

Abstract
Cigarette smoke, which contains several carcinogens known to initiate and promote tumorigenesis and metastasis, is the major cause of oral cancer. Lysosomal cathepsin proteases play important roles in tumor progression, invasion and metastasis. In the present work we investigated the effects of cigarette smoke condensate (CSC) on cathepsin (B, D and L) expression and protease-mediated invasiveness in human oral squamous cell carcinoma (OSCC) cells. Our results show that treatment of OSCC cells (686Tu and 101A) with CSC activated cathepsins B, D and L in a dose-dependent manner. Both expression and activity of these cathepsins were up-regulated in CSC-exposed versus non-exposed cells. Although cathepsin L had the lowest basal level, it had the highest induction in exposed cells compared to cathepsins B and D. Suppression of CSC-induced cathepsin B and L activities by specific chemical inhibitors decreased the invasion process, suggesting that these proteases are involved in the invasion process. Overall, our results indicate that CSC activates cathepsin B and L proteolytic activity and enhances invasiveness in OSCC cells, a response that may play a role in CSC-mediated tumor progression and metastasis dissemination.
Keywords: Cathepsins, oral cancer, invasion, cigarette smoke condensate
1. Introduction
Oral cancer is the eighth most common malignancy worldwide, with an estimated 405,300 newly diagnosed cases of oral cancer occurring in 2002 (Ferlay, 2004). Tobacco is a major independent risk factor for the development of oral cancer and the overall risk of oral cancer among smokers is 7–10 times higher than for non-smokers (Warnakulasuriya et al., 2005). Cigarette smoke is a complex chemical mixture containing ~4800 different compounds, of which ~100 are known carcinogens, co-carcinogens, mutagens and/or tumor promoters (Hoffmann et al., 2001). The increased presence of tobacco-related carcinogen-DNA adducts in oral squamous cells of smokers compared with nonsmokers has demonstrated a strong link between smoking and the development of oral cancer (Besaratinia et al., 2000).
Tumor cell invasion and metastasis are associated with the proteolytic activity of various types of proteinases, which induce the escape of cancer cells from the primary site, breaking down connective tissue barriers of the extracellular matrix and basement membrane (Nomura and Katunuma, 2005). In addition to serine and metallo-proteinases, such as MMP-1, MMP-2, or MMP-9 well known to be secreted outside cells, there is increasing evidence that lysosomal proteinases, especially cathepsins B, L and D, may also play important roles in the development and progression of malignant tumors (Lah et al., 2000; Joyce et al., 2004; Mohamed and Sloane, 2006). Since elevated expressions of cathepsins and diminished levels of their endogenous cystatin inhibitors have been observed in several human cancers, including breast, gastric, prostate and oral, cathepsins have been suggested as biological markers of malignant tumors and have been proven useful for disease prognosis (Koblinski et al., 2000; Vigneswaran et al., 2000; Nomura and Katunuma, 2005; Mohamed and Sloane, 2006).
Cathepsins are lysosomal cysteine (cathepsin B and L) or aspartic (cathepsin D) proteinases distributed in almost all mammalian cells. They undergo post-translational modification of mannose-6-phosphate (M6P) residues and are translocated to lysosomal vesicles via the M6P receptor, which is the main pathway of intracellular protein turnover (Nomura and Katunuma, 2005). Recent evidence has shown that the expression and activity of cathepsins B, D and L increase in breast cancer (Lah et al., 2000; Leto et al., 2004), lung cancer (Vasiljeva et al., 2006), colorectal cancer (Troy et al., 2004; Sebzda et al., 2005), prostate cancer (Nomura and Katunuma, 2005) and oral cancer (Vigneswaran et al., 2000; Kawasaki et al., 2002). These changes in expression and activity may have diagnostic and prognostic value for cancers. Cathepsins B and L have been shown to play an important role in matrix degradation and cell invasion, and administration of their inhibitors prevents invasion of cancer cells (Bervar et al., 2003; Mohamed and Sloane, 2006; Nomura and Katunuma, 2005; Wickramasinghe et al., 2005; Hashimoto et al., 2006). Furthermore, the roles of cathepsins in tumor spread and metastasis has been shown by downregulating individual cysteine cathepsins in tumor cells and in transgenic mouse models of human cancer (Mohamed and Sloane, 2006). These results indicate that cancer cells employ various cathepsins to progress malignant disease, and that cathepsins may be potential targets for cancer therapy (Nomura and Katunuma, 2005).
