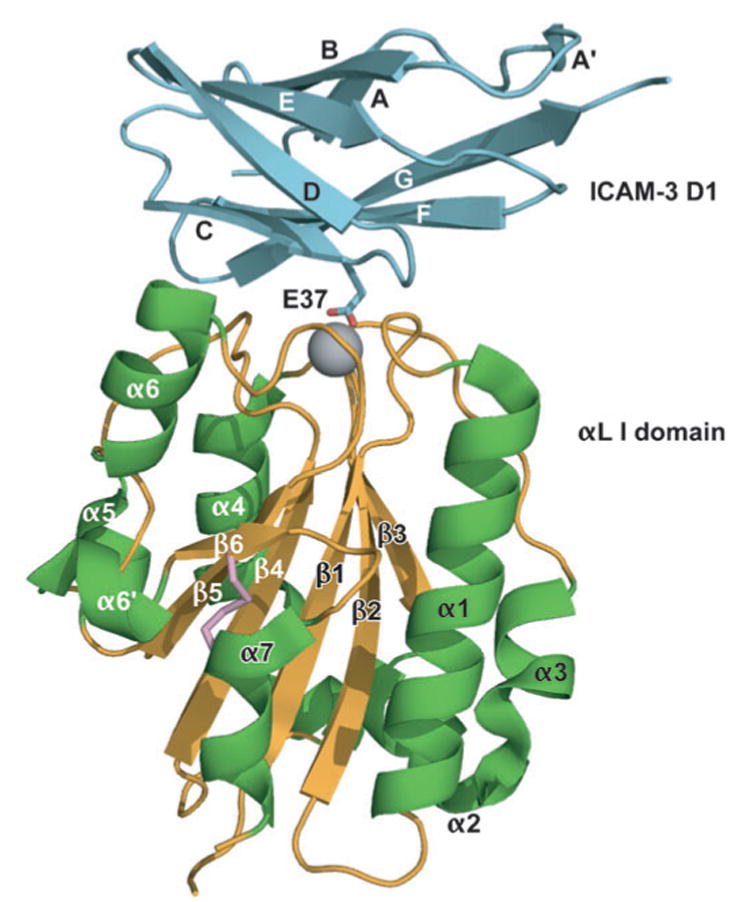
A mutant, high-affinity αL I domain (gold β-sheet and coil and green α-helices) in complex with domain 1 of ICAM-3 (cyan). The Mg2+ is shown as a gray sphere. The side chain of the key integrin-binding residue, Glu37 of ICAM-3, is shown. The mutationally introduced K287C/K294C disulfide bond that stabilizes the open conformation is shown in pink. ICAM-3 domain 2 is omitted for clarity. [From Protein Data Bank (PDB) ID code 1T0P (7).]
Figure 3.
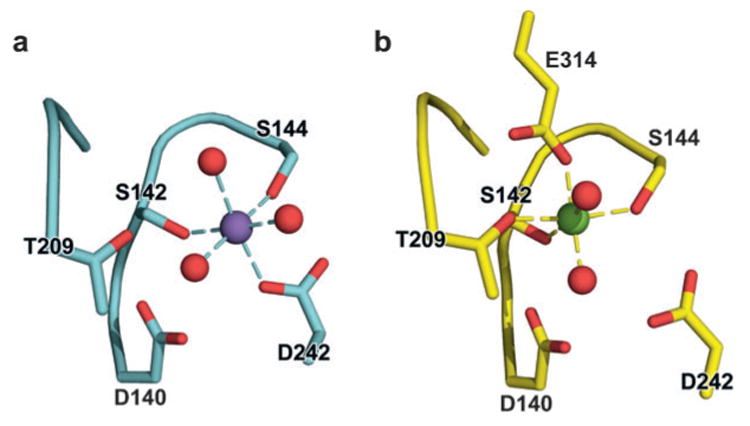
Structural rearrangement of the αM I domain MIDAS. (a) Structure of the closed αM I domain MIDAS. (b) Structure of the open α I domain MIDAS. Glu-314 from a neighboring αM I domain coordinates with the MIDAS magnesium. Purple and green spheres are Mn2+ and Mg2+ ions, respectively, and red spheres are coordinating water-molecule oxygens. [From PDB ID codes 1JLM and 1IDO (4, 8).]
Conformational Regulation of α I Domains
Structural studies of α I domains in the presence and absence of ligand, and with mutations that stabilize distinct affinity states, have provided a mechanistic understanding of conformational regulation during both priming and ligand binding. I domains have been crystallized in three distinct conformations, termed closed, intermediate, and open (4–6, 8). These demonstrate distinct coordination of the metal in the MIDAS, arrangement of the β6-α7 loop, and axial disposition of the C-terminal α7-helix along the side of the I domain (5, 6, 8) (Figure 4a). At the α I domain MIDAS, five residues and several water molecules contribute oxygen atoms to the primary and secondary coordination spheres surrounding the metal (Figure 3). In the open conformation of the MIDAS, two serines and one threonine are in the primary coordination sphere, whereas two aspartic acid residues are in the secondary coordination sphere (Figure 3b). Notably, the glutamic acid residue, which is contributed by the ligand or ligand-mimetic lattice contact, donates the only negatively charged oxygen to the primary coordination sphere in the open conformation (E314 in Figure 3b). The lack of any charged group in the primary coordination sphere donated by the I domain is hypothesized to enhance the strength of the metal-ligand bond. In the closed conformation of the α I domain (Figure 3a), the threonine moves from the primary to the secondary coordination sphere, and one of the aspartic acid residues moves from the secondary to the primary coordination sphere. The backbone and side chain rearrangements in the α I domain are accompanied by a 2.3 Å “sideways” movement of the metal ion away from the threonine and toward the aspartic acid on the opposite side of the coordination shell. The closed and open structures are consistent with the idea that an energetically favorable MIDAS requires at least one primary coordination to a negatively charged oxygen. In the absence of a ligand, pseudoligand, and the remainder of the integrin ectodomain, wild-type α I domains crystallize in the closed conformation. The closed conformation therefore appears to be the low-energy conformation, as verified computationally (9). However, with an engineered disulfide bond that was designed to stabilize the open conformation, an αL I domain crystallized in the open conformation in the absence of a ligand-mimetic lattice contact (6). This demonstrates that, in principle, interactions with other integrin domains might be capable of stabilizing an unliganded I domain in the open conformation and activating or priming it for ligand binding.
Figure 4.
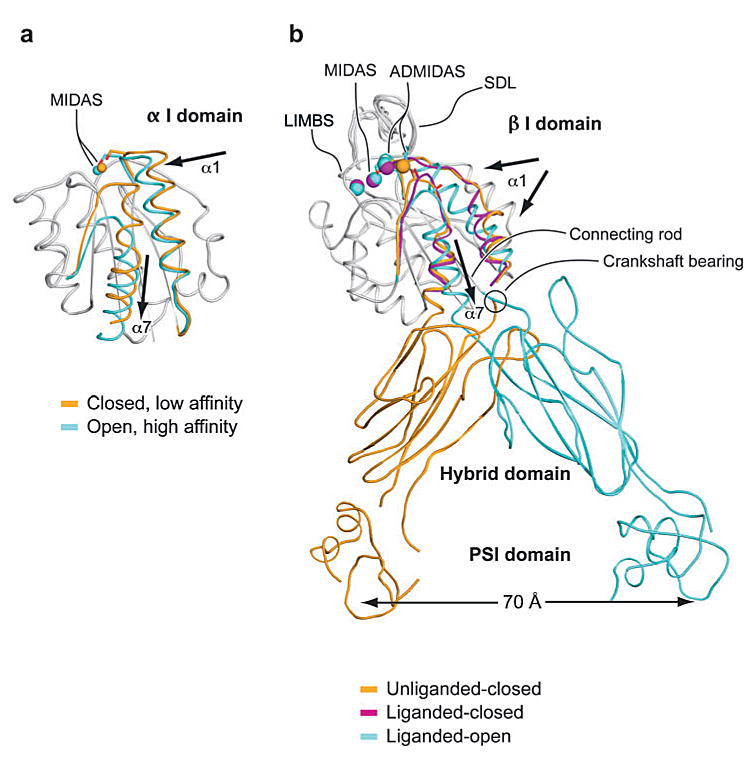
Conformational change and transmission of allostery by α and β I domains. (a) The α I domain. Nonmoving segments of the backbone are shown as a gray worm. The moving segments of the backbone and the MIDAS metal ions are closed (gold ) and open (cyan). The direction of movement is indicated with arrows. [From PDB ID codes 1JLM and 1IDO (4, 8).] (b) The β I domain and its linkage to the hybrid and plexin/semaphorin/integrin (PSI) domain. Nonmoving segments of the β I backbone are shown as a gray worm. Moving segments and metal ions are color coded as shown. Directions of α1- and α7-helix movements are shown with arrows. [PDB ID codes are 1U8C, 1L5G, and 1TXV (32, 36, 40).]
Change in coordination at the MIDAS of α I domains is coupled to backbone movements of loops that bear the coordinating residues. Several of these loops, including the β1-α1 and β4-α5 loops, also bear residues that directly contact ligand, and thus their movement increases complementarity to ligand. To accommodate these rearrangements, the β6-α7 loop undergoes the largest shift of all, although it is not a MIDAS loop nor does it contact ligand. Coupled to the β6-α7 loop rearrangement, the C-terminal α7-helix moves 7 Å down the side of the domain (Figure 4a). The axial displacement of the α7-helix represents the critical linkage for transmission of conformational signals between the MIDAS of the α I domain and other integrin domains, as discussed below. Engineered disulfide bonds that stabilize the α7-helix in intermediate and open conformations (shifted axially downward by approximately 3 Å or 7 Å relative to the closed conformation, respectively) induced rearrangements in the MIDAS and surrounding loops that were coupled to 500- and 10,000-fold increases in affinity for ICAM-1, respectively (6). As mentioned above, the downward movement of the α7-helix is sufficient for priming the α I domain into higher-affinity states. Crystal structures have been obtained of intermediate- and high-affinity mutant αL I domains both in the absence and presence of ligands (6, 7). For example, Figure 2 shows a complex between ICAM-3 and a high-affinity αL I domain mutant with a disulfide bond introduced into the β6-α7 loop to stabilize the open conformation. Conversely, binding of wild-type I domains to ligand at the extremely high, ∼mM, concentrations used in crystallization can induce MIDAS rearrangements and downward displacement of the α7-helix (5). Thus, the transmission of inside-out and outside-in signaling within the I domain occurs along the same pathway but flows in opposite directions.
The ability to modulate affinity by 10,000-fold demonstrates the exquisite efficiency of the α I domain in coupling change in conformation to change in affinity. Remarkably, through directed evolution an engineered αL I domain mutant (F265S/F292G) was recently obtained with an increase of 200,000-fold in affinity for ICAM-1 (10). However, whether α I domains achieve such high affinities for ligands under physiological conditions is unknown. A high-affinity state of integrins on intact cells can be induced by addition of Mn2+, which, as reviewed below, increases integrin affinity by replacing Ca2+ at the ADMIDAS site of the β I domain. Interestingly, activation of integrin adhesiveness on intact cells by physiologic stimuli appears to result in lower affinity than that induced by Mn2+. Therefore, investigators have hypothesized that an intermediate conformational state with intermediate affinity for ligand is important for physiologic, fine-tuned regulation of αLβ2 adhesiveness (11). Molecular dynamic studies showed that the intermediate conformation of the α I domain is on the pathway from the closed to the open conformation of the αL and αM I domains (12).
A monoclonal antibody (mAb), AL-57, has been developed by phage display that selectively targets the high-affinity open conformation of the αL I domain (13, 14). AL-57 does not bind the low-affinity state of leukocyte function-associated antigen (LFA)-1 (αLβ2) but does bind the intermediate- and high-affinity states of the αL I domain with KD of 4.7 μM and 23 nM, respectively. AL-57 is ligand-mimetic because it binds only upon activation and requires Mg2+ for binding. Interestingly, monovalent Fab AL-57 demonstrates affinity increases on a subset (∼10%) of lymphocyte cell surface LFA-1 molecules upon stimulation with chemokine CXCL-12 and PMA (phorbol 12-myristate 13-acetate). These results are consistent with previous observations on Mac-1 on neutrophils (15) and suggest that after physiologic activation a subset of cell surface Mac-1 molecules on neutrophils and LFA-1 molecules on lymphocytes are converted to a higher-affinity state. This active subset of molecules mediates adhesion because the antibodies specific for this subset of 10% to 30% of surface molecules completely inhibit cell adhesion.
