

Abstract
Recombinant proteins are being evaluated as smallpox and monkeypox vaccines because of their perceived safety compared to live vaccinia virus. Previously, we demonstrated that three or more injections of a Ribi-type adjuvant with a combination of three proteins from the outer membranes of intracellular (L1 protein) and extracellular (A33 and B5 proteins) forms of vaccinia virus protected mice against a lethal intranasal challenge with vaccinia virus. Here, we compared several adjuvants and found that QS-21 and to a lesser extent alum + CpG oligodeoxynucleotides accelerated and enhanced neutralizing antibody responses to a mixture of L1 and A33 proteins, provided the highest ratio of IgG2a to IgG1 isotype response, and protected mice against disease and death after only two immunizations 3 weeks apart. In addition, monkeys immunized with recombinant vaccinia virus proteins and QS-21 developed neutralizing antibody to monkeypox virus and had reduced virus load, skin lesions, and morbidity compared to the non-immunized group following monkeypox virus challenge.
Keywords: Smallpox, Monkeypox, Vaccinia virus
1. Introduction
The eradication of smallpox, through the administration of a vaccine comprised of live vaccinia virus (VACV), saved many millions of lives [1]. In addition to ending the mortality and morbidity of smallpox, eradication of this disease permitted the cessation of vaccination and the associated expense and adverse reactions, which can be life threatening particularly for the immunocompromised [2]. Nevertheless, having stopped vaccination, the human population is now more susceptible to smallpox, as well as monkeypox, than it was 30 years ago [3]. The decline in immunity and apprehension regarding the reintroduction of variola virus, the causative agent of smallpox, as a biological weapon have stimulated efforts to modernize and stockpile the conventional smallpox vaccine and to develop safer vaccine candidates. Highly attenuated strains of VACV and recombinant proteins and DNA provide the basis for alternative smallpox vaccines that might be used for those most vulnerable to the side effects of the standard vaccine.
The rational selection of immunogens for recombinant vaccines is dependent on knowledge of poxvirus structure and function [4]. Several studies demonstrated that antibodies to the extracellular (EV) form of VACV, in addition to the intracellular mature form (MV), provide superior protection to orthopoxviruses in small animal models [5], [6], [7]. MVs comprise the most basic form of the infectious particle and are released as such upon cell lysis. EVs, which have undergone exocytosis from cells at the plasma membrane to enhance spread within the host, are essentially MVs with an additional membrane [8], [9], [10]. Importantly, the viral protein constituents of the MV and outer EV membrane are entirely different and therefore present different immune targets. Several MV and EV proteins have been identified as protective immunogens in orthopoxvirus infections of experimental animals. Individual recombinant A27, L1 and H3 MV proteins [11], [12], [13] and A33 and B5 EV proteins [11], [14], [15] can induce partial protection. However, both DNA [16] and protein [11] immunization studies indicated that multicomponent vaccines eliciting antibodies to MV and EV proteins provided better protection against VACV than single component vaccines. In addition, monkeypox virus (MPXV) DNA priming and protein boosting provided better protection in a MPXV model than either alone [17]. Results obtained by passive administration of polyclonal or monoclonal IgGs to A33, B5 and L1 suggested that antibodies are important for the protection achieved by protein vaccines [18], [19].
Immunostimulatory adjuvants have been developed to enhance immune responses to weak protein immunogens. In our previous study [11], we used a non-toxic derivative of the Gram negative bacterial lipopolysaccharide monophosphoryl lipid A (MPL), which retains the ability to stimulate the innate immune response via the toll-like receptor TLR4 [20], [21], in conjunction with trehalose dicoyrnomycolate (TDM) from the cord factor of the tubercle bacillus to enhance the adjuvant effect [22]. MPL has been safely used as a vaccine adjuvant in animal models and in human clinical trials against several infectious diseases and has been effective in shifting immune responses to some antigens from a Th2-dominant to a Th1-dominant response [23]. The combination of MPL with TDM is often used as an alternative to Freund's complete adjuvant. Despite the use of this potent adjuvant system, three or four immunizations were needed to obtain good antibody responses to the VACV proteins and protection against VACV challenge [11]. Goals of the present study were to accelerate and enhance the immune response in order to reduce the number of immunizations and recombinant proteins necessary for full protection and to extend the work to non-human primates.
To achieve our goals, we compared the efficacy of the adjuvant system used in our prior study with several others. QS-21 is a water-soluble saponin extracted from the bark of the Quillaja saponaria molina tree that has been developed as an adjuvant [24]. QS-21 can enhance both humoral and cell-mediated immune responses and has been used in human clinical trials [25], [26], [27]. Another emerging adjuvant strategy employs synthetic oligodeoxynucleotides (ODNs) with unmethylated CpG motifs. Bacterial DNA contains a high frequency of unmethylated CpG dinucleotides, which have been shown to stimulate the innate immune response through recognition by the TLR 9 receptor [28], [29], [30]. CpG ODNs have been used in experimental vaccines and can induce a shift towards Th1-polarized responses in both animal models and humans [31] and can be combined with both mineral-based adjuvants like aluminum hydroxide gel (alum) as well as emulsion adjuvants like MPL. Protein–alum complexes form a depot at the site of injection, which enhances uptake by antigen presenting cells [32] and activates cytokines and specific T-cell subpopulations [33], [34].
In the present study, we compared the immunogenicity and protection induced by two recombinant VACV proteins A33 and L1 without adjuvant or combined with alum, alum + CpG ODNs, MPL + TDM or QS-21 in the VACV murine pneumonia model [35], [36]. We also describe an initial determination of the protective immunity induced by recombinant VACV proteins in conjunction with QS-21, determined to be the most effective of the adjuvant formulations in mice, in a MPXV cynomolgous monkey model [37].
2. Materials and methods
2.1. Viruses and cells
BS-C-1 monolayer cells (ATCC CCL-26) were maintained at 37 °C and 5% CO2 in modified Eagle's minimal essential medium (EMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Hyclone, Logan, UT), 2 mM l-glutamine (Invitrogen, Carlsbad, CA), 10 U/ml penicillin and 10 μg/ml streptomycin (Invitrogen). HeLa S3 suspension cells (ATCC CCL-2.2) were maintained at 37 °C in modified Eagle medium for spinner cells supplemented with 5% heat-inactivated equine serum (Hyclone). VACV strain Western Reserve (WR) (ATCC VR-1354), VV-NP-SIIINFEKL-EGFP [38], [39] and IHD-J (S. Dales, Rockefeller University, NY), were grown in HeLa S3 cells, purified by sucrose density centrifugation, and titered by plaque assay on BS-C-1 cells [40].
MPXV strain Zaire 79 (V-79-I-005) originally isolated from the scab of an infected human by incubation in LLC-MK2 cells and passaged twice in BS-C-40 cells was obtained from J. Esposito (Centers for Disease Control and Prevention, Atlanta, GA) and propagated in MA-104 cells. A titered clarified lysate was used for the virus challenge.
2.2. Recombinant proteins and ODNs
Soluble forms of the VACV proteins A33, B5 and L1 were prepared in insect cells infected with recombinant baculoviruses and purified from the medium by nickel affinity chromatography as previously described [41], [42]. A mixture of two CpG ODNs (GCTAGACGTTAGCGT and TCAACGTTGA) with phosphorothioate backbones was used as vaccine adjuvants [28]. Neither endotoxin (measured by chromogenic Limulus amoebocyte lysate assay) nor protein (measured by bicinchoninic acid protein assay kit, Pierce Chemicals) was detected in the ODN preparations.