It is not well understood whether cigarette smoke or its components cause tumor development through the cathepsin activation pathway, and the effects of cigarette smoke condensate (CSC) on human OSCC cells to promote tumorigenesis, invasion, and metastasis have not been previously reported. We postulated that the effects of CSC on tumor malignancy is mediated through cathepsin activation for several reasons: 1) cigarette smoking is known to activate TNF-α; 2) cathepsins B and D mediate TNF-α-induced tumor cell death; 3) activation of cathepsins B, D and L are involved in tumor promotion and metastasis. In the present report we demonstrate that exposure of OSCC cell lines to CSC activates cathepsins B and L, and that this activation leads to enhanced tumor cell invasiveness.
2. Materials and Methods
2.1. Materials
Rabbit polyclonal antibodies for human cathepsin B and D were obtained from Athens Research and Technology (Athens, GA), and HRP-conjugated goat anti-rabbit or anti-mouse antiserum and mouse monoclonal anti-cathepsin L antibody were from BD Biosciences (San Diego, CA). Rabbit polyclonal IgGs for caspase-3 and Bid were from Santa Cruz Biotechnology (Santa Cruz, CA), and rabbit polyclonal IgG for poly-(ADP-ribose)-polymerase (PARP) from Cell Signaling Technology (Beverly, MA). Protease inhibitor CA-074, cell permeable CA-074Me, and z-Arg-Arg-NHMec were obtained from Peptides International (Louisville, KY); G418 sulphate was from Mediatech Inc. (Herndon, VA); DEVD-AFC and DEVD-CHO were purchased from Alexis (San Diego, CA). Mouse monoclonal β-actin antibody, phenylmethylsulfonyl fluoride, aprotinin, leupeptin, pepstatin A, z-Phe-Arg-NHMec, and MTT (1-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazoliumbromide) were purchased from Sigma (St. Louis, MO). Bradford protein assay kit was from Bio-Rad (Hercules, CA), and M-PER Mammalian Protein Extraction Reagent from Pierce (Rockford, IL). Protease inhibitor cocktail was from Roche (Indianapolis, IN), and enhanced chemiluminescence ECL substrate from Amersham Biosciences (Piscataway, NJ). Cathepsin L inhibitors z-FF-FMK and z-FY-CHO were purchased from Calbiochem (San Diego, CA). Cell culture reagents were obtained form the following suppliers: Dulbecco Modified Eagle’s Medium (DMEM), DMEM/F12, Trypsin (0.25%)-EDTA, Penicillin-Streptomycin (10 mg/ml), Amphotericin B (250 mg/ml), HEPES Buffer IM, L-Glutamine (200 mM) from Cellgro (Herndorn, VA); fetal bovine serum (Premium) from Atlanta Biologicals (Atlanta, GA). Dimethylsulfoxide was from Fisher Biotech (Fair Lawn, NJ), and DEPC-treated water from Ambion (Austin, TX). TriZol™ reagent, SuperScript™ First-Strand Synthesis kit, and DNA primers for real-time PCR were from Invitrogen Corporation (Carlsbad, CA).
CSC was purchased from Murty Pharmaceuticals Inc. (Lexington, KY) and was prepared using a Phipps-Bird 20-channel smoking machine designed for Federal Trade Commission testing. The particulate matter from Kentucky standard cigarettes (1R3F; University of Kentucky, Lexington, KY) was collected on Cambridge glass fiber filters and the amount of CSC obtained was determined by weight increase of the filter. CSC was prepared by dissolving the collected smoke particulates in dimethylsulfoxide (DMSO) to yield a 4% solution (w/v); the average yield of CSC was 18.1 mg/cigarette. The CSC was diluted into DMSO and aliquots were stored at −80 °C.