Allosteric α I Domain Inhibitors
Small molecule allosteric inhibitors provide further support for the role of the α7-helix in α I domain regulation. One class of small molecule inhibitors, termed α I allosteric antagonists, binds underneath the C-terminal α-helix of the αL I domain (16–18). Such antagonists stabilize the closed conformation of the I domain by preventing downward axial shift of the α7-helix and thereby preventing MIDAS rearrangements necessary for efficient ligand binding. The mode of action of these antagonists is confirmed by the finding that a mutant αL I domain that stabilizes the high-affinity, open conformation of the C-terminal α7-helix with an engineered disulfide bond is resistant to inhibition by α I allosteric antagonists (11, 19, 20).
Ligand Recognition by α I Domains
Ligand recognition by α I domains has been elucidated by crystal structures of the α2 I domain in complex with a triple-helical collagenous peptide (5) and the αL I domain in complex with ICAM-1 and ICAM-3 (6, 7). ICAM-1, -2, -3, -4, and -5 are cell surface molecules with 2 to 9 IgSF domains (Figure 5). They share much more sequence identity with one another (30% to 50%) than with other IgSF molecules and thus comprise a subfamily of the Ig superfamily. Except for ICAM-4, they all bind to αLβ2 through a key glutamic acidic residue in domain 1 (21). The order of binding affinities for αLβ2 is ICAM-1 > ICAM-2 > ICAM-3 (11). In the structure of the αL I domain bound to ICAM-1 (6) or ICAM-3 (7), Glu-34 (ICAM-1) or Glu-37 (ICAM-3) at the end of the β-strand C of domain 1 forms a direct coordination to the Mg2+ in the α I domain MIDAS (Figure 2). This metal-coordination bond is surrounded by a ring of hydrophobic residues in both the α I domain and ICAM-1. The surrounding nonpolar environment strengthens the Coulombic interaction between the Glu and Mg2+. Surrounding the hydrophobic ring are polar interactions involving hydrogen bonds and salt bridges. The nonpolar region in ICAM-1 is more polar in ICAM-3 and appears to account for the lower affinity of αLβ2 for ICAM-3 than for ICAM-1 (7). In support of this finding, increasing the hydrophobicity of the nonpolar ring even further in ICAM-1 by structure-guided mutagenesis increases the affinity of αLβ2 for ICAM-1 (22). ICAM-1 and ICAM-3 dock in the same orientation to the αL I domain, and the structure of ICAM-2 (23) suggests an essentially identical docking mechanism.
Figure 5.
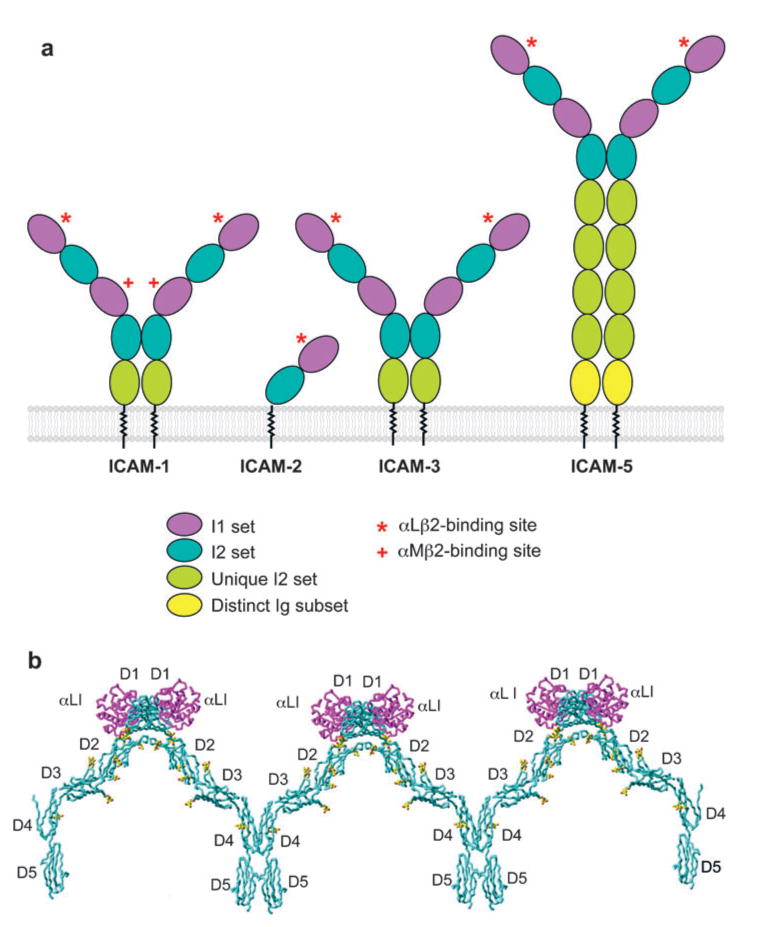
ICAM structure and integrin binding. (a) Schematic of ICAM-1, -2, -3, and -5. The domains are color coded, and integrin-binding sites are shown. (b) Structural model of ICAM-1 oligomers bound to αL I domain. The model was constructed from the structure of ICAM-1 D1-D2 in complex with αL I domain (PDB ID code 1MQ8) and from the structure of ICAM-1 D3-D5 (PDB ID code 1P53) (6, 24). ICAM-1 is cyan, with the first carbohydrate residue at each site in yellow; the αL I domain is purple.
The structure of the αL I domain bound to a portion of ICAM-1 (6) combined with a complementary structure containing the remaining portion of ICAM-1 (24) have provided a topological view of the αLβ2-ICAM-1 interaction as it might take place during cell-cell interactions (Figure 5b). αLβ2- and αMβ2-binding sites on ICAM-1 are located on D1 and D3, respectively. ICAM-1 exists on the cell surface predominantly as a homodimer (25, 26). Relatively strong but reversible dimerization takes place in D4 by merging of the β-sheets of two D4 domains into β-supersheets, as revealed by an ICAM-1 D3-D5 crystal structure (24). An apparently weaker hydrophobic dimerization interface in D1 has been revealed in different crystal structures, including the ICAM-1 D1-D2 complex with the αL I domain (6). Together, the αL I domain-ICAM-1 D1-D2 and the ICAM-1 D3-D5 structures show that ICAM-1 dimers are Y-shaped and that the dimeric interface at D4 and D5 provides a rigid stem to orient D1-D3 optimally for binding integrins on opposing cell surfaces (24) (Figure 5a). Furthermore, dimerization at D1 could link neighboring Y-shaped dimers yielding a one-dimensional array of ICAM-1 molecules on the cell surface (Figure 5b), which has an architecture appropriate for displaying the αLβ2-binding site in D1 and the αMβ2-binding site in D3 for recognition by integrins on an opposing cell (24).
In contrast to αLβ2, the αMβ2 and αXβ2 integrins bind to a range of diverse ligands, including ICAMs, fibrinogen, iC3b, heparin, and denatured and proteolyzed proteins (27–29). Proteolysis and denaturation enhance binding of αM and especially of αX I domains to fibrinogen, and investigators have proposed that αXβ2 functions as a danger receptor for proteolyzed and denatured proteins (29). In marked contrast to αL, the αX and αM I domains show a KD for small molecules with carboxyl groups of ∼100 μM. This relaxed ligand specificity is consistent with the ability of the wild-type αM I domain to engage in ligand-mimetic lattice contacts in crystals. In these contacts, the MIDAS binds to a glutamic acid residue in a neighboring αM I domain in the crystal lattice (Figure 3b), and the I domain crystallizes in the open conformation (4).
Integrin Global Topology
A complete understanding of integrin regulation requires knowledge of how conformational information is transmitted through the many domains that link the ligand-binding domains to the transmembrane and cytoplasmic domains. Both the integrin α and β subunits are type I transmembrane glycoproteins with large extracellular domains, single-spanning transmembrane domains, and, with the exception of β4, short cytoplasmic domains (Figure 6a,b). From electron microscopy (EM), investigators have known for years that the overall topology of integrins included an extracellular, globular, N-terminal ligand-binding head domain, representing a critical α and β subunit interface, standing on two long and extended C-terminal legs or stalks, which connect to the transmembrane and cytoplasmic domains of each subunit (30). However, X-ray crystal structures of the extracellular domain of the integrin αVβ3 provided the surprising finding that the legs were severely bent, generating a V-shaped topology in which the head domain was closely juxtaposed to the membrane-proximal portions of the legs (31, 32) (Figure 6c and Figure 7a). Since the elucidation of these initial structures, an increasing number of studies have together established that the bent conformation represents the physiological low-affinity state, whereas priming and ligand binding are associated with a large-scale global conformational rearrangement in which the integrin extends with a switchblade-like motion (33–36) (Figure 6c,d). The most recent studies have elucidated the detailed mechanisms for linking these global rearrangements to intradomain conformational changes that are associated with affinity modulation and ligand binding.
Figure 6.
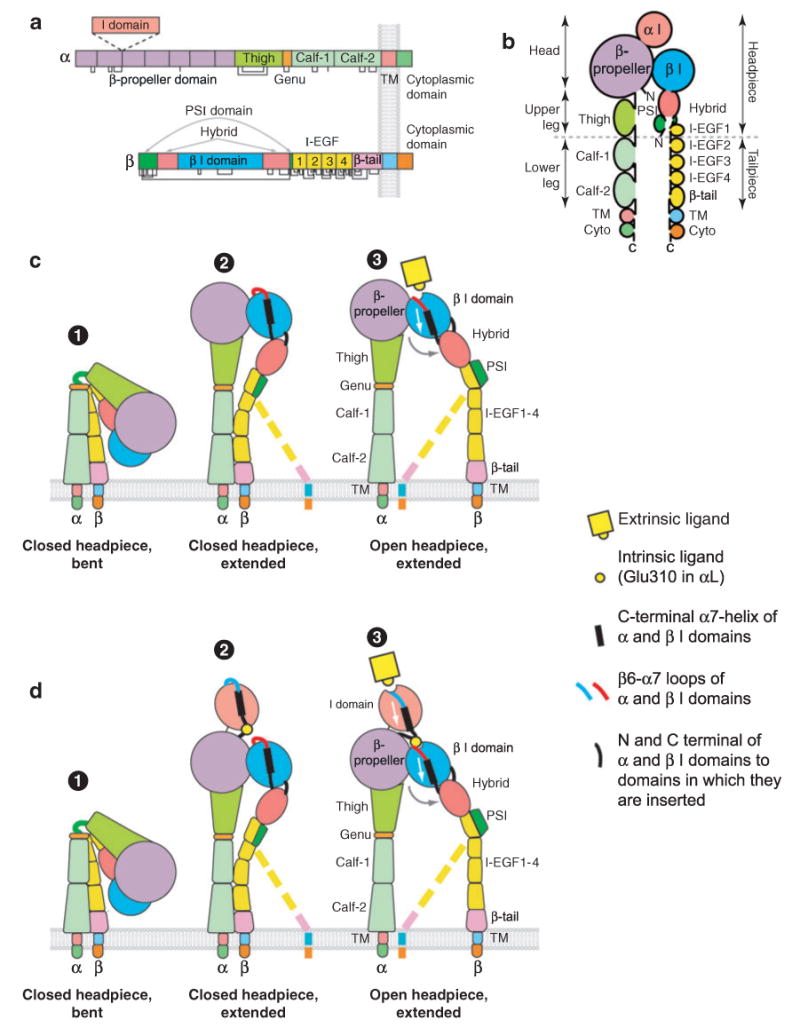
Integrin architecture. (a) Organization of domains within the primary structures. Some α subunits contain an I domain inserted in the position denoted by the dashed lines. Cysteines and disulfides are shown as lines below the stick figures. (b) Schematic of the course of the α and β subunit polypeptide chains through domains from the N to C termini. (c-d ) Rearrangement of domains during activation of integrins that lack (c) or contain (d ) an α I domain. The β subunit lower legs are flexible and are therefore shown in what may be the predominant (solid representation) and less predominant (dashed lines) orientations.
Figure 7.