2.3. Mouse immunization and challenge protocol
Five to 6-week-old female BALB/c mice (n = 4–5 mice/group) were purchased from Taconic (Germantown, NY) and were maintained in a pathogen-free environment in sterile microisolator cages at an NIAID animal facility. Mice were immunized subcutaneously and boosted 3 weeks later with 10 μg each of A33 and L1 proteins in phosphate buffered saline (PBS) or with alum, alum and 50 μg of phosphorothioate ODNs containing CpG motifs, a Ribi-adjuvant system (MPL + TDM; Sigma–Aldrich, St. Louis, MO), or a saponin adjuvant QS-21 (Antigenics Inc., New York, NY). Proteins or proteins and CpG ODNs were adsorbed to alum (protein/alum ratio = 2:1, w/w) by vortexing tubes containing immunogens while adding alum in a dropwise manner and then adding PBS to dilute mixtures to the appropriate concentration. MPL + TDM was solubilized in PBS to 2× concentration and combined with immunogens and PBS and vortexed to create a stable oil-in-water emulsion. QS-21 adjuvant (2 mg/ml stock in sterile water) was diluted with proteins and PBS to a final concentration of 15 μg/ml. All immunization mixtures were administered subcutaneously at a final volume of 100 μl. Mice were bled 1 day prior to each immunization and prior to challenge by tail bleed for serological analysis.
Three to 4 weeks following the second immunization, mice were challenged intranasally with VACV WR as previously described [11]. Briefly, a thawed aliquot of sucrose gradient purified VACV WR was sonicated and diluted in PBS to a final concentration of 106 pfu/20 μl. Mice were anesthetized with isoflurane and inoculated intranasally with 20 μl of the VACV preparation. The mice were weighed and observed daily for 2 weeks. Mice were terminated if they lost 30% of their initial weight according to a protocol approved by the NIAID Animal Care and Use Committee.
2.4. Monkey immunization and challenge protocol
Three female cynomolgous monkeys were immunized intramuscularly with 100 μg each of A33, B5 and L1 proteins combined with 50 μg of QS-21 adjuvant on days 0, 28, 57 and 251 of the study. At the same times, one monkey was immunized with 50 μg of QS-21 alone and two monkeys remained unimmunized. One day prior to each immunization or challenge, monkeys were bled and serum was obtained for analysis. One month after the fourth immunization, monkeys were challenged intravenously with 5 × 107 pfu MPXV and monitored daily for signs of illness. Supportive care including subcutaneous fluids was provided as needed. Every 3 or 4 days following challenge, blood was collected for analysis. Monkeys were housed at Bioqual, Inc. (Rockville, MD) during the immunization period and transferred to US Army Research Institute of Infectious Diseases (USAMRIID, Ft. Detrick, Frederick, MD) at the time of challenge. The USAMRIID Animal Care and Use Committee approved the protocols.
2.5. Enzyme-linked immunosorbent assay (ELISA)
Polystyrene 96-well round bottom plates (model 3799, Corning, Corning, NY) were coated with recombinant A33, B5 or L1 proteins or a VACV-infected cell lysate as previously described [11]. Serum was heat-inactivated at 56 °C for 30 min prior to analysis and reciprocal endpoint titers were determined by serial two-fold dilution of individual or pooled mouse serum or individual monkey serum samples. Total mouse IgG was detected by addition of anti-mouse (γ-chain) horseradish peroxidase (HRP)-conjugated antibody (Roche Diagnostics, GmbH, Mannheim, Germany) and isotype-specific antibodies were distinguished by using horseradish peroxidase-conjugated antibodies against murine IgG1 or IgG2a (BD Pharmingen, San Diego, CA). Monkey antibodies were detected with an anti-monkey Fc-specific peroxidase-conjugated antibody used at a 1:4000 dilution (Nordic Immunology, Tilburg, The Netherlands). A ready-to-use solution of soluble 3,3′,5,5′-tetramethylbenzidine (BM Blue, POD substrate; Roche Diagnostics) was added to plates after removal of HRP-conjugated antibody and the A 370nm and A 492nm were measured with a spectrophotometer after incubation for 30 min at room temperature. Reciprocal endpoint titers were determined for mouse samples as the dilution with an absorbance of 0.1 after subtraction of background absorbance of serum samples incubated on plates not coated with protein or lysate. Titers of monkey samples were determined as the dilution with an absorbance two standard deviations above that measured in wells not treated with serum.
2.6. VACV MV neutralization and comet reduction assays
Two types of MV neutralization assays were performed. For the flow cytometric assay, HeLa S3 cells were infected with VV-NP-siiinfekl-EGFP in the presence of cytosine arabinoside and treated with serial two-fold dilutions of mouse or monkey serum. The cells were analyzed 18 h later for green fluorescence by flow cytometry in order to determine the 50% inhibitory concentration of each sample as previously described [43].
For the plaque reduction assay, a 96-well U-bottom cluster plate (Corning) was coated with 0.1% FBS in PBS (0.1 ml/well) and incubated overnight at 4 °C. The coating solution was removed and duplicate two-fold serial dilutions of serum were made in Dulbecco's modified EMEM supplemented with l-glutamine, antibiotics and 2.5% FBS in a final volume of 0.1 ml/well. A purified virus stock of VACV WR was diluted in the same medium and 200 pfu (in 0.1 ml) was mixed with diluted serum and incubated for 1 h at 37 °C. Confluent Vero cells were infected with the virus/serum mixtures (0.1 ml virus/serum and 0.4 ml of Dulbecco's modified EMEM/2.5% FBS) for 2 h at 37 °C, and cells were overlaid with EMEM/2% FBS/0.5% methyl cellulose following removal of the virus inoculum. Cells were incubated for 2 days at 37 °C and plaques were visualized by staining with crystal violet.
For the VACV comet reduction assay, six-well plates of confluent BS-C-1 cells were infected with 80 pfu/well of VACV IHD-J strain for 2 h at 37 °C. The virus inoculum was removed and cells were overlaid with EMEM supplemented with 2.5% fetal bovine serum, 2 mM l-glutamine, antibiotics and a dilution of mouse or monkey serum. Plates were incubated for 40 h at 37 °C and stained with crystal violet to enumerate plaques.
2.7. MPXV neutralization and comet reduction assays
For the plaque reduction neutralization assay, 50 pfu MPXV strain Zaire 79 (V-79-I-005) was incubated at 35.5 °C for 3 h with dilutions of monkey sera in RPMI containing 2% FBS. Confluent monolayers of E6 cells were infected with the virus/serum mixtures in six-well plates and incubated at 35.5 °C and 6% CO2 for 1 h. The inoculum was removed and the cells were incubated at 35.5 °C and 6% CO2 for 3 days in RPMI containing 2% FBS. Plaques were visualized by staining with crystal violet.
For the comet reduction assay, confluent monolayers of BS-C-40 cells in six-well cell culture plates were infected with MPXV strain Zaire 79 (V-79-I-005) at 50 pfu/well in RPMI medium containing 2% FBS. Plates were incubated at 35.5 °C and 6% CO2 for 1 h and rocked every 15 min to ensure even distribution of inoculum. Medium was aspirated; cells were washed twice and overlaid with RPMI containing 2% FBS and a dilution of heat-inactivated monkey serum. Each treatment was performed in duplicate. Plates were incubated at a fixed angle for 3 days at 35.5 °C and 6% CO2. Cells were fixed in 10% phosphate buffered formalin and treated with polyclonal rabbit anti-variola antibody [44]. Following incubation with peroxidase-labeled goat anti-rabbit IgG (KPL 074-1506), comets were visualized by addition of TrueBlue peroxidase substrate (KPL 50-78-02).
2.8. Determination of MPXV genomes in blood
Viral DNA was extracted from whole blood using the QIAGEN QIAamp DNA Mini Kit. A quantitative TaqMan-Minor Groove Binder polymerase chain reaction was set up with a pan-orthopoxvirus probe as previously described [37]. Each sample was run in duplicate and the limit of detection for this assay was 200 genomes/ml of blood.
2.9. Statistical analysis
The mouse weight loss data collected following intranasal challenge were analyzed statistically. To compare treatment groups, the area under the curve corrected for the follow-up period was calculated for each mouse for days 2–14 post-infection as a summary statistic with a trapezoidal rule using all available measurements [45]. Area under the curve values was compared between all treatment groups with the non-parametric Wilcoxon rank sum test adjusting p-values according to Holm [46] to control family wise error rate in the multiple tests. Monkey viral load and lesion count data were analyzed similarly for days 0–28 except that a t-test was used to compare animal groups. Significance was set at a p-value < 0.05.