2.2. Cell culture
The following tumor cell lines, established and characterized previously as described in the cited references, were used. UM-SCC-101A (101A) was derived from the primary tumor of a 65-year old female with oral squamous cell carcinoma (T3N3M0) involving the tonsillar area (gift from Dr. T. Carey, University of Michigan, Ann Arbor, MI) (Takebayashi et al., 2000; Nagaraj et al., 2006a). MDA-686Tu (686Tu) was derived from the primary tumor of oral squamous cell carcinoma involving the left tonsillar fossa and posterior portion of the tongue in a 49-year old male (tumor stage T3N3B) (gift from Dr. Peter Sacks, New York University, New York, NY) (Sacks, 1996; Vigneswaran et al., 2005). Both cell lines are of epithelial origin and are tumorigenic in nude mice. Cells were maintained in DMEM (101A cells, passage 23) or DMEM/F12 1:1 (v/v) mix (686Tu cells, passage 19) containing 2 mM L-glutamine, 100 μg/ml penicillin G, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B, 10% fetal bovine serum and 0.4 μg/ml hydrocortisone at 37 ºC in a humidified atmosphere with 5% CO2. Treatment with CSC (0 to 100 μg/ml in DMSO) was done for time points of 0, 1, 5, 24 or 36 hours; control cells were treated with the same volume of DMSO solvent only. Three biological replicates were used for each treatment regimen. All protocols for the use of human cell lines in this work were approved by the Institutional Review Board of The University of Louisville, Louisville, KY.
2.3. Cell viability assays
Viability of cells after CSC exposure was detected using MTT dye reduction assay. Briefly, 500 cells/well were seeded in a 96-well plate and cultured to 60% confluence. Serial dilutions of CSC were added, and cells were treated for 1, 5, 24 or 36 hours. After treatment, 50 μl of MTT solution (5 mg/ml in PBS) were added to each well and incubated for 4 h. The reaction was terminated by addition of 100 μl of 20% SDS in 50% dimethylformamide, and the plate was incubated overnight at 37 ºC for total solubilization of reduced MTT. Absorbance was measured at 570 nm in a microplate reader (PowerWave™ Microplate Spectrophotometer, Bio-Tek Instruments Inc., Winooski, VT), and viable cell numbers determined based on a standard curve.
2.4. RNA isolation and cDNA synthesis
Cells were grown to approximately 75% confluence in 6-well plates, with or without CSC treatment (0–100 μg/ml for 24 h), and total RNA was isolated with TriZol™ reagent. The purity and integrity of each RNA preparation was evaluated by using RNA Nano-Chips on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). First strand cDNA was synthesized using the SuperScript™ First-Strand Synthesis kit. DNA contamination in RNA samples was monitored by ‘no reverse transcriptase’ reactions performed in parallel with cDNA synthesis reaction.
2.5. Quantitative real-time PCR
Quantitative real-time reverse transcriptase PCR (qRT-PCR) for cathepsins and β-actin was done using an ABI PRISM 7700 Sequence Detection System (Applied Biosystems, Foster City, CA). The cDNA was diluted to 1 ng/μL, and 2 μL used as template in a 25-μL reaction. Forward and reverse primer mix was added (3 μL of a 1:1 mix, 0.3 μM each) in SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA). Each reaction was performed in triplicate and ‘no-template’ controls were included in each experiment. Dissociation curves were run to eliminate non-specific amplification or primer-dimers. The cycle threshold (CT) values were normalized to the house keeping gene β-actin and the fold change was calculated using 2−ΔΔCT method.
DNA primer sequences for qRT-PCR were obtained from the cited reference for the following primer pairs: cathepsin B forward (5′-GATCTGCATCCACACCAATG-3′) and reverse (5′-AACCAGGCCTTTTCTTGTCC-3′) (Wickramasinghe et al., 2005); cathepsin L forward (5′-GAGGCAACAGAAGAATCCTGTAAGT-3′) and reverse (5′-AGGGCCTTCTCCTGCTTAGG-3′) (Schedel et al., 2004). Cathepsin D forward (5′-CACCACAAGTACAACAGCGAC-3′) and reverse (5′-CTTGGCTGCGATGAAGGTGA-3′), and control β-actin forward (5′-AGAAAATCTGGCACCACACC-3′) and reverse (5′-GGGGTGTTGAAGGTCTCAAA-3′) primers were designed using PrimerBank software (http://pga.mgh.harvard.edu/primerbank/).