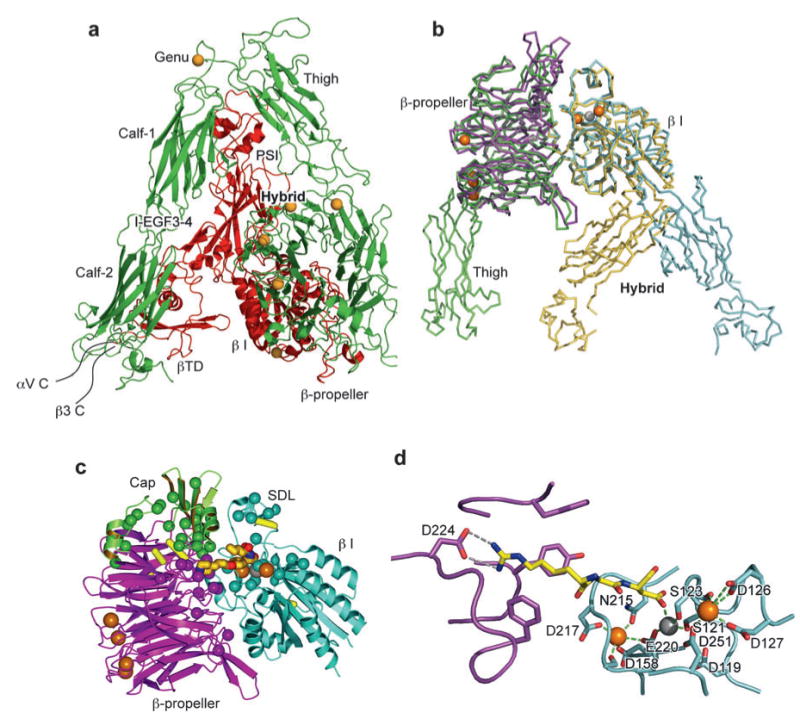
Crystal structures of integrins αVβ3 and αIIbβ3. (a) The structure of αVβ3 in the bent conformation. The αV and β3 subunits are colored in green and red, respectively. (b) Superposition of liganded-open αIIbβ3 and unliganded-closed αVβ3 headpieces. The α and β subunits are colored in green and yellow in αVβ3 and in purple and light blue in αIIbβ3. Calcium and magnesium ions in αIIbβ3 only are gold and gray spheres, respectively. (c) The drug tirofiban is shown bound to the αIIbβ3 head, and mapping is shown of fibrinogen binding-sensitive mutations. The clinically approved antagonist tirofiban is shown with yellow carbons, blue nitrogens, and red oxygens. The cap subdomain of the β-propeller is in green. Ca2+ and Mg2+ ions are large gold and gray spheres, respectively. Cβ atoms of fibrinogen binding-sensitive residues are shown as small spheres in the same color as the domains in which they are present. Disulfide bonds are yellow cylinders. (d ) The binding of eptifibatide to αIIbβ3 interface is depicted. The fragment of eptifibatide that mimics RGD is shown as a stick model with carbon, nitrogen, and oxygen colored yellow, blue, and red, respectively. The binding pocket is shown with αIIb and β3 in purple and light blue, respectively. Hydrogen bonds are shown as gray dashed lines. Ca2+ and Mg2+ are gold and gray spheres, respectively. The coordinations to the metal ions are shown as green dashed lines. [Structure PDB ID codes are, for αVβ3, 1U8C (40); for αIIbβ3 bound to eptifibatide, 1TY6; and for αIIbβ3 bound to tirofiban, 1TY5 (36).]
The Ligand-Binding Head
The N-terminal region of the integrin α subunit contains seven segments of about 60 amino acids, each with weak sequence similarity to one another. These were initially predicted (37) and later confirmed by crystal structures (31, 36) to fold into a seven-bladed β-propeller domain. When present, the α I domain is inserted between β-sheets 2 and 3 of the β-propeller (Figure 6a,b). An inserted domain in the β subunit, the β I domain, is homologous to the α I domain, except that it contains two additional segments; one of these helps form the interface with the β-propeller and the other is known as the specificity-determining loop (SDL) because of its role in ligand binding (Figure 6a,b and Figure 7c). One side of the β I domain binds to the upper hub of the β-propeller domain directly over the pseudosymmetry axis of the β-propeller (Figure 7c). The extensive interface, which buries 1700 Å2 of solvent-accessible surface area on each side, is much greater than any other domain-domain interface in integrins, including interfaces between domains that are contiguous in sequence in the α and β subunits. Mutations in the β2 I domain that disrupt this interface cause leukocyte adhesion deficiency (38, 39). The opposite, lower face of the α subunit β-propeller domain is stabilized by Ca2+ ions that bind to Ca2+-binding β-hairpin motifs (Figure 7b,c). Like the α I domain, the β I domain contains a MIDAS for binding negatively charged residues, which physiologically binds Mg2+ (36). Additionally, there are two adjacent metal ion-binding sites, which physiologically bind Ca2+ (36), share some coordinating residues in common with the MIDAS, and are known as the LIMBS (ligand-induced metal ion-binding site) and ADMIDAS (adjacent to metal ion-dependent adhesion site) (31, 32) (Figure 4b and Figure 7b).
Structures of αVβ3 (32) and αIIbβ3 (36) show that peptides containing ligand-mimetic Arg-Gly-Asp (RGD) sequences bind across the α-β subunit interface in the head (Figure 7c,d). The Asp carboxyl group directly coordinates the β I domain MIDAS Mg2+ ion, whereas the Arg side chain hydrogen binds to Asp residues in the αV or α IIb β-propeller domains (Figure 7d). The binding site for macromolecular ligands is larger. Residues shown by mutagenesis to be important for binding to fibrinogen (smaller spheres, Figure 7c) decorate the cap subdomain of the β-propeller (in Figure 7c, green), the remainder of the β-propeller (magenta), and the β I domain (cyan). The cap subdomain is formed by several insertions that are unusually long in α IIb in β-propeller domain β-sheets (blades) 2 and 3. The ligand-binding site in the β-propeller is formed largely by β-sheets 2 and 3, which lie opposite the ligand-binding MIDAS and SDL of the β I domain.
The α and β Subunit Legs
In the α subunit, the region C-terminal to the β-propeller comprises the leg of the α subunit and contains three β-sandwich domains. The upper leg contains the thigh domain and the lower leg consists of the calf-1 and calf-2 domains. A small Ca2+-binding loop located between the thigh and calf-1 domains represents the α subunit genu, the key pivot point for switchblade extension in the α subunit (Figure 6 and Figure 7a).
The topology of the β subunit is more complex. The β I domain is inserted in the hybrid domain, which forms the upper portion of the upper β leg (Figure 6a,b). In turn, the hybrid domain is inserted in the plexin/semaphorin/integrin (PSI) domain (Figure 6a,b). The second segment of the PSI domain is very short but can be assigned as part of the PSI domain because it contains β3-Cys435, which is involved in a long-range disulfide bond to β3-Cys11 in the first segment of the PSI domain, and this disulfide is structurally conserved in other PSI domains (36, 40, 41).
The remainder of the β leg is built from four integrin epidermal growth factor-like (I-EGF) domains and a β tail domain. I-EGF domains 1 and 2 were not resolved in the αVβ3 crystal structure. However, an NMR structure of β2 I-EGF3 and studies on I-EGF2 and I-EGF3 established an extended orientation between these domains. Furthermore, a structure of the β2 PSI, hybrid, and I-EGF1 domains has been solved (41). Superposition of these structures on the bent αVβ3 structure establishes that the bend in the β leg, or knee, is located between I-EGF domains 1 and 2, as suggested earlier (42). The PSI and I-EGF1 domains are located side by side (Figure 6b). The bends in the α leg at the genu and in the β leg between I-EGF1 and I-EGF2 are located close to one another and in a geometry appropriate so that extension can occur by pivoting of the headpiece about an axis through the α and β subunit knees (Figure 6c,d), as shown by EM studies (33, 43). Consistent with these findings, many antibodies that either activate or report activation in cell surface integrins map to the PSI and β I-EGF domains (44–47). Furthermore, αL antibodies that report extension map to the inner face of the thigh domain and require genu Ca2+-coordinating residues for binding and thereby provide evidence that integrin extension occurs by a rearrangement at the thigh/genu interface (48).
Conformational Regulation of Integrin Extracellular Domains
Conformational Activation of β I Domains
In the bent conformation, the ligand-binding site is not in an optimal orientation for binding macromolecular ligands in the extracellular matrix or on the surface of other cells (Figure 6c,d). However, integrins in the bent conformation can bind ligands, as clearly shown by soaking an RGD ligand-mimetic peptide into preformed crystals (32). Ligand binding induced movements in the β1-α1 and β6-α7 loops (liganded-closed conformation, Figure 4b). However, downward displacement of the α7-helix was not seen (32).
When an αIIbβ3 headpiece was first mixed with ligand-mimetic drugs, and then crystals were allowed to form, a different conformation, termed liganded-open, was obtained (36). In the high-affinity, liganded-open β I domain, compared with the low-affinity, unliganded-closed β I domain, marked movements occur of the β1-α1 and β6-α7 loops and of the α1- and α7-helices (Figure 4b). Coordination of the Met335 backbone carbonyl in the β6-α7 loop to the ADMIDAS Ca2+ ion in the low-affinity, unliganded conformation is broken in the high-affinity, liganded conformation. This enables the ADMIDAS metal and residues in the β1-α1 loop that coordinate to both the ADMIDAS and MIDAS metals to shift markedly, remodel the ligand-binding site, and increase affinity for ligand. These movements are tightly coupled, so that reshaping to the high-affinity, ligand-binding site is allosterically linked to downward movement of the α7-helix. This linkage is critical for propagation of conformational signals from the ligand-binding pocket to the other integrin domains and vice versa (Figure 4b). When the RGD-mimetic is soaked into preformed crystals (liganded-closed, Figure 4b), the β1-α1 loop moves almost as much but does not have as optimal an interaction with ligand as in the liganded-open structure, and the remaining movements are frustrated by crystal lattice contacts.
Effects of Mn2+ and Ca2+
Compared with results in the physiologic divalent cations Mg2+ and Ca2+, which are present at ∼1 mM in body fluids, addition of Mn2+ or removal of Ca2+ increases ligand-binding affinity and adhesiveness of almost all integrins. Recent studies show that binding of metal ions to the LIMBS and ADMIDAS sites can explain these effects (49, 50). Mutational studies show that the LIMBS functions as a positive regulatory site, and the ADMIDAS functions as a negative regulatory site (49–51). Additionally, in α5β1 and αLβ2 integrins, the ADMIDAS functions in transmission of allostery between the β I domain and other domains (50, 51). For most integrins, Ca2+ has both positive and negative regulatory effects. High concentrations of Ca2+ inhibit adhesion, whereas low concentrations of Ca2+ synergize with suboptimal Mg2+ concentrations to support adhesion. The LIMBS mediates the synergistic effects of low Ca2+ concentrations (49, 50), whereas the ADMIDAS mediates the negative regulatory effects of higher Ca2+ concentrations, which are competed by Mn2+ (49).
Communication Between the α I and β I Domains
Conformational regulation of integrins containing an α I domain requires the additional step of transmission of allostery from the β I domain to the α I domain (Figure 6d). An invariant Glu residue, E-310 in αL, in the linker between the C-terminal α7-helix of the α I domain and β-sheet 3 of the β-propeller domain is required for α I domain activation (52, 53). It has been proposed that this invariant Glu residue acts as an intrinsic ligand and binds to the β I MIDAS when it is activated and that it exerts a downward pull on the α7-helix and activates the α I domain (53, 54) (Figure 8a). Yang et al. (54) provided direct evidence for an activating interaction between αL residue 310 and the β2 MIDAS. Individual mutation of the αL linker residue Glu-310 or β2 MIDAS residue Ala-210 to cysteine abolishes I domain activation, whereas the double mutation of αL-E310C and β2-A210C results in formation of a disulfide bond that constitutively activates ligand binding (54) (Figure 8b).
Figure 8.