3. Results
3.1. Effect of adjuvants on antibody responses to VACV A33 and L1 proteins
We previously reported that a vaccine, composed of recombinant forms of the L1 MV protein and the A33 and B5 EV proteins secreted from insect cells, combined with MPL + TDM, administered three or four times protected mice against a lethal VACV intranasal challenge [11]. An objective of the present study was to determine if protection of mice from disease and death could be attained with fewer immunizations and proteins than were previously required. The number of protein immunogens was reduced from three to two as protection with L1 and A33 was nearly as good as that achieved with the combination of L1, A33 and B5 [11]. In addition, we thought that differences in adjuvants might be more easily discerned with fewer immunogens as well as fewer vaccinations. Female BALB/c mice were immunized subcutaneously and boosted 3 weeks later with recombinant forms of the VACV proteins A33 and L1 (the combination henceforth abbreviated as AL) with or without adjuvant. The following adjuvants were used: alum, alum + CpG ODNs, MPL + TDM or QS-21. An additional group of mice was immunized with alum + CpG ODNs without AL as a negative control. Serum samples were collected prior to each immunization to determine A33 and L1 binding antibodies. A strong anti-A33 response after a single vaccination was only obtained using QS-21 as the adjuvant for AL, and this response was boosted 3 weeks later (Fig. 1A). The latter titer was comparable to what was achieved after three immunizations with MPL + TDM in a previous study [11]. The boosted A33 titers obtained with AL + alum + CpG ODNs or MPL + TDM were similar to each other and higher than the titers achieved with AL and alum or no adjuvant but less than with AL and QS-21 (Fig. 1A). Although L1 is less immunogenic than A33 [11], significant antibody titers were achieved after boosting and were highest with AL and QS-21 or alum + CpG ODNs, somewhat lower with MPL + TDM, and undetectable with alum or no adjuvant (Fig. 1B). Overall, the effectiveness of the adjuvants in inducing antibody responses to AL was QS-21 > alum + CpG ODNs > MPL + TDM > alum = no adjuvant. The antibody titers following challenge, which appear in the non-shaded areas of Fig. 1A and B, will be discussed in Section 3.3.
Fig. 1.
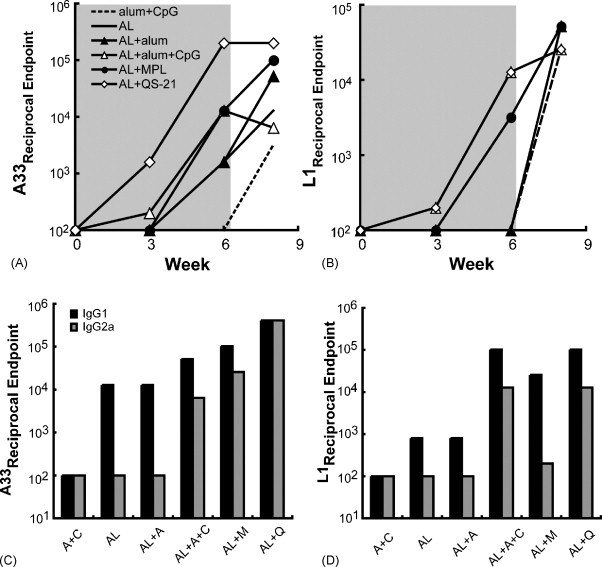
ELISA values of pooled mouse sera following immunizations with A33 and L1 proteins combined with different adjuvants. Mice (n = 5) were immunized twice with a mixture containing 10 μg each of A33 and L1 proteins (AL) alone or combined with alum, alum + CpG, MPL + TDM (MPL) or QS-21 and challenged with VACV strain WR at 3 weeks after the last immunization. Serum was collected prior to immunization (week 0), 3 weeks following immunizations 1 and 2 (shaded area), and 2 weeks after challenge (unshaded area). Antibodies to A33 (A) and L1 (B) were determined on pooled sera by ELISA and reciprocal endpoint values are plotted. In panel B, the plots for AL + QS-21 and for AL + alum + CpG are superimposed. Serum collected 3 weeks after the second immunization was re-analyzed for IgG1 or IgG2a isotype antibodies to A33 (C) and L1 (D). In the latter panels, alum and CpG are abbreviated as A and C, respectively.
The data shown in Fig. 1 were obtained with sera pooled from animals within each group. We also analyzed the sera from individual mice collected after the boost in order to perform a statistical analysis (Table 1 ). The A33 ELISA titers of sera from mice immunized with AL + QS-21 were significantly higher than the titers of mice of any other group (p < 0.00002). The titers obtained after immunization with AL + alum and CpG ODNs or with AL + MPL + TDM were significantly higher than after AL without adjuvant or with alum but did not differ significantly from each other. Similar results were obtained from the L1 ELISA determinations, except that the difference between the titers obtained with AL + QS-21 or with AL and alum + CpG ODNs did not reach statistical significance.
Table 1.
Reciprocal endpoint dilution ELISA titers to A33 and L1 (italic values) of sera from individual mice immunized with AL plus various adjuvants
| Immunogen | Reciprocal endpoint dilution ELISA titer A33/L1a |
|---|---|
| AL | 100/200, 200/200, 100/400, 200/200, 400/100 (200)/(220) |
| AL + A | 400/100, 400/100, 400/100, 400/100 (400)/(100) |
| AL + A + C | 800/1600, 25,600/800, 1600/3200, 12,800/12,800, 3200/12,800 (8800)/(6240) |
| AL + M | 12,800/800, 6400/6400, 3200/800, 3200/1600, 51,200/800 (15,360)/(2080) |
| AL + Q | 204,800/3200, 409,600/51,200, 409,600/25,600, 204,800/6400, 409,600/3200 (327,680)/(17,920) |
Abbreviations: AL, A33 + L1; AL + A, A33 + L1 + alum; AL + A + C, A33 + L1 + alum + CpG ODNs; AL + M, A33 + L1 + MPL + TDM; AL + Q, A33 + L1 + QS-21.
Average of values in parentheses.
Immunizations with protein, in contrast to live virus, typically induce a predominant Th2 response with IgG1 as the dominant antibody isotype in Balb/c mice. However, the Th1 response as revealed by IgG2a antibody can be enhanced by some adjuvants. Fig. 1C and D shows the results of isotype-specific ELISAs that detect antibodies against A33 and L1 in sera collected 3 weeks after the second immunization. The anti-A33 antibodies from mice immunized with AL alone or with alum were exclusively IgG1 as the IgG2a titers were no higher than the adjuvant alone control. Some IgG2a in addition to IgG1 was made after AL + alum + CpG ODNs or MPL + TDM (Fig. 1C). However, the highest total IgG2a as well as the highest ratio of IgG2a to IgG1 occurred with AL + QS-21. Isotype-specific titers against L1 were IgG1-dominant in all groups, but IgG2a titers above the control were observed with sera from mice immunized with AL + QS-21 or alum + CpG ODNs (Fig. 1D). These results suggested that the magnitude and isotype of the antibody responses were influenced by both the nature of the protein as well as the adjuvant. Overall with the two protein immunogens, QS-21 induced the most IgG2a with alum + CpG ODNs and MPL + TDM next.
3.2. Induction of VACV neutralizing and comet-reducing antibodies
A flow cytometry-based GFP assay was used to detect MV neutralizing antibody induced by L1. Neutralizing antibody was not detected after the primary immunization but was present after the boost. The highest neutralizing titers were measured in the AL + QS-21 and the AL + alum + CpG ODNs groups (Fig. 2 ). These neutralization values were similar to that of vaccinia immune globulin, although the latter targeted additional MV proteins (data not shown). Significant neutralization was achieved with sera from mice immunized with AL and MPL + TDM but not with AL and alum or no adjuvant (Fig. 2). The post-challenge titers will be discussed in Section 3.3.
Fig. 2.
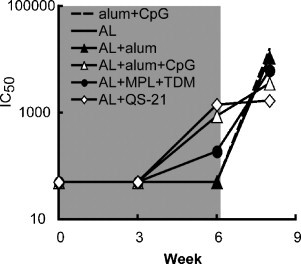
Induction of neutralizing antibody. The sera described in Fig. 1 obtained from mice immunized with AL plus the indicated adjuvants were used. MV neutralizing antibodies were measured with a flow cytometry-based GFP assay and the 50% inhibitory concentration (IC50) was determined for each pool of mouse sera.