2.6. Cathepsin activity assays
Cells were grown in 6-well plates to 60–70 % confluence, and treated for 24 h with CSC (0–100 μg/ml). After treatment, the cells were rinsed twice with ice-cold PBS, treated with lysis buffer (400 mM Na-phosphate, 75 mM NaCl, 4 mM EDTA, 0.25 % Triton-X 100, pH 6.0) for 1 h on ice, ultrasonicated at 40 W for 1 min (1.0 sec on/0.5 sec off pulses) (Model 550 Sonic Dismembrator, Fisher Scientific, Pittsburgh, PA), and centrifuged at 25,000 × g (10 min, 4 °C) to remove cell debris. Total protein amounts were determined using the Bradford protein assay kit. Cathepsin B and L activity was determined fluorimetrically using the methyl-coumarylamide substrate z-Arg-Arg-NHMec at pH 6.0 and z-Phe-Arg-NHMec at pH 5.5, respectively, with excitation at 360 nm and emission at 460 nm, as described (Nagaraj et al., 2007). The cathepsin B substrate was used in conjunction with the cathepsin B inhibitor CA-074 (50 μM) in all control assays; the difference between values without and with CA-074 corresponded to cathepsin B activity. To detect cathepsin L activity, the substrate was used in conjunction with the cathepsin L inhibitor z-FY-CHO (25 μM) as control assays; the difference between values without and with z-FY-CHO corresponded to cathepsin L activity. One unit of enzyme activity was defined as the release of 1 μmol of product/min, specific activity as units/mg protein. Cathepsin D was determined using acid-denatured hemoglobin (16.6 g/l) as substrate in ammonium acetate buffer, pH 3.5, as described (Nagaraj et al., 2007). The reaction was stopped with TCA and the absorbance at 750 nm was determined after addition of Folin-Ciocalteau reagent.
2.7. Protein extractions and Western blotting
Treated or control cells were rinsed with ice-cold PBS, scraped into 1 ml of PBS, and centrifuged at 4,000 rpm for 3 min. The pellets were resuspended into RIPA buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 % Triton X-100, 0.1 % SDS, and 1 mM EDTA) containing protease inhibitors (0.5 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, and 2 μg/ml of both leupeptin and pepstatin). Then, cell extracts were sonicated as described above and cell debris was removed by centrifugation. Cells were washed in ice-cold PBS, and extracted using M-PER Reagent with protease inhibitor cocktail. Proteins were quantified using the Bradford protein assay kit based on a γ-globulin standard curve. Equal amounts of total proteins were separated on a SDS-polyacrylamide gel and transferred onto nitrocellulose membranes by electroblotting overnight at 20 V. Membranes were blocked in TBS-T (10 mM Tris-HCl, 150 mM NaCl, 0.25 % Tween 20, pH 7.5) with 5 % fat-free powdered milk at room temperature for 1 h. After rinsing membranes in TBS-T, the following primary antibodies were used: rabbit polyclonal antibodies for human cathepsin B and D, mouse monoclonal antibody for cathepsin L, or mouse monoclonal β-actin antibody. After incubation overnight at 4 °C or 1 h at room temperature, the membranes were washed four times, 10 min each, in TBS-T. Secondary antibodies used were either horseradish peroxidase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG, followed by five washes with TBS-T. Bands were detected using ECL substrate. For β-actin detection, previously probed membranes were soaked in stripping buffer (70 mM Tris-HCl, pH 6.8, 2 % SDS, 0.1 % β-mercaptoethanol) at 60 °C for 30 min, and incubated as above.