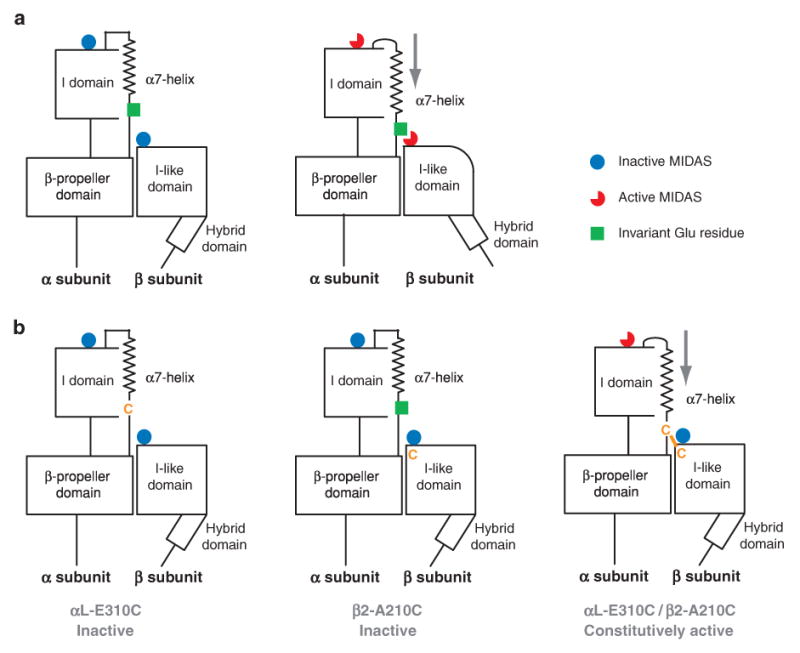
Communication between α I and β I domains. (a) It has been proposed that αL-Glu-310 acts as an intrinsic ligand that binds to the β2 I domain MIDAS and, thus, axially displaces the αL I domain α7-helix in the C-terminal direction, reshapes the β6-α7 loop, and activates the αL I domain MIDAS. (b) Individual mutation of αL-Glu-310 or β2-Ala-210 to cysteine abolishes I domain activation, whereas the double mutation of αL-E310C with β2-A210C forms a disulfide bond that constitutively activates ligand binding (104).
α/β I Domain Allosteric Antagonists
Small molecule integrin antagonists have yielded further insight into the mechanisms for α I-β I communication. A class of αLβ2 and αMβ2 small molecule antagonists perturbs the interface between the α I domain and the β I domain (55–57). These antagonists do not inhibit binding of isolated wild-type or mutant intermediate- or high-affinity α I domains to ICAM-1 (56). Furthermore, these inhibitors bind to αLβ2 even when the α I domain is deleted, but do not bind when the β I domain MIDAS is mutated (56). Some but not all compounds of the series exhibit α subunit selectivity, suggesting that a portion of the α subunit nearby the β I domain, likely the β-propeller domain or its linkers to the α I domain, is involved in binding. Therefore, these inhibitors have been designated as α/β I domain allosteric antagonists.
These antagonists apparently bind to the MIDAS of the β2 I domain, competitively inhibit binding of the intrinsic ligand in the α I domain linker, and thus leave the α I domain in its default low-energy, inactive, closed conformation. At the same time, the α/β I allosteric antagonists mimic intrinsic ligand binding and thereby stabilize the β I domain in its active configuration, as shown by induction of activation epitopes in the β2 I domain, as well as the αL and β2 legs (56). The antagonists induce integrin extension as shown in EM studies (43). In in vitro shear flow assays and in vivo, the antagonists enhance rolling of leukocytes and inhibit firm adhesion (57). These results on ICAM-1 substrates suggest that the postulated αL Glu-310-β2 MIDAS interaction is not required for rolling adhesion, in agreement with the ability of isolated, wild-type αL I domains to support rolling adhesion (58, 59), but is required for firm adhesion. LFA-1 containing an αL Glu-310 → Ala mutation shows lowered expression of activation epitopes in the β2 I domain and leg, demonstrating cooperativity between the postulated αL Glu-310-β2 MIDAS interaction and conformational rearrangements elsewhere in the LFA-1 molecule. This mutant is also deficient in supporting rolling interactions on ICAM-1 substrates. However, treatment of αL Glu-310 LFA-1 mutants with α/β I allosteric antagonists induces epitope exposure and renders them competent to support rolling, consistent with the hypothesis that these antagonists bind to the same site to which αL Glu-310 binds (59).
Hybrid Domain Swing-Out and Integrin Extension
The orientation between the β I and hybrid domains appears to be the critical translator converting global conformational change into local intradomain conformational changes that regulate integrin affinity for ligand (see Figure 4b and Figure 6c,d ). As a consequence of the topology of insertion of the β I domain in the hybrid domain, the piston-like displacement of the α7-helix in the high-affinity, liganded structure results in complete remodeling of the interface between these domains, leading to the swing-out of the hybrid domain (36) (Figure 4b). Actually, the α7-helix functions more like a connecting rod than a piston because as it moves downward, its angle changes (Figure 4b), like a connecting rod between a piston and a crankshaft. This forces rotation about a crankshaft bearing (circled in Figure 4b) between the last β-strand of the hybrid domain and the first β-strand of the β I domain. Note the structural design of this machine: Hydrogen bonds in α-helices are all internal, allowing them to move independently of other structural units, whereas the three other connecting units between the β I and hybrid domains are β-strands, which are fixed in position within β-sheets by hydrogen bonds. Therefore, there is little compliance in the central β I domain β-sheet or the two hybrid domain β-sheets. This enables the rearrangement of the loops around the β I domain MIDAS to be transmitted as a 60° swing of the hybrid domain away from the α subunit and a 70 Å movement of the rigidly connected PSI domain, i.e., a 70 Å separation of the integrin knees (36, 41) (Figure 4b).
Three major integrin conformations have been resolved by crystal and EM studies (Figure 6c,d). The bent conformation contains a closed headpiece (31). The hybrid domain is highly buried in the interfaces that stabilize the bent structure, and therefore its swing-out destabilizes the bent conformation (33). By contrast, the extended integrin conformation is compatible with both closed and open headpiece conformations (33, 43) (Figure 6c,d and Figure 9a,b). The influence on equilibration between these states has been studied of a flexible, C-terminal clasp fused to the C termini of the α and β subunit ectodomains, which mimics association between the α and β subunit transmembrane domains (33, 43). Whereas clasped αVβ3 or αXβ2 particles are predominantly in the bent conformation (see panel 1 of both Figure 9a and b), unclasped particles are predominantly extended. For αVβ3 and αXβ2, about half of unclasped, extended particles have the closed headpiece (see panel 2 of both Figure 9a and b) and half have the open headpiece (see panel 3 of both Figure 9a and b) (33, 43). Therefore, once these integrins extend, the energies of the closed and open headpiece conformations must be comparable. However, the energetics for conformational transitions appear to vary among integrins, as exemplified with αLβ2. Thus, clasped αLβ2 shows about equal proportions of bent and extended particles, and unclasped αLβ2 particles adopt predominantly the closed headpiece, with a smaller proportion of particles having the open headpiece (43).
Figure 9.
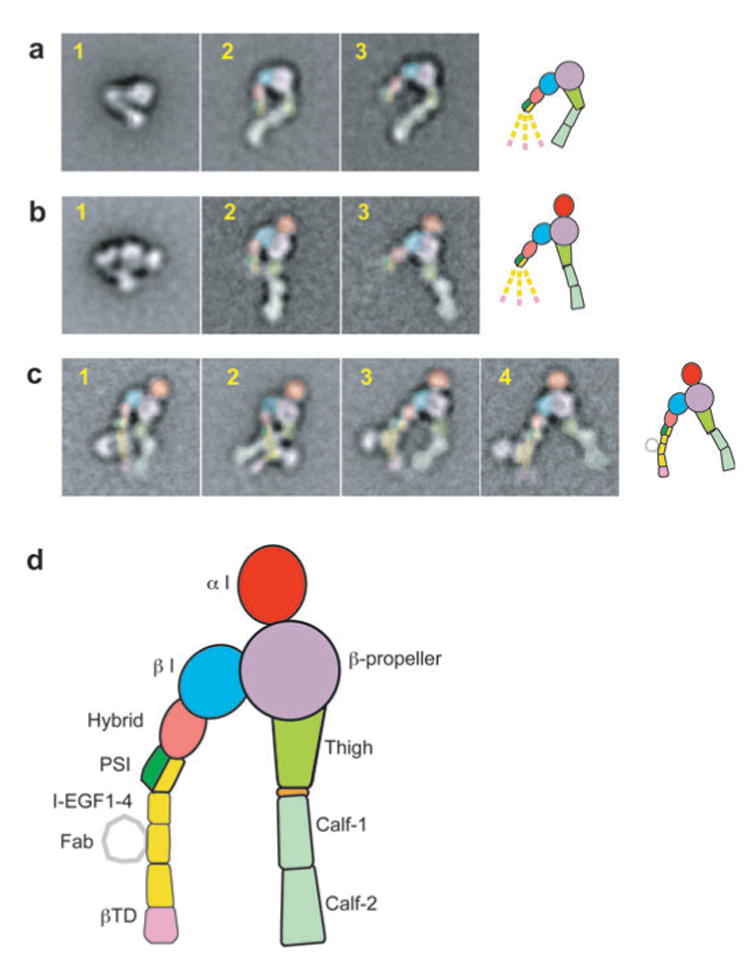
EM negative-stain class averages of integrins αVβ3 and αXβ2 in bent and extended conformations (33, 43). The EM images of the extended conformations only are colored according to the scheme shown in d. (a) αVβ3 in bent ( panel 1), extended with closed headpiece ( panel 2), and extended with open headpiece ( panel 3) conformations. (b) αXβ2 in bent ( panel 1), extended with closed headpiece ( panel 2), and extended with open headpiece ( panel 3) conformations. (c) αXβ2 in complex with CBR LFA-1/2 Fab illustrates flexibility of the β leg: panel 1, closed headpiece with parallel legs; panel 2, closed headpiece with crossed legs; panels 3 and 4, open headpiece. Panels 1–3 are with clasped αXβ2, and panel 4 is with unclasped αXβ2. In a-c, a schematic in the same orientation as the right-most panel is shown to the right; the dashed lower β legs symbolize flexibility and averaging-out.
A large number of studies are in agreement with the three integrin conformational states described above and support the importance of hybrid domain swing-out in inducing high affinity for ligand. EM studies of the α5β1 headpiece show that hybrid domain swing-out occurs upon binding fibronectin fragments (34). Electron tomography of negatively stained, active, detergent-soluble α IIbβ3 purified on an RGD peptide affinity column reveals an extended conformation with >90% of particles showing an open headpiece structure that matches perfectly (60) the open, liganded α IIbβ3 headpiece crystal structure (36). In addition to structural investigations (33, 34, 36, 43, 60, 61), integrin hybrid domain swing-out is supported by a range of other studies. Stabilizing the open headpiece by mutationally introducing an N-glycosylation site into the hybrid-β I domain interface increases ligand-binding affinity (62, 63). As shown by epitope mapping and EM, an allosteric β1 antibody that inhibits ligand binding restricts the swing-out of the hybrid domain (63). The functional properties of an inhibitory β2 mAb suggest it also inhibits by blocking signal transmission at the β I-hybrid domain interface (64). Moreover, activation-dependent mAbs map to the inner face of the hybrid domain, consistent with exposure of this face after swing-out (65, 66), and specific mutations of the β I domain α7-helix facilitate hybrid domain swing-out (65).
In contrast to the consensus in the above studies, an alternative deadbolt model (67) has received little experimental support, as reviewed in more detail elsewhere (68). The presumed deadbolt interface between β3 Val332 and Ser674 is extremely small, at 60 Å2 (67), and a three-residue deletion of β3 residues 672–674 that removes this interface has no effect on ligand binding to cell surface integrins αVβ3 and αIIbβ3 ( J. Zhu, B. Luo & T.A. Springer, unpublished observation). One negative-stain EM study found that ligand binding to αVβ3 did not induce extension (69); however, much greater particle aggregation was present than in other studies and must have complicated the analysis.