The IHD-J strain of VACV produces large numbers of satellite plaques known as comets when cells are infected in liquid medium. Antibodies to certain EV proteins such as A33 but not MV proteins such as L1 can suppress the formation of comets. Pre-challenge sera from animals receiving alum + CpG alone or proteins without adjuvant had no evident effect on comet size, whereas pre-challenge sera from animals immunized with proteins and adjuvants reduced the sizes of comets to various extents (Fig. 3 ). The greatest reduction occurred with AL + QS-21, consistent with the highest anti-A33 ELISA titer (Fig. 1A). The comet-reducing activity of post-challenge sera will be discussed in Section 3.3.
Fig. 3.

Induction of comet-reducing antibody. The sera described in Fig. 1 obtained from mice immunized with A33 and L1 (AL) plus the indicated adjuvants were used to detect antibodies that inhibit the formation satellite plaques due to spread of EV in liquid medium. BS-C-1 cells were infected with VACV strain IHD-J (80 pfu/well), overlaid with medium containing a 1:50 dilution of pooled mouse serum, and 40 h later stained with crystal violet. The column labeled pre-challenge represents samples collected 3 weeks after the second immunization and 1 day prior to intranasal virus challenge. The post-challenge column shows samples collected from surviving mice 2 weeks following challenge. The well shown in the upper left corner shows the typical formation of comet-shaped plaques in the absence of serum.
3.3. Effects of adjuvants on the induction of protective immune responses to A33 and L1 proteins
The protective effects of immunization with AL combined with different adjuvants were determined in mice that were infected intranasally with the WR strain of VACV [35], [36]. Weight loss, which occurs during the first week following challenge, is directly correlated with virus replication in the lung and provides an objective, non-invasive way of following disease [7], [47]. According to our animal protocol, mice were terminated if they lost greater than 30% of their initial weight. Three to 4 weeks following the second protein immunization with various adjuvants, mice were challenged intranasally with 5 LD50 of VACV WR. The majority of mice died or was sacrificed by 10 days post-infection in the control group immunized with adjuvant without protein or with AL in the absence of adjuvant (Fig. 4A). Half of the mice immunized with AL and alum and 80% of those immunized with AL + MPL + TDM survived. Most impressively, all mice immunized with AL combined with either QS-21 or alum + CpG ODNs survived challenge (Fig. 4A). Dramatic weight loss of surviving mice was observed over the first 6 days following challenge in most groups, except for mice immunized with AL + QS-21 (Fig. 4B). The AL + QS-21 group showed little fluctuation in weight and no visible signs of illness throughout the observation period. Mice immunized with AL and alum + CpG ODNs were next best and showed less weight loss overall than the other immunized groups. It is important to note that Rees et al. [48] reported that CpG ODNs alone could protect against VACV challenge through the upper respiratory route by stimulating innate immunity. However, the protective action of CpG ODNs was short lived and gone by 21 days, which was the minimum time that we waited before challenge. Moreover, our control group received alum + CpG ODNs.
Fig. 4.
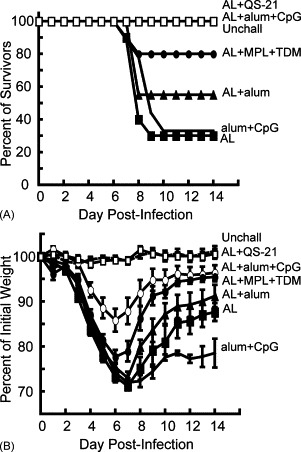
Survival and weight changes in mice immunized with AL proteins and different adjuvants followed by intranasal VACV challenge. Mice were immunized as described in Fig. 1, and 3 weeks following the second immunization, were challenged intranasally with 106 pfu VACV strain WR. The unchallenged (Unchall) mice were not immunized or challenged. The alum + CpG control group received no recombinant protein prior to challenge. Mice were weighed daily for 2 weeks and sacrificed if their weight fell below 70% of the initial value. The percent of survivors (A) and the percent of initial weight of surviving mice (B) are shown for each group. The data shown here represent two independent experiments and each group had 4–5 mice/group. Each data point is the average weight ± S.E.M. of mice in each group from the two challenge experiments.
Statistical analysis of the weight loss data was achieved by calculating the area under the curve as a summary statistic for each animal and using the non-parametric Wilcoxon rank sum test with the Holm p-value adjustment method for multiple tests to compare the animal groups. The resulting p-values are shown in Table 2 . Challenged mice that were immunized with AL + QS-21 lost significantly less weight than mice given any of the other immunizations; furthermore the weight loss in this group was not significantly different from unchallenged mice. Mice given AL and alum + CpG ODNs had significantly less weight loss than mice immunized with AL alone or with alum alone. Mice immunized with AL and MPL + TDM showed significantly less weight loss than mice immunized with AL alone.
Table 2.
p-Values calculated with area under the curve analysis of weight loss data in Fig. 4B followed by the Wilcoxan rank sum test using the Holm p-value adjustment
| Alum + CpG | AL | AL + alum | AL + alum + CpG | AL + MPL + TDM | |
|---|---|---|---|---|---|
| ALa + alum | 0.4 | 0.4 | – | – | – |
| AL + alum + CpG | 0.002b | 0.002 | 0.02 | – | – |
| AL + MPL + TDM | 0.02 | 0.02 | 0.4 | 0.4 | – |
| AL + QS-21 | 0.0005 | 0.0003 | 0.0005 | 0.0008 | 0.0003 |
AL, A33 + L1 proteins.
Bold numbers indicate significant p-values.
This statistical analysis confirmed that mice immunized with AL and QS-21 were the best protected, in addition to inducing the highest overall antibody response. Binding the proteins to alum did not significantly enhance protection, but addition of CpG ODNs to AL + alum provided significant protection from weight loss. The weight loss data paralleled the number of survivors, since the groups with more survivors showed less weight loss.
Boosting of antibody to the viral immunogens following challenge is an indirect measure of virus replication. Hence, surviving animals that were least well protected were anticipated to show highest boosting. Convalescent serum, collected from surviving mice 2 weeks after challenge, showed a boost in A33 antibody except for the AL + QS-21 or alum + CpG ODNs groups (Fig. 1A), which had been the best protected. The same two groups showed the least boosting of anti-L1 antibody after challenge (Fig. 1B). Although prior to challenge, MV neutralization and EV comet reduction were exclusively due to anti-L1 and anti-A33 antibodies, respectively, antibodies to additional proteins may have contributed to inhibition after challenge. Thus, sera from all groups, except those receiving AL + QS-21, showed a dramatic rise in neutralizing antibodies 2 weeks post-challenge (Fig. 2), likely targeting additional MV proteins. Similarly, all of the post-challenge sera showed strong comet-inhibiting activity (Fig. 3).
3.4. Binding and VACV neutralizing antibodies following immunization of cynomolgous monkeys with recombinant A33, B5 and L1 combined with QS-21
Prior to the adjuvant comparison studies described above, we initiated an experiment to test the efficacy of a recombinant protein vaccine in a non-human primate. Three cynomolgous monkeys (030, 770, 974) were immunized three times at 1-month intervals with 100 μg each of recombinant A33, B5 and L1 proteins with QS-21 adjuvant. One control monkey (026) was immunized with the QS-21 adjuvant alone and two additional controls (398, 419) received neither adjuvant nor protein. The average ELISA titers for the three immunized monkeys are shown at each time point (Fig. 5A). Antibodies specific to A33, B5 and L1 were detected 1 month after the first immunization and were boosted by the second immunization. The titers fell somewhat between the second and third immunizations but were boosted again by the latter. The reciprocal ELISA titers declined gradually over the next 6 months and at the end of this time were more than a log lower than the peak values. However, the titers rebounded again after another immunization. A similar response pattern was obtained using a VACV-infected cell lysate for the ELISA in order to detect antibodies capable of recognizing non-recombinant VACV antigens (Fig. 5B).
Fig. 5.