2.8. Cell invasion assay
Invasion assays were carried out using a modification of a published procedure (Wickramasinghe et al., 2005). After trypsinization, 2.5 × 104 cells were suspended in 500 μl serum-free medium containing 0.1% bovine serum albumin and seeded on Matrigel-coated membrane inserts; uncoated inserts were used as controls for determination of motility. Cells were grown for 24 h with or without CSC (25 μg/ml or up to 500 μg/ml), and with or without pretreatment of cathepsin inhibitors CA-074Me (25 μM), pepstatin A (5 μM), or zFF-FMK (10 μM). The bottom chamber contained 0.75 ml of a 50:50 mixture of normal growth media and NIH3T3 conditioned media as chemoattractant, either with or without CSC and cathepsin inhibitors as described above. After incubation for 24 h at 37 °C, the cells remaining inside the insert were removed with a cotton swab, and cells that had moved to the lower membrane surface were fixed in methanol and stained using Diff-quick reagent kit. After air-drying the membrane, the cells were counted at a magnification of 20x under a Nicon Inverted Microscope Eclipse TE300, and images were captured by a cool snap HQ digital B/W CCD camera (Roper Scientific, Trenton, NJ). Assays were performed two times in triplicate wells, and cells present in nine fields covering the center of each membrane were counted. To determine differences in migration for various CSC concentrations, the cell numbers for untreated controls (solvent only) were set as 100%. For differences in migration in the presence of inhibitors, the cell numbers for wells without inhibitor present were set as 100%. Percent invasion (invasion index) for each condition was calculated as (M/C)×100, with M as mean number of cells invading through Matrigel insert and C as mean number of cells migrating through control insert.
2.9. Tests for apoptosis marker activation
Cells were grown in 6-well plates to 60–70 % confluence, treated for 24 h with CSC (0–500 μg/ml) or DMSO solvent as control, and cellular proteins were extracted as described above. Western blots for apoptosis marker proteins were performed according to our previously published procedures; the polyclonal antibodies used here bind to both full-length proteins and their cleavage products (Nagaraj et al, 2006b; Nagaraj et al, 2007). Enzymatic activity for processed caspase-3 was determined as described (Nagaraj et al, 2006b; Nagaraj et al, 2007), using the caspase-3-specific fluorescent peptide substrate Ac-DEVD-AFC; the caspase-3-specific inhibitor DEVD-CHO was used in control reactions.
2.10. Statistical analysis
Data for cell viability, real time RT-PCR, Western blots, enzyme activity and invasion assays were derived from at least three independent experiments. Statistical analyses were conducted using the Prism4 and InStat3 GraphPad software and values were represented as means ± SD. Significance level was calculated using one-way ANOVA and Tukey-Kramer post-test to assess the differences between experimental groups.
3. Results
3.1. Effect of CSC treatment on cell viability
To address whether CSC affected the growth of tumor cells, the 686Tu and 101A cells were treated with different concentration of CSC (from 0 to 100 μg/ml) for time periods up to 36 hours, and viabilities were determined by MTT dye reduction assay (Fig. 1). We observed decreased viabilities in both cells with increasing concentration of CSC at 24- and 36-hour treatments. For early time periods at 1 and 5 hours, 686Tu cells (Fig. 1A) increased in growth rate compared to 101A cells (Fig. 1B). Based on these data, we used 25 μg/ml of CSC for 24 hours in invasion studies since this dose showed only low toxicity against longer CSC exposure. The 24-hour time point showed only gradual decreases in the number of viable cells with increased CSC concentration, and we used this time point for all other assays. We believe these changes are due to altered cell proliferation, since MTT assays measure metabolic activity of live cells, and since we did not observe measurable apoptosis induction under these conditions (Section 3.5).
Fig. 1.