The use of functionally well-characterized antibodies in EM experiments has provided definitive evidence that integrin extension occurs on intact cells in response to physiologic stimuli and is sufficient to activate integrin adhesiveness (43). Extensive, physiologically relevant studies of β2 integrins on intact cells have shown that CBR LFA-1/2 mAb induce the high-affinity state and that, depending on the experimental system, KIM127 mAb can either stabilize or report the high-affinity state (35, 45, 54, 57, 70–76). The binding sites for KIM127 and CBR LFA-1/2 antibodies have been mapped to the I-EGF2 and I-EGF3 domains, respectively (45, 77). Although clasped αXβ2 was >95% in the bent conformation, binding of CBR LFA-1/2 Fab induced complete conversion to the extended conformation (43) (Figure 9c, panels 1–3). Furthermore, KIM127 Fab bound only when extension was induced by another agent such as CBR LFA-1/2 Fab or an α/β allosteric antagonist. In combination with the following cited functional studies, the EM study (43) established that (a) extension is sufficient to activate ligand-binding competence by β2 integrins (35, 45, 54, 72–76, 78), (b) ligand-bound β2 integrins on cell surfaces are extended (70, 71), (c) binding to soluble ligand induces extension (57), and (d ) extracellular activation of integrins by Mn2+ and inside-out activation of integrins stimulated by protein kinase C or cytoplasmic domain mutations induce the extended conformation in the absence of ligand binding (35, 45, 75).
When viewed in combination, the crystal and EM studies demonstrate two structurally linked mechanisms for activating integrin adhesiveness. First, extension moves the ligand-binding head 100 Å to 200 Å further away from the cell surface and orients it optimally for adhesion to another cell or to the extracellular matrix. Second, extension enables hybrid domain swing-out, which induces increased affinity for ligand.
The Compliant Integrin Legs
The design of the connecting rod and crankshaft bearing between the β I and hybrid domains and the rigidity of the hybrid domain/PSI domain unit amplify reshaping of the ligand-binding site into a 70 Å separation at the integrin knees. Such a large movement appears to be important for transmission of conformational change to the transmembrane and cytoplasmic domains because the β leg in particular is highly compliant, i.e., flexible. Below, we discuss the role of integrin α and β subunit transmembrane domain separation in activation. Transmembrane domain separation, extension, and hybrid domain swing-out are linked; however, this linkage is not tight because of the flexibility of the lower β leg. When extended αVβ3 or αXβ2 particles are imaged and class averaged, the domains in the lower β leg tend to disappear because they appear in different orientations and are averaged out (33, 43) (panels 2 and 3 in both Figure 9a and b). Fab binding results in better resolution of the lower β leg, both in clasped (Figure 9c, panels 1–3) and unclasped preparations (Figure 9c, panel 4). Both parallel and crossed orientations of the α and β legs are seen (Figure 9c, panels 1 and 2, respectively), and the β leg is clearly flexible above the Fab-binding sites in I-EGF domains 2 and 3, i.e., at the knee between I-EGF1 and I-EGF2, and appears to be flexible at other locations as well. This flexibility is symbolized with the dashed β legs in Figure 6c,d. In αVβ3 the α leg snaps into a preferred orientation when it is extended (33) (Figure 9a, panels 2 and 3). In αXβ2 the extended α leg is flexible at the genu (43) (Figure 9b,c).
Conformational Change of the Integrin Cytoplasmic and Transmembrane Domains
In the bent αVβ3 crystal structure, the α and β subunit ectodomain C termini are a few angstroms apart (31), consistent with association of the α and β subunit transmembrane domains. Ectodomain constructs with the C termini clasped have lower affinity for ligand than unclasped constructs (79). Many studies show that deletions or mutations in the α and β subunit transmembrane and cytoplasmic domains, which are expected to destabilize α/β association, activate integrins (80–83). Furthermore, replacement of the αL and β2 cytoplasmic domains with ACID/BASE peptides that form a heterodimeric α-helical coiled-coil stabilizes αLβ2 in an inactive state, whereas replacement with similar peptides that do not heterodimerize causes constitutive activation of αLβ2 (84). Fluorescent proteins were fused to the αL and β2 cytoplasmic domains for fluorescent resonance energy transfer (FRET) studies. These studies on live cells show that in the resting state the integrin α and β subunit cytoplasmic domains are close to one another (35). However, they undergo significant spatial separation upon inside-out activation induced by stimulation of protein kinase C, stimulation by a chemoattractant of a G protein-coupled receptor, or transfection with the talin head domain, which binds the integrin β cytoplasmic domain. Furthermore, extracellular addition of Mn2+ and soluble ICAM-1, which induces integrin extension as shown by exposure of the KIM127 epitope, also induces α and β subunit cytoplasmic domain separation (35).
NMR studies of the integrin cytoplasmic tails suggest that their association is weak, with significant differences between published structures of associated cytoplasmic domains (85, 86) or with undetectable association between α and β subunit cytoplasmic domains (87, 88). These studies imply that the cytoplasmic interaction is modest and/or transient and that other domains are required for stable α and β association. Binding of intracellular proteins such as RAPL (89) and the talin head domain (90–92) to the integrin cytoplasmic tails activates integrins for ligand binding, presumably by disrupting α and β association. Other proteins also bind to the cytoplasmic tails, including filamin, which competes with talin for binding to the β tail and modulates cell migration (93), and ICAP-1, which binds to the same motif as talin and has a related fold (94). The structural basis for talin and filamin binding to the integrin β cytoplasmic domain has been demonstrated by NMR and crystal studies (91, 95, 96).
Mutational studies have defined interfaces on the integrin α and β subunit transmembrane domains that, when substituted, result in activation (97–100). Furthermore, disulfide scanning of the exofacial portions of the transmembrane domains revealed a specific α-helical interface between the α and β transmembrane domains in the resting state (97). Disulfide scanning also revealed that after activation from inside the cell, the α and β subunits moved apart in the membrane instead of rearranging into a distinct α/β interface.
A Model for Bidirectional Signal Transmission by Integrins Across the Plasma Membrane
Based on the preponderance of the results described above, the following model is evident. Integrins are in equilibrium between different conformational states (Figure 6c,d). The bent conformation is stabilized by interfaces between the headpiece and the lower legs, between the lower α and β legs (33), and between the α and β transmembrane and cytoplasmic domains. However, none of these interfaces is tight, and small perturbations can readily shift the equilibrium toward extension and separation. Perturbation of the cytoplasmic domains by mutation or by binding of the talin head domain or other effector proteins induces separation of the cytoplasmic and transmembrane domains. This in turn results in separation of the α and β lower legs. Lower leg separation destabilizes the interface between the lower legs and the headpiece and results in integrin extension. Transmembrane domain separation would favor the open over the closed headpiece because the upper α and β legs are 70 Å further apart in the open than the closed headpiece. However, because the lower β leg is highly flexible, transmembrane domain and lower leg separation and extension would not be sufficient to enforce hybrid domain swing-out [compare panel 2 of Figure 6c or d (with dashed β leg) with panel 3 of Figure 6c or d (with solid β leg)]. EM studies and results with activation-dependent antibodies demonstrate that extension is sufficient to induce integrin adhesiveness and to enable a substantial proportion of integrin molecules to equilibrate to the high-affinity, open headpiece conformation. The set point for the equilibria between bent and extended conformations and between extended open headpiece and extended closed headpiece conformations is integrin dependent (43) and may help explain differences between integrins in their susceptibility to activation (101).
The mechanochemical design of integrins favors extension and hybrid domain swing-out when integrins function in adhesion. The distance in the β subunit between the ligand-binding site and the I-EGF1 domain is 20 Å further (Δx) in the open than in the closed headpiece conformation (see Figure 4b). Therefore, in cell migration or as a consequence of cytoskeleton contraction, when tensile force (F) is exerted on a ligand-bound integrin and resisted by cytoskeletal proteins bound to the β subunit cytoplasmic domain, the open headpiece conformation will be stabilized relative to the closed headpiece by approximately FΔx. Notably, the extended conformation would similarly be favored over the bent conformation and has a substantially greater Δx. Thus, a mechanochemical switch favors the high-affinity state when tensile force is applied to integrins, and this is expected to be of great importance for force resistance and mechanotransduction by integrins during cell adhesion and migration (102). This mechanochemical design stabilizes the high-affinity state when tensile force is applied to selectins and integrin α I domains as well, and the importance of how force is linked to allostery has been experimentally demonstrated for α I domains (103).
Conformational change can also be transmitted from the integrin ligand-binding site to the cytoplasm, as demonstrated with FRET (35). Which integrin conformation first binds ligand is unknown and may depend on (a) the rate of equilibration between different conformational states, (b) the population of the different states, and (c) the binding kinetics and affinities of the different states. However, the preponderance of EM and crystal structure studies demonstrates that once ligand is bound, it stabilizes the extended conformation with the open headpiece. Swing-out of the hybrid domain would favor, but seems unlikely to enforce, transmembrane domain separation because of the flexibility of the lower β leg (compare integrins with dashed and solid β legs in panel 3 of Figure 6c or d ). In agreement, a disulfide bond between the exofacial portions of the α and β subunit transmembrane domains does not prevent extracellular agents such as Mn2+ and antibodies from activating ligand binding, although it does prevent intracellular signals from activating ligand binding (97). It seems likely that the stability of α and β subunit transmembrane and cytoplasmic domain association is low, and that in the absence of close association between the α and β ectodomain C-terminal segments, the transmembrane and cytoplasmic domains spontaneously dissociate and thereby transmit signals into the cell.
Role and Regulation of Integrin Lateral Association
As discussed above, conformational mechanisms for regulating integrin affinity have become relatively well established. However, the role and regulation of integrin lateral redistribution on the cell surface, often referred to as clustering, has remained unclear and controversial (104). Several early studies suggested a dominant and proactive role for integrin redistribution in the initiation or priming of cells for efficient ligand binding (105). In practice, such valency regulation (104) has usually been inferred when activators promote cell adhesion without promoting detectable soluble ligand binding. However, this appears to reflect a lack of sensitivity of assays to intermediate levels of affinity, rather than a lack of affinity regulation. Recent improvements in soluble ligand-binding assays and studies with Fabs specific for the high-affinity conformation have clearly demonstrated rapid and transient integrin affinity regulation in response to chemokines (14, 106–108). Furthermore, sensitive assays often demonstrate that physiological stimuli normally induce markedly less soluble ligand binding than Mn2+ (109), which has been commonly employed as a positive control for affinity regulation.
The idea of clustering as a mode of priming implies proactive and directed mechanisms for lateral redistribution (110). Vesicular trafficking (111, 112) and Rap1- and RAPL-driven polarization of integrins to the lamellapodium (89, 113) represent important active modes of integrin reorganization that take place during cell migration. However, mechanistic support for active reorganization of integrins during the initial stages of priming remains tenuous. On the basis of the observation that peptides containing integrin α and β subunit transmembrane domains form homodimers and homotrimers in detergent, investigators have proposed that homomeric association between the transmembrane domains can induce integrin clustering (114). However, several subsequent studies in intact cells have shown that homomeric α- α or β-β association does not occur as a consequence of integrin priming and dissociation of αβ transmembrane domain heterodimers (75, 98). Other studies have implied a role for cholesterol-rich lipid rafts in driving integrin clustering, but this has remained controversial (104).
Many of the ideas on activation-induced integrin clustering have been replaced by an emerging model of multivalent ligand-dependent, mass-action-driven integrin redistribution that is modulated by the cytoskeleton (104). On resting cells β2 integrin mobility is confined by cytoskeletal interactions with the cytoplasmic tail (115). Cell activation by PMA or chemokine increases LFA-1 diffusion on the membrane (106, 116). Moreover, artificially increasing LFA-1 diffusiveness by actin cytoskeleton disruption enhances both mobility and adhesion (116). However, redistribution or clustering of integrins was not induced directly by treatments that increased membrane mobility alone, and redistribution was, instead, dependent on the presence of multivalent ligand substrates, suggesting a ligand- and mass-action-driven redistribution model that functions in adhesion strengthening rather than in priming (75, 117). Complexity is added to this model by findings that LFA-1 (118, 119) and other integrins (120) become confined upon ligand binding or stabilization of the open integrin conformation and that diffusion rates may depend on affinity states (121).