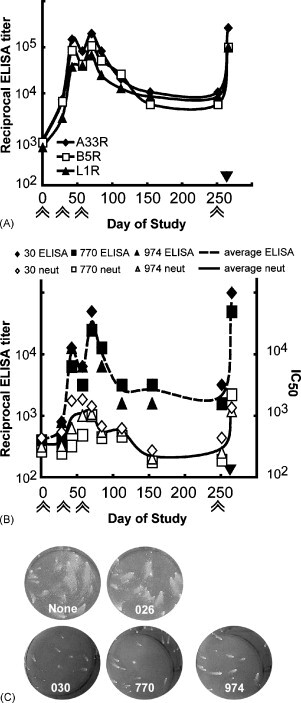
Analysis of monkey sera following immunizations with A33, B5 and L1 combined with QS-21. Cynomolgous monkeys were immunized with 100 μg each of recombinant A33, B5 and L1 proteins combined with the adjuvant QS-21 at days 0, 28, 57 and 251 as indicated by double arrows below the x-axis. Four weeks after the fourth immunization, monkeys were challenged intravenously with 5 × 107 pfu MPXV as indicated by the solid black triangle above the x-axis. (A) Averages of the ELISA values specific for A33, B5 and L1 are shown. (B) Reciprocal ELISA values determined against an infected cell lysate and flow cytometry VACV neutralization titers of individual monkeys and averages are presented. (C) The presence of EV neutralizing antibodies in sera collected prior to challenge was determined using the comet reduction assay. Key: None, no serum; 026, adjuvant only monkey serum; 030, 770, 974, sera from immunized monkeys.
Neutralizing antibodies were detected using the flow cytometry assay following protein immunization and were boosted following each immunization as shown in Fig. 5B. Only L1-specific antibodies neutralize MVs since antibodies against A33 and B5 only target the EV form of the virus. IC50 titers fell to background levels between the third and the fourth immunization, but were boosted to peak levels after the fourth. The sera obtained after the fourth immunization were also tested by a VACV plaque reduction assay, which gives lower titers than the flow cytometry assay (Fig. 6A).
Fig. 6.
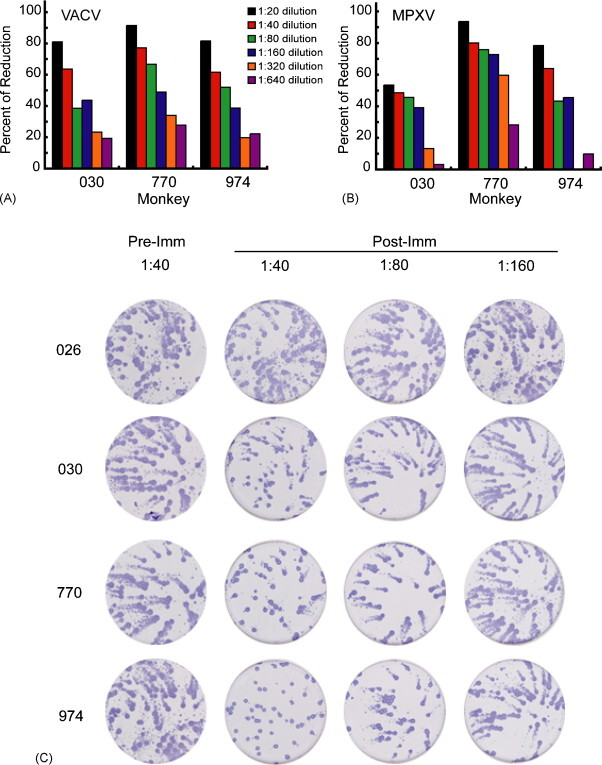
VACV and MPXV plaque reduction and MPXV comet inhibition. (A) Pre-challenge serum samples were tested in a VACV plaque reduction assay. Data are from three separate analyses each done in duplicate and percentage reductions were determined with respect to serum from adjuvant only monkey (026). (B) Pre-challenge serum samples were tested in a MPXV plaque reduction assay. Analyses were done in duplicate and the percentage reduction determined as above. (C) Sera (1:40 dilution) obtained prior to immunization (Pre-Imm) and after the final immunization (Post-Imm) were tested in a MPXV comet reduction assay. The plates were stained with anti-variola rabbit antibody followed by peroxidase-labeled goat anti-rabbit IgG; comets were visualized by adding peroxidase substrate accounting for their dark color. Experiment was done in duplicate and photographs of one set are shown.
Antibodies to both A33 and B5 can reduce the release of extracellular virus from cells, which is responsible for comet formation. Pre-immune sera (not shown) and serum from the control monkey receiving adjuvant alone (026) did not cause any comet reduction compared to the no serum control. Serum from each of the three immunized monkeys prior to challenge dramatically reduced VACV comet size as shown in the bottom three wells of Fig. 5C.
3.5. MPXV neutralizing antibodies following immunization of cynomolgous monkeys with recombinant A33, B5 and L1 combined with QS-21
Sera from the immunized monkeys also neutralized MPXV. The MPXV plaque reduction titer (Fig. 6B) was similar to the titer determined with VACV (Fig. 6A), consistent with the conserved protein sequences. The immune sera also inhibited MPXV comet formation whereas the serum from the monkey receiving adjuvant alone (026) did not (Fig. 6C). Thus, antibodies to VACV L1 neutralized MPXV MVs and antibody to VACV B5 or A33 prevented spread of MPXV.
3.6. Protection of monkeys from severe disease and death following an intravenous MPXV challenge
Although we had originally intended to challenge the monkeys with MPXV after the third immunization, logistical problems prevented this. An opportunity to challenge arose about 6 months later leading us to give an additional protein immunization. Four weeks after that, the monkeys receiving proteins and QS-21 (030, 770, 974), QS-21 alone (026) and no vaccine (398, 419) were challenged intravenously with 5 × 107 pfu MPXV. The monkeys were monitored for 1 month following challenge during which the immunized monkeys appeared healthy. In contrast, the non-immunized monkeys were severely ill with fever and weight loss as previously described [37] and one (026) died on day 12, despite supportive efforts including subcutaneous fluids. The number of virus-induced skin lesions was counted every 3–4 days. The monkeys that were not immunized with protein developed 575–820 skin lesions each, which peaked at 12 days post-infection (Fig. 7A). There were fewer lesions in immunized monkeys (maximum of 65–140 each) and they were generally smaller and atypical compared to those observed in naïve monkeys, developed less synchronously, and healed rapidly. The difference in lesion number between immunized and control animals was statistically significant (p = 0.02).
Fig. 7.
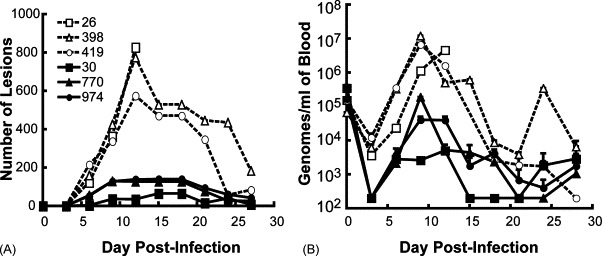
Protein immunization reduces skin lesions and circulating virus in macaques. (A) Skin lesions. Monkeys immunized as described in the legend to Fig. 5 were challenged intravenously with 5 × 107 pfu MPXV and skin lesions were counted at 3–4-day intervals. (B) Blood samples were collected at 3–4-day intervals and the number of viral genomes was determined in duplicate by real-time quantitative PCR. The average ± standard deviation is shown for each time point. The limit of sensitivity was 200 genomes/ml. Key to monkeys: 026 (QS-21 adjuvant only); 398 and 419 (no immunization); 030, 770 and 974 (QS-21 and A33, B5 and L1 proteins). Note that the dashed and solid lines are from control and protein immunized animals, respectively.
A real-time PCR assay detected viral genomes in the blood of all monkeys immediately after challenge (day 0), followed by a decline (Fig. 7B). On day 6, however, an increase in viral genomes was detected with peak values generally present at day 9. The viral loads were higher in the controls than in the immunized monkeys with an average difference of about 2.5 logs, which was statistically significant (p = 0.04) despite the small number of animals.