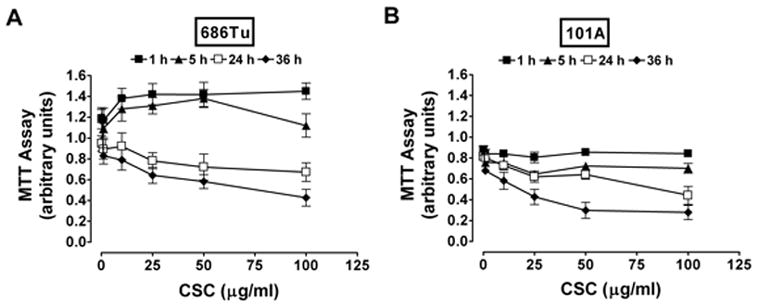
Viability of OSCC cells treated with CSC. Cells 686Tu (A) and 101A (B) were treated with 0 to 100 μg/ml of CSC for 1, 5, 24 and 36 hours. After each indicated time/concentration treatment, the growth of cells was evaluated by MTT assay as described in Materials and Methods. Results are the mean ± SD of three independent measurements for each point.
3.2. Cathepsin RNA expression in CSC-treated OSCC cells
During our previous and current work, analysis of cathepsin B, D and L mRNA baseline expression was performed on two matched pairs of primary (686Tu, 101A) and metastatic (686Ln, 101B) OSCC cells without CSC treatment (unpublished results). Interestingly, cathepsin transcripts were elevated in both metastatic (686Ln, 101B) compared to their corresponding primary tumor (686Tu, 101A) cell lines (2- to 4-fold for cathepsins B and L, and 1.5- to 2-fold for cathepsin D), in agreement with our earlier observations on tumor tissues using immunohistochemistry to determine correlations of cathepsin protein expression with metastasis (Vigneswaran et al., 2000). Considering the significant role played by cathepsins in cigarette smoke-treated macrophages and dendritic cells (Chang et al., 1989; Gairola et al., 1989; Bracke et al., 2005), we investigated whether CSC stimulates cathepsin RNA expression in OSCC cells. Real-time quantitative RT-PCR analyses using gene-specific primers were performed after exposure of 686Tu and 101A cells to CSC (0–100 μg/ml) for 24 h (Fig. 2). Cathepsins B, D and L were all induced by the CSC treatment in a dose-dependent manner. Interestingly, cathepsin L mRNA with the lowest baseline expression was highly up-regulated in both 686Tu and 101A cells (8.3–9.2 fold) at 100 μg/ml CSC treatment compared to moderate changes for cathepsin B (2.8–4.2 fold) and cathepsin D (2.3–3.2 fold). At 25 μg/ml of CSC stimulation, an approximately 1–1.8 fold increase in cathepsin B and D mRNA levels was observed, and the increase in cathepsin L was approximately 3–3.2 fold (Fig. 2). Thus, cathepsin L had the highest increase in mRNA expression among the three cathepsins analyzed.
Fig. 2.
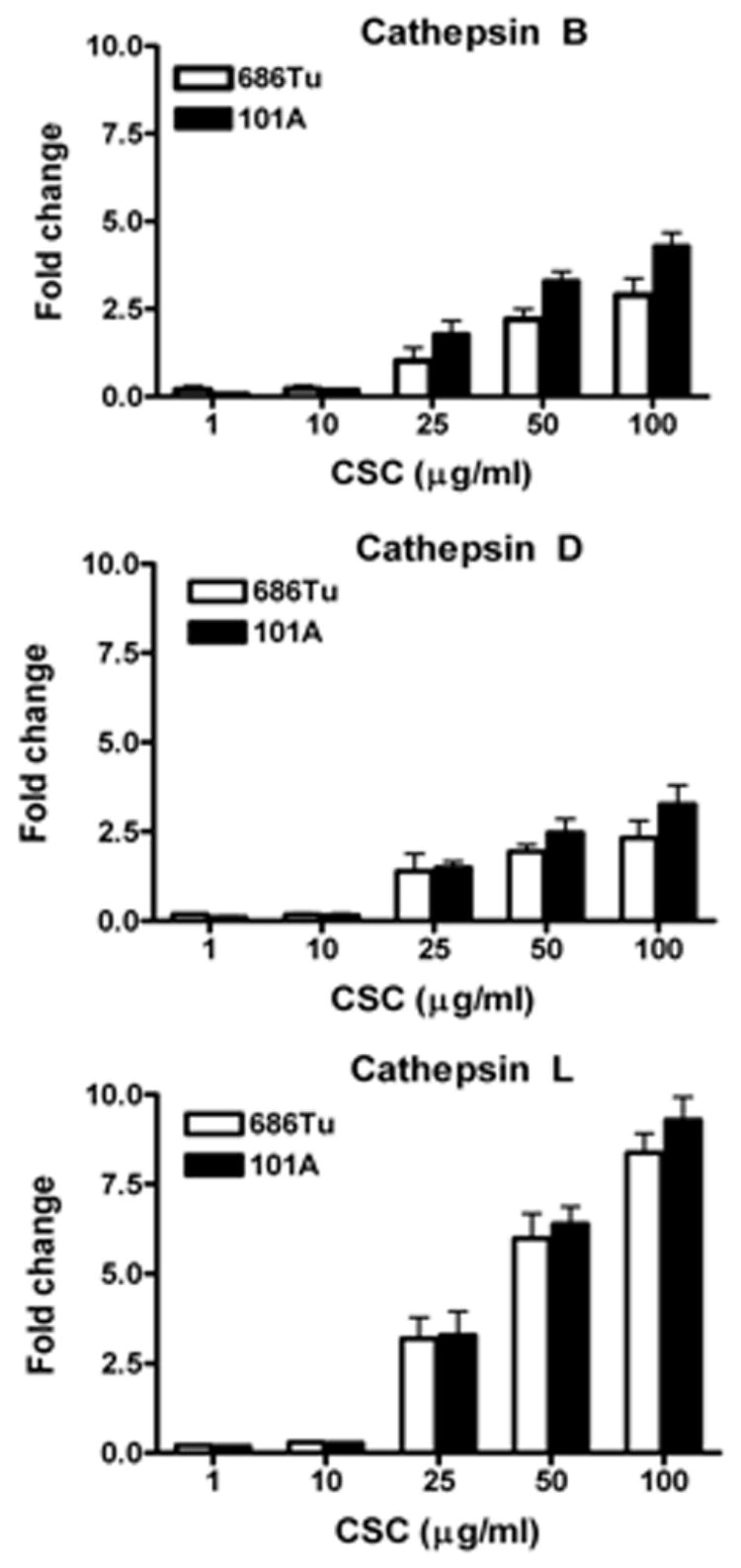