Integrin Outside-In Signaling
Binding of extracellular ligands by integrins results in signal transduction across the plasma membrane that regulates cell shape, migration, growth, and survival, a process termed outside-in signaling. Details of the many signaling pathways emanating from integrins are beyond the scope of this review, and readers are referred to several recent and extensive reviews (122–125). Investigators widely believe that lateral association (i.e., clustering) of integrin heterodimers, which occurs as a consequence of multivalent ligand binding (75, 117), plays a major role in outside-in signaling (see review in Reference 122). However, ligand binding can also directly lead to and stabilize separation of integrin cytoplasmic domains (35). To characterize the role of integrin conformational change (e.g., separation of the transmembrane and cytoplasmic domain interfaces) in outside-in signaling, a mutant with an intersubunit disulfide bond between the α and β transmembrane domains (97) was studied. The α IIbβ3 mutant retains Mn2+-stimulated ligand binding as described above and mediates adhesion to fibrinogen substrates. However, this mutant exhibits a profound defect in adhesion-induced outside-in signaling as measured by cell spreading, actin stress fiber, focal adhesion formation, and focal adhesion kinase activation ( J. Zhu, C. Carman, M. Kim, M. Shimaoka, T.A. Springer & B. Luo, unpublished observations). These defects in outside-in signaling were rescued by reduction of the intersubunit disulfide bond. Thus, separation of transmembrane domains is an important component of integrin outside-in signal transduction. The role of clustering might then be to facilitate interactions among different integrin-bound and focal adhesion-associated kinases to promote transphosphorylation/activation events in a fashion loosely analogous to the well-characterized paradigm of receptor tyrosine kinase subunit-subunit transactivation.
Concluding Remarks
Recent structural, biochemical, and biophysical studies have greatly advanced our understanding of the mechanisms of integrin bidirectional signaling across the plasma membrane. Indeed, perhaps more is known about how integrins transmit signals across the membrane than for any other receptors with two transmembrane domains, including receptor kinases. Accumulating evidence demonstrates that conformational affinity regulation plays a dominant role in integrin priming (inside-out signaling), whereas lateral redistribution (clustering) functions in adhesion strengthening, and both integrin conformational change and clustering are required for outside-in signaling. The many different conformational states of integrins are in dynamic equilibrium. Intracellular signals or ligand binding act by shifting the equilibrium, not by locking integrins in one specific state. Furthermore, inside-out signals activate only a subset of integrin molecules on the cell surface, and these may have a localized cell surface distribution. Much more remains to be learned about integrin structure, dynamics, and linkage to the cytoskeleton and both downstream and upstream effectors.
Acknowledgments
The authors are supported by the American Heart Association (0535403T to B.H.L.), the Arthritis Foundation (C.V.C.), and NIH grants (HL48675, CA31798, and CA31799 to T.A.S.). We thank Can Xie and Nori Nishida for help with figures.
- MIDAS
metal ion-dependent adhesion site
- ADMIDAS
adjacent to metal ion-dependent adhesion site
- I-EGF domain
integrin epidermal growth factor-like domain
Literature Cited
- 1.Pribila JT, Quale AC, Mueller KL, Shimizu Y. Integrins and T cell-mediated immunity. Annu Rev Immunol. 2004;22:157–80. doi: 10.1146/annurev.immunol.22.012703.104649. [DOI] [PubMed] [Google Scholar]
- 2.Kinashi T. Intracellular signaling controlling integrin activation in lymphocytes. Nat Rev Immunol. 2005;5:546–59. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- 3.Anderson DC, Springer TA. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med. 1987;38:175–94. doi: 10.1146/annurev.me.38.020187.001135. [DOI] [PubMed] [Google Scholar]
- 4.Lee JO, Rieu P, Arnaout MA, Liddington R. Crystal structure of the A domain from the α subunit of integrin CR3 (CD11b/CD18) Cell. 1995;80:631–38. doi: 10.1016/0092-8674(95)90517-0. [DOI] [PubMed] [Google Scholar]
- 5.Emsley J, Knight CG, Farndale RW, Barnes MJ, Liddington RC. Structural basis of collagen recognition by integrin α2β1. Cell. 2000;101:47–56. doi: 10.1016/S0092-8674(00)80622-4. [DOI] [PubMed] [Google Scholar]
- 6.Shimaoka M, Xiao T, Liu JH, Yang Y, Dong Y, et al. Structures of the αL I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell. 2003;112:99–111. doi: 10.1016/s0092-8674(02)01257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song G, Yang Y, Liu JH, Casasnovas JM, Shimaoka M, et al. An atomic resolution view of ICAM recognition in a complex between the binding domains of ICAM-3 and integrin αLβ2. Proc Natl Acad Sci USA. 2005;102:3366–71. doi: 10.1073/pnas.0500200102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JO, Bankston LA, Arnaout MA, Liddington RC. Two conformations of the integrin A-domain (I-domain): a pathway for activation? Structure. 1995;3:1333–40. doi: 10.1016/s0969-2126(01)00271-4. [DOI] [PubMed] [Google Scholar]
- 9.Shimaoka M, Shifman JM, Jing H, Takagi J, Mayo SL, Springer TA. Computational design of an integrin I domain stabilized in the open, high affinity conformation. Nat Struct Biol. 2000;7:674–78. doi: 10.1038/77978. [DOI] [PubMed] [Google Scholar]
- 10.Jin M, Song G, Kim YS, Astrof N, Shimaoka M, et al. Directed evolution to probe protein allostery and integrin I domains of 200,000-fold higher affinity. Proc Natl Acad Sci USA. 2006;103:5758–63. doi: 10.1073/pnas.0601164103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimaoka M, Lu C, Palframan RT, von Andrian UH, McCormack A, et al. Reversibly locking a protein fold in an active conformation with a disulfide bond: integrin αL I domains with high affinity and antagonist activity in vivo. Proc Natl Acad Sci USA. 2001;98:6009–14. doi: 10.1073/pnas.101130498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin M, Andricioaei I, Springer TA. Conversion between three conformational states of integrin I domains with a C-terminal pull spring studied with molecular dynamics. Structure. 2004;12:2137–47. doi: 10.1016/j.str.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Huang L, Shimaoka M, Rondon I, Roy I, Chang Q, et al. Identification and characterization of a human monoclonal antagonistic antibody AL-57 that preferentially binds the high-affinity form of lymphocyte function-associated antigen-1. J Leukoc Biol. 2006;80:905–14. doi: 10.1189/jlb.1105649.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimaoka M, Kim M, Cohen EH, Yang W, Astrof N, et al. AL-57, a ligand-mimetic antibody to integrin LFA-1, reveals chemokine-induced affinity up-regulation in lymphocytes. Proc Natl Acad Sci USA. 2006;103:13991–96. doi: 10.1073/pnas.0605716103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diamond MS, Garcia-Aguilar J, Bickford JK, Corbi AL, Springer TA. The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J Cell Biol. 1993;120:1031–43. doi: 10.1083/jcb.120.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kallen J, Welzenbach K, Ramage P, Geyl D, Kriwacki R, et al. Structural basis for LFA-1 inhibition upon lovastatin binding to the CD11a I-domain. J Mol Biol. 1999;292:1–9. doi: 10.1006/jmbi.1999.3047. [DOI] [PubMed] [Google Scholar]
- 17.Last-Barney K, Davidson W, Cardozo M, Frye LL, Grygon CA, et al. Binding site elucidation of hydantoin-based antagonists of LFA-1 using multidisciplinary technologies: evidence for the allosteric inhibition of a protein-protein interaction. J Am Chem Soc. 2001;123:5643–50. doi: 10.1021/ja0104249. [DOI] [PubMed] [Google Scholar]
- 18.Liu G, Huth JR, Olejniczak ET, Mendoza R, DeVries P, et al. Novel p-arylthio cinnamides as antagonists of leukocyte function-associated antigen-1/intracellular adhesion molecule-1 interaction. 2. Mechanism of inhibition and structure-based improvement of pharmaceutical properties. J Med Chem. 2001;44:1202–10. doi: 10.1021/jm000503f. [DOI] [PubMed] [Google Scholar]
- 19.Lu C, Shimaoka M, Zang Q, Takagi J, Springer TA. Locking in alternate conformations of the integrin αLβ2 I domain with disulfide bonds reveals functional relationships among integrin domains. Proc Natl Acad Sci USA. 2001;98:2393–98. doi: 10.1073/pnas.041618598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu C, Shimaoka M, Ferzly M, Oxvig C, Takagi J, Springer TA. An isolated, surface-expressed I domain of the integrin αLβ2 is sufficient for strong adhesive function when locked in the open conformation with a disulfide. Proc Natl Acad Sci USA. 2001;98:2387–92. doi: 10.1073/pnas.041606398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gahmberg CG, Tolvanen M, Kotovuori P. Leukocyte adhesion: structure and function of human leukocyte β2-integrins and their cellular ligands. Eur J Biochem. 1997;245:215–32. doi: 10.1111/j.1432-1033.1997.00215.x. [DOI] [PubMed] [Google Scholar]
- 22.Song G, Lazar GA, Kortemme T, Shimaoka M, Desjarlais JR, et al. Rational design of ICAM-1 variants for antagonizing integrin LFA-1-dependent adhesion. J Biol Chem. 2006;281:5042–49. doi: 10.1074/jbc.M510454200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casasnovas JM, Springer TA, Liu J, Harrison SC, Wang J. The crystal structure of ICAM-2 reveals a distinctive integrin recognition surface. Nature. 1997;387:312–15. doi: 10.1038/387312a0. [DOI] [PubMed] [Google Scholar]
- 24.Yang Y, Jun CD, Liu J, Zhang RG, Jochimiak A, et al. Structural basis for dimerization of ICAM-1 on the cell surface. Mol Cell. 2004;14:269–76. doi: 10.1016/s1097-2765(04)00204-7. [DOI] [PubMed] [Google Scholar]
- 25.Reilly PL, Woska JR, Jr, Jeanfavre DD, McNally E, Rothlein R, Bormann BJ. The native structure of intercellular adhesion molecule-1 (ICAM-1) is a dimer: correlation with binding to LFA-1. J Immunol. 1995;155:529–32. [PubMed] [Google Scholar]
- 26.Miller J, Knorr R, Ferrone M, Houdei R, Carron CP, Dustin ML. Intercellular adhesion molecule-1 dimerization and its consequences for adhesion mediated by lymphocyte function associated-1. J Exp Med. 1995;182:1231–41. doi: 10.1084/jem.182.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altieri DC, Morrissey JH, Edgington TS. Adhesive receptor Mac-1 coordinates the activation of factor X on stimulated cells of monocytic and myeloid differentiation: an alternative initiation of the coagulation protease cascade. Proc Natl Acad Sci USA. 1988;85:7462–66. doi: 10.1073/pnas.85.20.7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diamond MS, Staunton DE, de Fougerolles AR, Stacker SA, Garcia-Aguilar J, et al. ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18) J Cell Biol. 1990;111:3129–39. doi: 10.1083/jcb.111.6.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vorup-Jensen T, Carman CV, Shimaoka M, Schuck P, Svitel J, Springer TA. Exposure of acidic residues as a danger signal for recognition of fibrinogen and other macromolecules by integrin αXβ2. Proc Natl Acad Sci USA. 2005;102:1614–19. doi: 10.1073/pnas.0409057102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nermut MV, Green NM, Eason P, Yamada SS, Yamada KM. Electron microscopy and structural model of human fibronectin receptor. EMBO J. 1988;7:4093–99. doi: 10.1002/j.1460-2075.1988.tb03303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiong J-P, Stehle T, Diefenbach B, Zhang R, Dunker R, et al. Crystal structure of the extracellular segment of integrin αVβ3. Science. 2001;294:339–45. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, et al. Crystal structure of the extracellular segment of integrin αVβ3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296:151–55. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 33.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–611. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 34.Takagi J, Strokovich K, Springer TA, Walz T. Structure of integrin α5β1 in complex with fibronectin. EMBO J. 2003;22:4607–15. doi: 10.1093/emboj/cdg445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–25. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 36.Xiao T, Takagi J, Wang J, Coller BS, Springer TA. Structural basis for allostery in integrins and binding of ligand-mimetic therapeutics to the platelet receptor for fibrinogen. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Springer TA. Folding of the N-terminal, ligand-binding region of integrin α-subunits into β-propeller domain. Proc Natl Acad Sci USA. 1997;94:65–72. doi: 10.1073/pnas.94.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bilsland CA, Diamond MS, Springer TA. The leukocyte integrin p150,95 (CD11c/CD18) as a receptor for iC3b: activation by a heterologous β subunit and localization of a ligand recognition site to the I domain. J Immunol. 1994;152:4582–89. [PubMed] [Google Scholar]