4. Discussion
An important role of antibody in protection against secondary orthopoxvirus infections has been well documented. Studies using B- and T-cell depletion [49], [50] or gene knockouts [51] demonstrated the pivotal role of the CD4(+) T-cell-driven antibody response for protection against VACV infection in mice. The fundamental role of B-cells was also shown in ectromelia virus infections of mice [15], [52] and in a macaque MPXV model [53]. These findings suggest that a protein subunit vaccine, which primarily induces an antibody response, may be sufficient to defend against smallpox, if the animal models are predictive of disease protection in humans.
Our previous study [11] showed that a combination of soluble recombinant forms of VACV MV and EV membrane proteins could protect as well as live VACV in the VACV WR mouse pneumonia model. However, the requirement for three or four protein immunizations would be impractical for vaccine delivery and we therefore investigated the use of other adjuvants. We now show that the combination of QS-21 with only the A33 and L1 proteins completely prevented weight loss as well as death after only two vaccinations at 3-week intervals. Using the same vaccination regimen, the combination of the two proteins with alum + CpG ODNs was next best, followed by MPL + TDM. Alum + CpG ODNs was clearly superior to alum alone. The degree of protection in the mouse model with different adjuvants correlated with antibody levels, whether determined by ELISA, neutralization of MVs, or reduction in spread of satellite plaques.
Analysis of convalescent serum from surviving mice showed a consistent pattern with respect to ELISA and neutralizing antibody titers. Groups of mice that had higher titers against A33 and L1 prior to challenge and the best protection, namely those receiving AL + QS-21 or AL + alum + CpG ODNs, showed the lowest boosting of A33 and L1 antibodies. Conversely, groups that had the lowest A33 and L1 titers prior to challenge and the poorest protection had the greatest boosting of antibody responses following challenge. This trend was observed with both MV and EV antigens, and likely reflects the degree of replication of the challenge virus in poorly immunized animals.
Protein immunogens typically induce a predominantly Th2-type response that is characterized by activation of B-cells and production of antibodies, especially IgG1. QS-21 and to a lesser extent alum + CpG ODNs and MPL + TDM increased the IgG2a antibody, indirectly suggesting an enhancement of the Th1 response. This effect of immunostimulatory adjuvants has been noted previously for other immunogens. Both QS-21 [54], [55], [56], [57], [58] and CpG ODNs [59], [60] can also augment the production of antigen-specific cytotoxic T-cells, though this was not evaluated in the present study.
The monkey study described here provides a preliminary evaluation of the immune response to recombinant VACV proteins in a primate and provides evidence that they can induce protection against severe disease following a MPXV challenge. This study mimicked our earlier mouse experiments [11], which used B5 in addition to A33 and L1. Recombinant B5 protein [11], [14] as well as antibodies to B5 [18], [19] can provide protection in the VACV mouse pneumonia model. In addition, B5 is the major target of EV neutralizing antibody in human VACV immune globulin [61], though antibody to B5 is not necessarily the most protective in vivo. Also, VACV B5 has a slightly greater amino acid identity (96%) to the MPXV homolog than VACV A33, which has 93% identity. L1 is the most highly conserved with 98% identity between the VACV and MPXV homologs. Therefore, we anticipated that antibodies to these VACV proteins would cross react with the MPXV homologs and provide at least partial protection. We chose to use QS-21, based on our previous experience with recombinant HIV proteins in a monkey model [62], even though we had not yet determined the superiority of this adjuvant for inducing VACV protection in mice. The recombinant VACV proteins were immunogenic in monkeys and binding antibodies were detected after the first immunization and boosted by a second, at which time VACV neutralizing and comet-reducing antibodies were also found. Between immunizations, the antibody levels dropped but were boosted again even after 7 months. At this time, we also demonstrated MPXV neutralizing and comet-reducing antibodies. A previously described intravenous MPXV challenge was used because of its consistency [37], which was particularly important with a small number of animals. There were several indicators of protection: each unimmunized animal developed approximately 700 typical pustular skin lesions, whereas the vaccinated ones developed about 100 smaller atypical lesions; the virus load was reduced by about 2.5 logs; and most importantly the vaccinated animals appeared healthy whereas the unvaccinated were gravely ill and one died. Based on the number of skin lesions, the protection was less than achieved with modified VACV Ankara or the licensed smallpox vaccine [37] and similar to that obtained with a four-component DNA vaccine (VACV L1R, A27L, A33R and B5R) administered percutaneously with a gene gun [63]. In a recently published study [17], it was reported that a four-component DNA vaccine composed of MPXV orthologs of the same VACV genes administered intramuscularly did not induce neutralizing antibody or protect monkeys against a MPXV challenge. In contrast, the corresponding recombinant MPXV proteins with alum or CPG ODNs provided partial protection, which was enhanced by prior DNA vaccinations. Although VACV and MPXV membrane proteins are very closely related to the variola virus orthologs, it would seem that the latter would be most appropriate for a smallpox vaccine.
Acknowledgements
This work was done to partially fulfill the Ph.D. thesis requirements of C.F. at the University of Maryland. The authors would like to thank Gary Cohen and Roselyn Eisenberg for purified proteins, the production of which was supported by USPHS Grant NIH RCE-U54-AI57168 from the NIAID, NIH. Antigenics Inc. kindly provided QS-21 adjuvant, and Norman Cooper supplied cells and virus stocks. Thanks also to the NIAID Animal Care Branch. The study was partially supported by intramural funds from the NIAID, NIH and the Office for Chemical and Biological Defense of DTRA.
References
- 1.Fenner F., Henderson D.A., Arita I., Jezek Z., Ladnyi I.D. 1st ed. World Health Organization; Geneva: 1988. Smallpox and its eradication. [Google Scholar]
- 2.Fulginiti V.A., Papier A., Lane J.M., Neff J.M., Henderson D.A. Smallpox vaccination: a review, part II. Adverse events. Clin Inf Dis. 2003;37(July (2)):251–271. doi: 10.1086/375825. [DOI] [PubMed] [Google Scholar]
- 3.Henderson D.A. The looming threat of bioterrorism. Science. 1999;283(February (5406)):1279–1282. doi: 10.1126/science.283.5406.1279. [DOI] [PubMed] [Google Scholar]
- 4.Moss B. In: Fields virology. 4th ed. Knipe D.M., Howley P.M., editors. Lippincott Williams & Wilkins; Philadelphia: 2001. Poxviridae: the viruses and their replication; pp. 2849–2883. [Google Scholar]
- 5.Boulter E.A., Zwartouw H.T., Titmuss D.H.J., Maber H.B. The nature of the immune state produced by inactivated vaccinia virus in rabbits. Am J Epidemiol. 1971;94:612–620. doi: 10.1093/oxfordjournals.aje.a121360. [DOI] [PubMed] [Google Scholar]
- 6.Boulter E.A., Appleyard G. Differences between extracellular and intracellular forms of poxvirus and their implications. Prog Med Virol. 1973;16:86–108. [PubMed] [Google Scholar]
- 7.Law M., Putz M.M., Smith G.L. An investigation of the therapeutic value of vaccinia-immune IgG in a mouse pneumonia model. J Gen Virol. 2005;86(April (Pt 4)):991–1000. doi: 10.1099/vir.0.80660-0. [DOI] [PubMed] [Google Scholar]