CSC-induced cathepsin mRNA expression in OSCC cells. The 686Tu and 101A cells were treated with different concentration of CSC for 24 hours. Extracted RNAs were subjected to real-time quantitative RT-PCR analysis using primers specific for cathepsin B (CB), cathepsin D (CD), cathepsin L (CL); β-actin primers were used as control to normalize to equal RNA amounts. The fold change levels for CB, CD and CL are relative to the expression in untreated cells; results are the mean ± SD of three independent assays. Open bars, 686Tu cells; solid bars, 101A cells.
3.3. Cathepsin protein expression and activity in CSC-treated OSCC cells
Cathepsins B and L were suggested as markers for malignant oral tumors and as prognostic factors for disease relapse, because their expression correlated with the invasiveness of oral carcinomas (Kawamata et al., 1997; Wickramasinghe et al., 2005). It is not known whether their enzymatic activities increase concurrently with protein levels in cigarette smokers. We measured cathepsin B, D and L protein expression and enzyme activity in cell lysates with or without CSC treatment (Fig. 3). Cathepsin induction was observed in Western blot bands, which showed estimated increases of up to approximately 10-fold for cathepsins B and L, and approximately 2- to 4-fold for cathepsin D, at 100 μg/ml CSC compared to basal levels (Fig. 3A). Also, the specific activities increased 19- or 16-fold for cathepsin B, 190- or 221-fold for cathepsin L, and 28- or 20-fold for cathepsin D in 686Tu or 101A cells, respectively (Fig. 3B), in good agreement with the induction of mRNA levels (Fig. 2). Again, the enzyme activity of cathepsin L in both 686Tu and 101A cells had the highest up-regulation at any CSC concentration compared to cathepsins B and D (Fig. 3B). To demonstrate specificity for each of the cathepsins, protease-specific inhibitors and substrates were used. The cell lysates were treated with z-Arg-Arg-NHMec and CA-074 (as substrate and inhibitor for cathepsin B), z-Phe-Arg-NHMec and z-FY-CHO (as substrate and inhibitor for cathepsin L), and hemoglobin and pepstatin A (as substrate and inhibitor for cathepsin D). These protease inhibitors decreased enzyme activities by 83–97% for CA-074 ( ≥ 50 μM), z-FY-CHO ( ≥ 25 μM), and pepstatin A ( ≥ 5 μM) compared to the non-inhibited control values (data not shown).