- 39.Huang C, Zang Q, Takagi J, Springer TA. Structural and functional studies with antibodies to the integrin β2 subunit: a model for the I-like domain. J Biol Chem. 2000;275:21514–24. doi: 10.1074/jbc.M002286200. [DOI] [PubMed] [Google Scholar]
- 40.Xiong JP, Stehle T, Goodman SL, Arnaout MA. A novel adaptation of the integrin PSI domain revealed from its crystal structure. J Biol Chem. 2004;279:40252–54. doi: 10.1074/jbc.C400362200. [DOI] [PubMed] [Google Scholar]
- 41.Shi M, Sundramurthy K, Liu B, Tan SM, Law SK, Lescar J. The crystal structure of the plexin-semaphorin-integrin domain/hybrid domain/I-EGF1 segment from the human integrin β2 subunit at 1.8-Å resolution. J Biol Chem. 2005;280:30586–93. doi: 10.1074/jbc.M502525200. [DOI] [PubMed] [Google Scholar]
- 42.Takagi J, Springer TA. Integrin activation and structural rearrangement. Immunol Rev. 2002;186:141–63. doi: 10.1034/j.1600-065x.2002.18613.x. [DOI] [PubMed] [Google Scholar]
- 43.Nishida N, Xie C, Shimaoka M, Cheng Y, Walz T, Springer TA. Activation of leukocyte β2 integrins by conversion from bent to extended conformations. Immunity. 2006;25:583–94. doi: 10.1016/j.immuni.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 44.Honda S, Tomiyama Y, Pelletier AJ, Annis D, Honda Y, et al. Topography of ligand-induced binding sites, including a novel cation-sensitive epitope (AP5) at the amino terminus, of the human integrin β3 subunit. J Biol Chem. 1995;270:11947–54. doi: 10.1074/jbc.270.20.11947. [DOI] [PubMed] [Google Scholar]
- 45.Lu C, Ferzly M, Takagi J, Springer TA. Epitope mapping of antibodies to the C-terminal region of the integrin β2 subunit reveals regions that become exposed upon receptor activation. J Immunol. 2001;166:5629–37. doi: 10.4049/jimmunol.166.9.5629. [DOI] [PubMed] [Google Scholar]
- 46.Mould AP, Travis MA, Barton SJ, Hamilton JA, Askari JA, et al. Evidence that monoclonal antibodies directed against the integrin β subunit plexin/semaphorin/integrin domain stimulate function by inducing receptor extension. J Biol Chem. 2005;280:4238–46. doi: 10.1074/jbc.M412240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peterson JA, Nyree CE, Newman PJ, Aster RH. A site involving the “hybrid” and PSI homology domains of GPIIIa (β3-integrin subunit) is a common target for antibodies associated with quinine-induced immune thrombocytopenia. Blood. 2003;101:937–42. doi: 10.1182/blood-2002-07-2336. [DOI] [PubMed] [Google Scholar]
- 48.Xie C, Shimaoka M, Xiao T, Schwab P, Klickstein LB, Springer TA. The integrin α subunit leg extends at a Ca2+-dependent epitope in the thigh/genu interface upon activation. Proc Natl Acad Sci USA. 2004;101:15422–27. doi: 10.1073/pnas.0406680101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen JF, Salas A, Springer TA. Bistable regulation of integrin adhesiveness by a bipolar metal ion cluster. Nat Struct Biol. 2003;10:995–1001. doi: 10.1038/nsb1011. [DOI] [PubMed] [Google Scholar]
- 50.Mould AP, Barton SJ, Askari JA, Craig SE, Humphries MJ. Role of ADMIDAS cation-binding site in ligand recognition by integrin α5β1. J Biol Chem. 2003;278:51622–29. doi: 10.1074/jbc.M306655200. [DOI] [PubMed] [Google Scholar]
- 51.Chen JF, Yang W, Kim M, Carman CV, Springer TA. Regulation of outside-in signaling and affinity by the β2 I domain of integrin αLβ2. Proc Natl Acad Sci USA. 2006;103:13991–96. doi: 10.1073/pnas.0605666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huth JR, Olejniczak ET, Mendoza R, Liang H, Harris EA, et al. NMR and mutagenesis evidence for an I domain allosteric site that regulates lymphocyte function-associated antigen 1 ligand binding. Proc Natl Acad Sci USA. 2000;97:5231–36. doi: 10.1073/pnas.97.10.5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alonso JL, Essafi M, Xiong JP, Stehle T, Arnaout MA. Does the integrin αA domain act as a ligand for its βA domain? Curr Biol. 2002;12:R340–42. doi: 10.1016/s0960-9822(02)00852-7. [DOI] [PubMed] [Google Scholar]
- 54.Yang W, Shimaoka M, Salas A, Takagi J, Springer TA. Inter-subunit signal transmission in integrins by a receptor-like interaction with a pull spring. Proc Natl Acad Sci USA. 2004;101:2906–11. doi: 10.1073/pnas.0307340101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Welzenbach K, Hommel U, Weitz-Schmidt G. Small molecule inhibitors induce conformational changes in the I domain and the I-like domain of lymphocyte function-associated antigen-1: molecular insights into integrin inhibition. J Biol Chem. 2002;277:10590–98. doi: 10.1074/jbc.M110521200. [DOI] [PubMed] [Google Scholar]
- 56.Shimaoka M, Salas A, Yang W, Weitz-Schmidt G, Springer TA. Small molecule integrin antagonists that bind to the β2 subunit I-like domain and activate signals in one direction and block them in another. Immunity. 2003;19:391–402. doi: 10.1016/s1074-7613(03)00238-3. [DOI] [PubMed] [Google Scholar]
- 57.Salas A, Shimaoka M, Kogan AN, Harwood C, von Andrian UH, Springer TA. Rolling adhesion through an extended conformation of integrin αLβ2 and relation to α I and β I-like domain interaction. Immunity. 2004;20:393–406. doi: 10.1016/s1074-7613(04)00082-2. [DOI] [PubMed] [Google Scholar]
- 58.Knorr R, Dustin ML. The lymphocyte function-associated antigen 1 I domain is a transient binding module for intercellular adhesion molecule (ICAM)-1 and ICAM-1 in hydrodynamic flow. J Exp Med. 1997;186:719–30. doi: 10.1084/jem.186.5.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salas A, Shimaoka M, Chen SQ, Carman CV, Springer TA. Transition from rolling to firm adhesion is regulated by the conformation of the I domain of the integrin LFA-1. J Biol Chem. 2002;277:50255–62. doi: 10.1074/jbc.M209822200. [DOI] [PubMed] [Google Scholar]
- 60.Iwasaki K, Mitsuoka K, Fujiyoshi Y, Fujisawa Y, Kikuchi M, et al. Electron tomography reveals diverse conformations of integrin α IIbβ3 in the active state. J Struct Biol. 2005;150:259–67. doi: 10.1016/j.jsb.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 61.Mould AP, Symonds EJ, Buckley PA, Grossmann JG, McEwan PA, et al. Structure of an integrin-ligand complex deduced from solution X-ray scattering and site-directed mutagenesis. J Biol Chem. 2003;278:39993–99. doi: 10.1074/jbc.M304627200. [DOI] [PubMed] [Google Scholar]
- 62.Luo BH, Springer TA, Takagi J. Stabilizing the open conformation of the integrin headpiece with a glycan wedge increases affinity for ligand. Proc Natl Acad Sci USA. 2003;100:2403–8. doi: 10.1073/pnas.0438060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo BH, Strokovich K, Walz T, Springer TA, Takagi J. Allosteric β1 integrin antibodies that stabilize the low affinity state by preventing the swing-out of the hybrid domain. J Biol Chem. 2004;279:27466–71. doi: 10.1074/jbc.M404354200. [DOI] [PubMed] [Google Scholar]
- 64.Tng E, Tan SM, Ranganathan S, Cheng M, Law SK. The integrin αLβ2 hybrid domain serves as a link for the propagation of activation signal from its stalk regions to the I-like domain. J Biol Chem. 2004;279:54334–39. doi: 10.1074/jbc.M407818200. [DOI] [PubMed] [Google Scholar]
- 65.Mould AP, Barton SJ, Askari JA, McEwan PA, Buckley PA, et al. Conformational changes in the integrin βA domain provide a mechanism for signal transduction via hybrid domain movement. J Biol Chem. 2003;278:17028–35. doi: 10.1074/jbc.M213139200. [DOI] [PubMed] [Google Scholar]
- 66.Tang RH, Tng E, Law SK, Tan SM. Epitope mapping of monoclonal antibody to integrin αLβ2 hybrid domain suggests different requirements of affinity states for inter-cellular adhesion molecules (ICAM)-1 and ICAM-3 binding. J Biol Chem. 2005;280:29208–16. doi: 10.1074/jbc.M503239200. [DOI] [PubMed] [Google Scholar]
- 67.Arnaout MA, Mahalingam B, Xiong JP. Integrin structure, allostery, and bidirectional signaling. Annu Rev Cell Dev Biol. 2005;21:381–410. doi: 10.1146/annurev.cellbio.21.090704.151217. [DOI] [PubMed] [Google Scholar]
- 68.Luo BH, Springer TA. Integrin structures and conformational signaling. Curr Opin Cell Biol. 2006;18:579–86. doi: 10.1016/j.ceb.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adair BD, Xiong JP, Maddock C, Goodman SL, Arnaout MA, Yeager M. Three-dimensional EM structure of the ectodomain of integrin αVβ3 in a complex with fibronectin. J Cell Biol. 2005;168:1109–18. doi: 10.1083/jcb.200410068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Robinson MK, Andrew D, Rosen H, Brown D, Ortlepp S, et al. Antibody against the Leu-CAM β-chain (CD18) promotes both LFA-1-and CR3-dependent adhesion events. J Immunol. 1992;148:1080–85. [PubMed] [Google Scholar]
- 71.Andrew D, Shock A, Ball E, Ortlepp S, Bell J, Robinson M. KIM185, a monoclonal antibody to CD18 which induces a change in the conformation of CD18 and promotes both LFA-1- and CR3-dependent adhesion. Eur J Immunol. 1993;23:2217–22. doi: 10.1002/eji.1830230925. [DOI] [PubMed] [Google Scholar]
- 72.Evangelista V, Manarini S, Rotondo S, Martelli N, Polischuk R, et al. Platelet/polymorphonuclear leukocyte interaction in dynamic conditions: evidence of adhesion cascade and cross talk between P-selectin and the β2 integrin CD11b/CD18. Blood. 1996;88:4183–94. [PubMed] [Google Scholar]
- 73.Takami M, Herrera R, Petruzzelli L. Mac-1-dependent tyrosine phosphorylation during neutrophil adhesion. Am J Physiol Cell Physiol. 2001;280:C1045–56. doi: 10.1152/ajpcell.2001.280.5.C1045. [DOI] [PubMed] [Google Scholar]
- 74.Petruzzelli L, Maduzia L, Springer TA. Activation of LFA-1 (CD11a/CD18) and Mac-1 (CD11b/CD18) mimicked by an antibody directed against CD18. J Immunol. 1995;155:854–66. [PubMed] [Google Scholar]
- 75.Kim M, Carman CV, Yang W, Salas A, Springer TA. The primacy of affinity over clustering in regulation of adhesiveness of the integrin αLβ2. J Cell Biol. 2004;167:1241–53. doi: 10.1083/jcb.200404160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shimaoka M, Lu C, Salas A, Xiao T, Takagi J, Springer TA. Stabilizing the integrin αM inserted domain in alternative conformations with a range of engineered disulfide bonds. Proc Natl Acad Sci USA. 2002;99:16737–41. doi: 10.1073/pnas.252633099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takagi J, Beglova N, Yalamanchili P, Blacklow SC, Springer TA. Definition of EGF-like, closely interacting modules that bear activation epitopes in integrin β subunits. Proc Natl Acad Sci USA. 2001;98:11175–80. doi: 10.1073/pnas.201420198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang W, Shimaoka M, Chen JF, Springer TA. Activation of integrin β subunit I-like domains by one-turn C-terminal α-helix deletions. Proc Natl Acad Sci USA. 2004;101:2333–38. doi: 10.1073/pnas.0307291101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takagi J, Erickson HP, Springer TA. C-terminal opening mimics “inside-out” activation of integrin α5β1. Nat Struct Biol. 2001;8:412–16. doi: 10.1038/87569. [DOI] [PubMed] [Google Scholar]