- 8.Payne L.G. Significance of extracellular virus in the in vitro and in vivo dissemination of vaccinia virus. J Gen Virol. 1980;50:89–100. doi: 10.1099/0022-1317-50-1-89. [DOI] [PubMed] [Google Scholar]
- 9.Blasco R., Moss B. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J Virol. 1992;66(7):4170–4179. doi: 10.1128/jvi.66.7.4170-4179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith G.L., Vanderplasschen A., Law M. The formation and function of extracellular enveloped vaccinia virus. J Gen Virol. 2002;83(December (Pt 12)):2915–2931. doi: 10.1099/0022-1317-83-12-2915. [DOI] [PubMed] [Google Scholar]
- 11.Fogg C., Lustig S., Whitbeck J.C., Eisenberg R.J., Cohen G.H., Moss B. Protective immunity to vaccinia virus induced by vaccination with multiple recombinant outer membrane proteins of intracellular and extracellular virions. J Virol. 2004;78(October (19)):10230–10237. doi: 10.1128/JVI.78.19.10230-10237.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai C.F., Gong S.C., Esteban M. The purified 14-kilodalton envelope protein of vaccinia virus produced in Escherichia coli induces virus immunity in animals. J Virol. 1991;65(10):5631–5635. doi: 10.1128/jvi.65.10.5631-5635.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies D.H., McCausland M.M., Valdez C., Huynh D., Hernandez J.E., Mu Y.X., et al. Vaccinia virus H3L envelope protein is a major target of neutralizing antibodies in humans and elicits protection against lethal challenge in mice. J Virol. 2005;79(September (18)):11724–11733. doi: 10.1128/JVI.79.18.11724-11733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galmiche M.C., Goenaga J., Wittek R., Rindisbacher L. Neutralizing and protective antibodies directed against vaccinia virus envelope antigens. Virology. 1999;254(1):71–80. doi: 10.1006/viro.1998.9516. [DOI] [PubMed] [Google Scholar]
- 15.Fang M., Cheng H., Dai Z.P., Bu Z.M., Sigal L.J. Immunization with a single extracellular enveloped virus protein produced in bacteria provides partial protection from a lethal orthopoxvirus infection in a natural host. Virology. 2006;345(February (1)):231–243. doi: 10.1016/j.virol.2005.09.056. [DOI] [PubMed] [Google Scholar]
- 16.Hooper J.W., Custer D.M., Thompson E. Four-gene-combination DNA vaccine protects mice against a lethal vaccinia virus challenge and elicits appropriate antibody responses in nonhuman primates. Virology. 2003;306(February (1)):181–195. doi: 10.1016/S0042-6822(02)00038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heraud J.M., Edghill-Smith Y., Ayala V., Kalisz I., Parrino J., Kalyanaraman V.S., et al. Subunit recombinant vaccine protects against monkeypox. J Immunol. 2006;177(August (4)):2552–2564. doi: 10.4049/jimmunol.177.4.2552. [DOI] [PubMed] [Google Scholar]
- 18.Lustig S., Fogg C., Whitbeck J.C., Eisenberg R.J., Cohen G.H., Moss B. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J Virol. 2005;79:13454–13462. doi: 10.1128/JVI.79.21.13454-13462.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z.C., Earl P., Americo J., Damon I., Smith S.K., Zhou Y.H., et al. Chimpanzee/human mAbs to vaccinia virus B5 protein neutralize vaccinia and smallpox viruses and protect mice against vaccinia virus. Proc Natl Acad Sci USA. 2006;103(February (6)):1882–1887. doi: 10.1073/pnas.0510598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poltorak A., Smirnova I., He X., Liu M.Y., Van Huffel C., McNally O., et al. Genetic and physical mapping of the Lps locus: identification of the toll-4 receptor as a candidate gene in the critical region. Blood Cells Mol Dis. 1998;24(September (3)):340–355. doi: 10.1006/bcmd.1998.0201. [DOI] [PubMed] [Google Scholar]
- 21.Takayama K., Ribi E., Cantrell J.L. Isolation of a nontoxic lipid A fraction containing tumor regression activity. Cancer Res. 1981;41(July (7)):2654–2657. [PubMed] [Google Scholar]
- 22.Masihi K.N., Lange W., Brehmer W., Ribi E. Immunobiological activities of nontoxic lipid A: enhancement of nonspecific resistance in combination with trehalose dimycolate against viral infection and adjuvant effects. Int J Immunopharmacol. 1986;8(3):339–345. doi: 10.1016/0192-0561(86)90116-5. [DOI] [PubMed] [Google Scholar]
- 23.Evans J.T., Cluff C.W., Johnson D.A., Lacy M.J., Persing D.H., Baldridge J.R. Enhancement of antigen-specific immunity via the TLR4 ligands MPL adjuvant and Ribi.529. Expert Rev Vaccines. 2003;2(April (2)):219–229. doi: 10.1586/14760584.2.2.219. [DOI] [PubMed] [Google Scholar]
- 24.Liu G., Anderson C., Scaltreto H., Barbon J., Kensil C.R. QS-21 structure/function studies: effect of acylation on adjuvant activity. Vaccine. 2002;20(21–22):2808–2815. doi: 10.1016/s0264-410x(02)00209-8. [DOI] [PubMed] [Google Scholar]
- 25.Evans T.G., McElrath M.J., Matthews T., Montefiori D., Weinhold K., Wolff M., et al. QS-21 promotes an adjuvant effect allowing for reduced antigen dose during HIV-1 envelope subunit immunization in humans. Vaccine. 2001;19(February (15–16)):2080–2091. doi: 10.1016/s0264-410x(00)00415-1. [DOI] [PubMed] [Google Scholar]
- 26.Livingston P.O., Adluri S., Helling F., Yao T.J., Kensil C.R., Newman M.J., et al. Phase 1 trial of immunological adjuvant QS-21 with a GM2 ganglioside-keyhole limpet haemocyanin conjugate vaccine in patients with malignant melanoma. Vaccine. 1994;12(November (14)):1275–1280. doi: 10.1016/s0264-410x(94)80052-2. [DOI] [PubMed] [Google Scholar]
- 27.Kashala O., Amador R., Valero M.V., Moreno A., Barbosa A., Nickel B., et al. Safety, tolerability and immunogenicity of new formulations of the Plasmodium falciparum malaria peptide vaccine SPf66 combined with the immunological adjuvant QS-21. Vaccine. 2002;20(May (17–18)):2263–2277. doi: 10.1016/s0264-410x(02)00115-9. [DOI] [PubMed] [Google Scholar]
- 28.Klinman D.M., Yi A.K., Beaucage S.L., Conover J., Krieg A.M. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc Natl Acad Sci USA. 1996;93(April (7)):2879–2883. doi: 10.1073/pnas.93.7.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krieg A.M., Yi A.K., Matson S., Waldschmidt T.J., Bishop G.A., Teasdale R., et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374(April (6522)):546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto S., Yamamoto T., Shimada S., Kuramoto E., Yano O., Kataoka T., et al. DNA from bacteria, but not from vertebrates, induces interferons, activates natural killer cells and inhibits tumor growth. Microbiol Immunol. 1992;36(9):983–997. doi: 10.1111/j.1348-0421.1992.tb02102.x. [DOI] [PubMed] [Google Scholar]
- 31.Klinman D.M. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol. 2004;4(April (4)):249–258. doi: 10.1038/nri1329. [DOI] [PubMed] [Google Scholar]
- 32.Bomford R. In: NATO Advanced Study Institute on Immunological Adjuvants and Vaccines. Gregoriadis G.A., Poste A.G., editors. Plenum Press; New York: 1988. Aluminium salts: perspectives in their use as adjuvants; pp. 35–41. [Google Scholar]
- 33.Jordan M.B., Mills D.M., Kappler J., Marrack P., Cambier J.C. Promotion of B cell immune responses via an alum-induced myeloid cell population. Science. 2004;304(June (5678)):1808–1810. doi: 10.1126/science.1089926. [DOI] [PubMed] [Google Scholar]
- 34.Lindblad E.B. Aluminium compounds for use in vaccines. Immunol Cell Biol. 2004;82(October (5)):497–505. doi: 10.1111/j.0818-9641.2004.01286.x. [DOI] [PubMed] [Google Scholar]
- 35.Turner G.S. Respiratory infection of mice with vaccinia virus. J Gen Virol. 1967;1(3):399–402. doi: 10.1099/0022-1317-1-3-399. [DOI] [PubMed] [Google Scholar]
- 36.Williamson J.D., Reith R.W., Jeffrey L.J., Arrand J.R., Mackett M. Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J Gen Virol. 1990;71(November):2761–2767. doi: 10.1099/0022-1317-71-11-2761. [DOI] [PubMed] [Google Scholar]