- 80.O'Toole TE, Mandelman D, Forsyth J, Shattil SJ, Plow EF, Ginsberg MH. Modulation of the affinity of integrin αIIbβ3 (GPIIb-IIIa) by the cytoplasmic domain of αIIb. Science. 1991;254:845–47. doi: 10.1126/science.1948065. [DOI] [PubMed] [Google Scholar]
- 81.O'Toole TE, Katagiri Y, Faull RJ, Peter K, Tamura R, et al. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994;124:1047–59. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hughes PE, Diaz-Gonzalez F, Leong L, Wu C, McDonald JA, et al. Breaking the integrin hinge. J Biol Chem. 1996;271:6571–74. doi: 10.1074/jbc.271.12.6571. [DOI] [PubMed] [Google Scholar]
- 83.Lu C, Springer TA. The α subunit cytoplasmic domain regulates the assembly and adhesiveness of integrin lymphocyte function-associated antigen-1 (LFA-1) J Immunol. 1997;159:268–78. [PubMed] [Google Scholar]
- 84.Lu C, Takagi J, Springer TA. Association of the membrane-proximal regions of the α and β subunit cytoplasmic domains constrains an integrin in the inactive state. J Biol Chem. 2001;276:14642–48. doi: 10.1074/jbc.M100600200. [DOI] [PubMed] [Google Scholar]
- 85.Vinogradova O, Velyvis A, Velyviene A, Hu B, Haas TA, et al. A structural mechanism of integrin αIIbβ3 “inside-out” activation as regulated by its cytoplasmic face. Cell. 2002;110:587–97. doi: 10.1016/s0092-8674(02)00906-6. [DOI] [PubMed] [Google Scholar]
- 86.Weljie AM, Hwang PM, Vogel HJ. Solution structures of the cytoplasmic tail complex from platelet α IIb- and β3-subunits. Proc Natl Acad Sci USA. 2002;99:5878–83. doi: 10.1073/pnas.092515799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ulmer TS, Yaspan B, Ginsberg MH, Campbell ID. NMR analysis of structure and dynamics of the cytosolic tails of integrin α IIbβ3 in aqueous solution. Biochemistry. 2001;40:7498–508. doi: 10.1021/bi010338l. [DOI] [PubMed] [Google Scholar]
- 88.Vinogradova O, Vaynberg J, Kong X, Haas TA, Plow EF, Qin J. Membrane-mediated structural transitions at the cytoplasmic face during integrin activation. Proc Natl Acad Sci USA. 2004;101:4094–99. doi: 10.1073/pnas.0400742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Katagiri K, Maeda A, Shimonaka M, Kinashi T. RAPL, a novel Rap1-binding molecule, mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat Immunol. 2003;4:741–48. doi: 10.1038/ni950. [DOI] [PubMed] [Google Scholar]
- 90.Calderwood DA, Zent R, Grant R, Rees DJ, Hynes RO, Ginsberg MH. The talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. J Biol Chem. 1999;274:28071–74. doi: 10.1074/jbc.274.40.28071. [DOI] [PubMed] [Google Scholar]
- 91.Garcia-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, et al. Structural determinants of integrin recognition by talin. Mol Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- 92.Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, et al. Talin binding to integrin β tails: a final common step in integrin activation. Science. 2003;302:103–6. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 93.Calderwood DA, Huttenlocher A, Kiosses WB, Rose DM, Woodside DG, et al. Increased filamin binding to β-integrin cytoplasmic domains inhibits cell migration. Nat Cell Biol. 2001;3:1060–68. doi: 10.1038/ncb1201-1060. [DOI] [PubMed] [Google Scholar]
- 94.Chang DD, Hoang BQ, Liu J, Springer TA. Molecular basis for interaction between Icap1α PTB domain and β1 integrin. J Biol Chem. 2002;277:8140–45. doi: 10.1074/jbc.M109031200. [DOI] [PubMed] [Google Scholar]
- 95.Ulmer TS, Calderwood DA, Ginsberg MH, Campbell ID. Domain-specific interactions of talin with the membrane-proximal region of the integrin β3 subunit. Biochemistry. 2003;42:8307–12. doi: 10.1021/bi034384s. [DOI] [PubMed] [Google Scholar]
- 96.Kiema T, Lad Y, Jiang P, Oxley CL, Baldassarre M, et al. The molecular basis of filamin binding to integrins and competition with talin. Mol Cell. 2006;21:337–47. doi: 10.1016/j.molcel.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 97.Luo BH, Springer TA, Takagi J. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol. 2004;2:776–86. doi: 10.1371/journal.pbio.0020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Luo BH, Carman CV, Takagi J, Springer TA. Disrupting integrin transmembrane domain heterodimerization increases ligand binding affinity, not valency or clustering. Proc Natl Acad Sci USA. 2005;102:3679–84. doi: 10.1073/pnas.0409440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li W, Metcalf DG, Gorelik R, Li R, Mitra N, et al. A push-pull mechanism for regulating integrin function. Proc Natl Acad Sci USA. 2005;102:1424–29. doi: 10.1073/pnas.0409334102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Partridge AW, Liu S, Kim S, Bowie JU, Ginsberg MH. Transmembrane domain packing stabilizes integrin α IIbβ3 in the low affinity state. J Biol Chem. 2005;280:7294–300. doi: 10.1074/jbc.M412701200. [DOI] [PubMed] [Google Scholar]
- 101.Bazzoni G, Ma L, Blue ML, Hemler ME. Divalent cations and ligands induce conformational changes that are highly divergent among β1 integrins. J Biol Chem. 1998;273:6670–78. doi: 10.1074/jbc.273.12.6670. [DOI] [PubMed] [Google Scholar]
- 102.Chen CS, Tan J, Tien J. Mechanotransduction at cell-matrix and cell-cell contacts. Annu Rev Biomed Eng. 2004;6:275–302. doi: 10.1146/annurev.bioeng.6.040803.140040. [DOI] [PubMed] [Google Scholar]
- 103.Astrof NS, Salas A, Shimaoka M, Chen JF, Springer TA. The importance of force linkage in mechanochemistry of adhesion receptors. Biochemistry. 2006;45:15020–28. doi: 10.1021/bi061566o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Carman CV, Springer TA. Integrin avidity regulation: Are changes in affinity and conformation underemphasized? Curr Opin Cell Biol. 2003;15:547–56. doi: 10.1016/j.ceb.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 105.Bazzoni G, Hemler ME. Are changes in integrin affinity and conformation overemphasized? Trends Biochem Sci. 1998;23:30–34. doi: 10.1016/s0968-0004(97)01141-9. [DOI] [PubMed] [Google Scholar]
- 106.Constantin G, Majeed M, Giagulli C, Piccib L, Kim JY, et al. Chemokines trigger immediate β2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity. 2000;13:759–69. doi: 10.1016/s1074-7613(00)00074-1. [DOI] [PubMed] [Google Scholar]
- 107.Chan JR, Hyduk SJ, Cybulsky MI. Chemoattractants induce rapid and transient upregulation of monocyte α4 integrin affinity for vascular adhesion molecule 1 which mediates arrest: an early step in the process of emmigration. J Exp Med. 2001;193:1149–58. doi: 10.1084/jem.193.10.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chan JR, Hyduk SJ, Cybulsky MI. Detecting rapid and transient upregulation of leukocyte integrin affinity induced by chemokines and chemoattractants. J Immunol Methods. 2003;273:43–52. doi: 10.1016/s0022-1759(02)00417-9. [DOI] [PubMed] [Google Scholar]
- 109.Lollo BA, Chan KW, Hanson EM, Moy VT, Brian AA. Direct evidence for two affinity states for lymphocyte function-associated antigen 1 on activated T cells. J Biol Chem. 1993;268:21693–700. [PubMed] [Google Scholar]
- 110.van Kooyk Y, Figdor CG. Avidity regulation of integrins: the driving force in leukocyte adhesion. Curr Opin Cell Biol. 2000;12:542–47. doi: 10.1016/s0955-0674(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 111.Lawson MA, Maxfield FR. Ca2+- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature. 1995;377:75–79. doi: 10.1038/377075a0. [DOI] [PubMed] [Google Scholar]
- 112.Tohyama Y, Katagiri K, Pardi R, Lu C, Springer TA, Kinashi T. The critical cytoplasmic regions of the αL/β2 integrin in Rap1-induced adhesion and migration. Mol Biol Cell. 2003;14:2570–82. doi: 10.1091/mbc.E02-09-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shimonaka M, Katagiri K, Kakayama T, Fujita N, Tsuruo T, et al. Rap1 translates chemokine signals to integrin activation, cell polarization, and motility across vascular endothelium under flow. J Cell Biol. 2003;161:417–27. doi: 10.1083/jcb.200301133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li R, Mitra N, Gratkowski H, Vilaire G, Litvinov SV, et al. Activation of integrin α IIbβ3 by modulation of transmembrane helix associations. Science. 2003;300:795–98. doi: 10.1126/science.1079441. [DOI] [PubMed] [Google Scholar]
- 115.Jin T, Li J. Dynamitin controls β2 integrin avidity by modulating cytoskeletal constraint on integrin molecules. J Biol Chem. 2002;277:32963–69. doi: 10.1074/jbc.M201525200. [DOI] [PubMed] [Google Scholar]
- 116.Kucik DF, Dustin ML, Miller JM, Brown EJ. Adhesion-activating phorbol ester increases the mobility of leukocyte integrin LFA-1 in cultured lymphocytes. J Clin Invest. 1996;97:2139–44. doi: 10.1172/JCI118651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Buensuceso C, De Virgilio M, Shattil SJ. Detection of integrin α IIbβ3 clustering in living cells. J Biol Chem. 2003;278:15217–24. doi: 10.1074/jbc.M213234200. [DOI] [PubMed] [Google Scholar]
- 118.Peters IM, van Kooyk Y, van Vliet SJ, de Grooth BG, Figdor CG, Greve J. 3D single-particle tracking and optical trap measurements on adhesion proteins. Cytometry. 1999;36:189–94. doi: 10.1002/(sici)1097-0320(19990701)36:3<189::aid-cyto7>3.3.co;2-v. [DOI] [PubMed] [Google Scholar]
- 119.Smith A, Carrasco YR, Stanley P, Kieffer N, Batista FD, Hogg N. A talin-dependent LFA-1 focal zone is formed by rapidly migrating T lymphocytes. J Cell Biol. 2005;170:141–51. doi: 10.1083/jcb.200412032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Felsenfeld DP, Choquet D, Sheetz MP. Ligand binding regulates the directed movement of β1 integrins on fibroblasts. Nature. 1996;383:438–40. doi: 10.1038/383438a0. [DOI] [PubMed] [Google Scholar]
- 121.Cairo CW, Mirchev R, Golan DE. Cytoskeletal regulation couples LFA-1 conformational changes to receptor lateral mobility and clustering. Immunity. 2006;25:297–308. doi: 10.1016/j.immuni.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 122.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104:1606–15. doi: 10.1182/blood-2004-04-1257. [DOI] [PubMed] [Google Scholar]
- 123.Grashoff C, Thievessen I, Lorenz K, Ussar S, Fassler R. Integrin-linked kinase: integrin's mysterious partner. Curr Opin Cell Biol. 2004;16:565–71. doi: 10.1016/j.ceb.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 124.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 125.Guo W, Giancotti FG. Integrin signaling during tumor progression. Nat Rev Mol Cell Biol. 2004;5:816–26. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 126.Van der Vieren M, Crowe DT, Hoekstra D, Vazeux R, Hoffman PA, et al. The leukocyte integrin αDβ2 binds VCAM-1: evidence for a binding interface between I domain and VCAM-1. J Immunol. 1999;163:1984–90. [PubMed] [Google Scholar]
- 127.Schlossman SF, Boumsell L, Gilks W, Harlan JM, Kishimoto TK, et al. White Cell Differentiation Antigens. New York: Oxford Univ. Press; 1995. [Google Scholar]
- 128.Kishimoto TK, Kikutani H, von dem Borne AEGK, Goyert SM, Mason DY, et al. White Cell Differentiation Antigens. New York: Garland; 1997. [Google Scholar]