- 37.Earl P.L., Americo J.L., Wyatt L.S., Eller L.A., Whitbeck J.C., Cohen G.H., et al. Immunogenicity of a highly attenuated MVA smallpox vaccine and protection against monkeypox. Nature. 2004;428:182–185. doi: 10.1038/nature02331. [DOI] [PubMed] [Google Scholar]
- 38.Anton L.C., Schubert U., Bacik I., Princiotta M.F., Wearsch P.A., Gibbs J., et al. Intracellular localization of proteasomal degradation of a viral antigen. J Cell Biol. 1999;146(July (1)):113–124. doi: 10.1083/jcb.146.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Norbury C.C., Malide D., Gibbs J.S., Bennink J.R., Yewdell J.W. Visualizing priming of virus-specific CD8(+) T cells by infected dendritic cells in vivo. Nat Immunol. 2002;3(March (3)):265–271. doi: 10.1038/ni762. [DOI] [PubMed] [Google Scholar]
- 40.Earl P.L., Cooper N., Wyatt S., Moss B., Carroll M.W. In: Current protocols in molecular biology. Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., et al., editors. John Wiley and Sons; New York: 1998. Preparation of cell cultures and vaccinia virus stocks; pp. 16.16.1–16.16.3. [Google Scholar]
- 41.Aldaz-Carroll L., Whitbeck J.C., Ponce de Leon M., Lou H., Hirao L., Isaacs S.N., et al. Epitope-mapping studies define two major neutralization sites on the vaccinia virus extracellular enveloped virus glycoprotein B5R. J Virol. 2005;79(May (10)):6260–6271. doi: 10.1128/JVI.79.10.6260-6271.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aldaz-Carroll L., Whitbeck J.C., Ponce de Leon M., Lou H., Pannell L.K., Lebowitz J., et al. Physical and immunological characterization of a recombinant secreted form of the membrane protein encoded by the vaccinia virus L1R gene. Virology. 2005;341(1):59–71. doi: 10.1016/j.virol.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 43.Earl P.L., Americo J.L., Moss B. Development and use of a vaccinia virus neutralization assay based on flow cytometric detection of green fluorescent protein. J Virol. 2003;77(October (19)):10684–10688. doi: 10.1128/JVI.77.19.10684-10688.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang H.L., Kim S.K., Kim M., Reche P.A., Morehead T.J., Damon I.K., et al. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J Clin Invest. 2005;115(February (2)):379–387. doi: 10.1172/JCI23220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Journot V., Chene G., Joly P., Saves M., Jacqmin-Gadda H., Molina J.M., et al. Viral load as a primary outcome in human immunodeficiency virus trials: a review of statistical analysis methods. Control Clin Trials. 2001;22(December (6)):639–658. doi: 10.1016/s0197-2456(01)00158-1. [DOI] [PubMed] [Google Scholar]
- 46.Holm S. A simple sequentialrejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- 47.Luker K.E., Hutchens M., Schultz T., Pekosz A., Luker G.D. Bioluminescence imaging of vaccinia virus: effects of interferon on viral replication and spread. Virology. 2005;341(October (2)):284–300. doi: 10.1016/j.virol.2005.06.049. [DOI] [PubMed] [Google Scholar]
- 48.Rees D.G.C., Gates A.J., Green M., Eastaugh L., Lukaszewski R.A., Griffin K.F., et al. CpG-DNA protects against a lethal orthopoxvirus infection in a murine model. Antiviral Res. 2005;65(February (2)):87–95. doi: 10.1016/j.antiviral.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 49.Belyakov I.M., Earl P., Dzutsev A., Kuznetsov V.A., Lemon M., Wyatt L.S., et al. Shared modes of protection against poxvirus infection by attenuated and conventional smallpox vaccine viruses. Proc Natl Acad Sci USA. 2003;100(August (16)):9458–9463. doi: 10.1073/pnas.1233578100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu R., Johnson A.J., Liggitt D., Bevan M.J. Cellular and humoral immunity against vaccinia virus infection of mice. J Immunol. 2004;172(May (10)):6265–6271. doi: 10.4049/jimmunol.172.10.6265. [DOI] [PubMed] [Google Scholar]
- 51.Wyatt L.S., Earl P.L., Eller L.A., Moss B. Highly attenuated smallpox vaccine protects mice with and without immune deficiencies against pathogenic vaccinia virus challenge. Proc Nat Acad Sci USA. 2004;101:4590–4595. doi: 10.1073/pnas.0401165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Panchanathan V., Chaudhri G., Karupiah G. Interferon function is not required for recovery from a secondary poxvirus infection. Proc Natl Acad Sci USA. 2005;102(September (36)):12921–12926. doi: 10.1073/pnas.0505180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edghill-Smith Y., Golding H., Manischewitz J., King L.R., Scott D., Bray M., et al. Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat Med. 2005;11(July (7)):740–747. doi: 10.1038/nm1261. [DOI] [PubMed] [Google Scholar]
- 54.Mikloska Z., Ruckholdt M., Ghadiminejad I., Dunckley H., Denis M., Cunningham A.L. Monophosphoryl lipid A and QS21 increase CD8 T lymphocyte cytotoxicity to herpes simplex virus-2 infected cell proteins 4 and 27 through IFN-gamma and IL-12 production. J Immunol. 2000;164(May (10)):5167–5176. doi: 10.4049/jimmunol.164.10.5167. [DOI] [PubMed] [Google Scholar]
- 55.Moore A., McCarthy L., Mills K.H. The adjuvant combination monophosphoryl lipid A and QS21 switches T cell responses induced with a soluble recombinant HIV protein from Th2 to Th1. Vaccine. 1999;17(June (20–21)):2517–2527. doi: 10.1016/s0264-410x(99)00062-6. [DOI] [PubMed] [Google Scholar]
- 56.Newman M.J., Wu J.Y., Gardner B.H., Munroe K.J., Leombruno D., Recchia J., et al. Saponin adjuvant induction of ovalbumin-specific CD8 + cytotoxic T lymphocyte responses. J Immunol. 1992;148(April (8)):2357–2562. [PubMed] [Google Scholar]
- 57.Wu J.Y., Gardner B.H., Murphy C.I., Seals J.R., Kensil C.R., Recchia J., et al. Saponin adjuvant enhancement of antigen-specific immune responses to an experimental HIV-1 vaccine. J Immunol. 1992;148(March (5)):1519–1525. [PubMed] [Google Scholar]
- 58.Newman M.J., Munroe K.J., Anderson C.A., Murphy C.I., Panicali D.L., Seals J.R., et al. Induction of antigen-specific killer T lymphocyte responses using subunit SIVmac251 gag and env vaccines containing QS-21 saponin adjuvant. AIDS Res Hum Retroviruses. 1994;10(July (7)):853–861. doi: 10.1089/aid.1994.10.853. [DOI] [PubMed] [Google Scholar]
- 59.Davis H.L., Weeratna R., Waldschmidt T.J., Tygrett L., Schorr J., Krieg A.M. CpG DNA is a potent enhancer of specific immunity in mice immunized with recombinant hepatitis B surface antigen. J Immunol. 1998;160(January (2)):870–876. [PubMed] [Google Scholar]
- 60.Oxenius A., Martinic M.M., Hengartner H., Klenerman P. CpG-containing oligonucleotides are efficient adjuvants for induction of protective antiviral immune responses with T-cell peptide vaccines. J Virol. 1999;73(May (5)):4120–4126. doi: 10.1128/jvi.73.5.4120-4126.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bell E., Shamim M., Whitbeck J.C., Sfyroera G., Lambris J.D., Isaacs S.N. Antibodies against the extracellular enveloped virus B5R protein are mainly responsible for the EEV neutralizing capacity of vaccinia immune globulin. Virology. 2004;325(August (2)):425–431. doi: 10.1016/j.virol.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 62.Earl P.L., Sugiura W., Montefiori D.C., Broder C.C., Lee S.A., Wild C., et al. Immunogenicity and protective efficacy of oligomeric human immunodeficiency virus type 1 gp140. J Virol. 2001;75(2):645–653. doi: 10.1128/JVI.75.2.645-653.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hooper J.W., Thompson E., Wilhelmsen C., Zimmerman M., Ichou M.A., Steffen S.E., et al. Smallpox DNA vaccine protects nonhuman primates against lethal monkeypox. J Virol. 2004;78(9):4433–4443. doi: 10.1128/JVI.78.9.4433-4443.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]