Fig. 3.
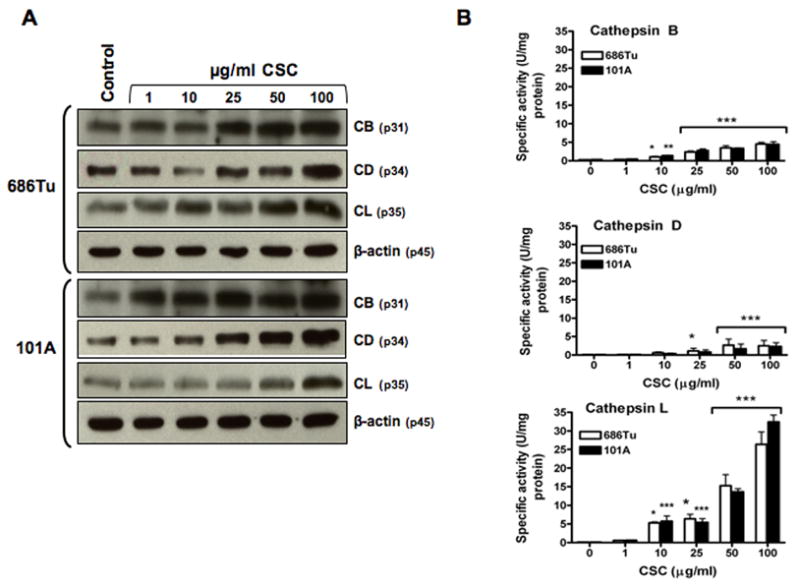
CSC-induced cathepsin protein expression and enzyme activity in OSCC cell extracts. OSCC cells were treated with different concentration of CSC for 24 hours. A. Cellular protein extracts from control and treated cells were subjected to immunoblot analysis using specific antibodies for cathepsin B (CB), cathepsin D (CD), cathepsin L (CL), and β-actin. The bands shown for CB (31 kDa), CD (34 kDa), and CL (35 kDa) are the mature forms. Probing for β-actin (45 kDa) was used as a control to normalize for equal protein amounts in each sample. The data shown are representative of three independent experiments. B. Specific activities of CB, CD and CL were quantitated for control and treated cells using z-Arg-Arg-NHMec, hemoglobin, and z-Phe-Arg-NHMec as a substrate, respectively. Open bars, 686Tu cells; and solid bars, 101A cells; significance was determined by ANOVA (mean ± SD, n=3): * p<0.05; ** p<0.01; *** p<0.001; ns, non-significant.
3.4. Involvement of cathepsin B and L in CSC-induced invasiveness of OSCC cells
As an ultimate functional test, the consequences of CSC-induced cathepsin B, D and L expression on the migration and invasiveness of 686Tu and 101A cells were determined (Fig. 4 and 5). CSC increased invasiveness of both cell lines at concentrations that also significantly increased cathepsin expression. Cell motilities were increased by 9.4–51.8% and 4.1–38.3% in 686Tu and 101A cells, respectively, after CSC exposure (25 μg to 500 μg) compared to control cells (Figs. 4A and 5A). More importantly, the number of cells that invaded through Matrigel increased (Figs. 4A and 5A), and invasion indices (Figs. 4B, C and 5B, C) increased significantly by 21.6–29% (p ≤ 0.01) at 50 μg/ml CSC and by 37.6–47.9% (p < 0.001) at 250 or 500 μg/ml CSC for treated compared to untreated cells.
Fig. 4.
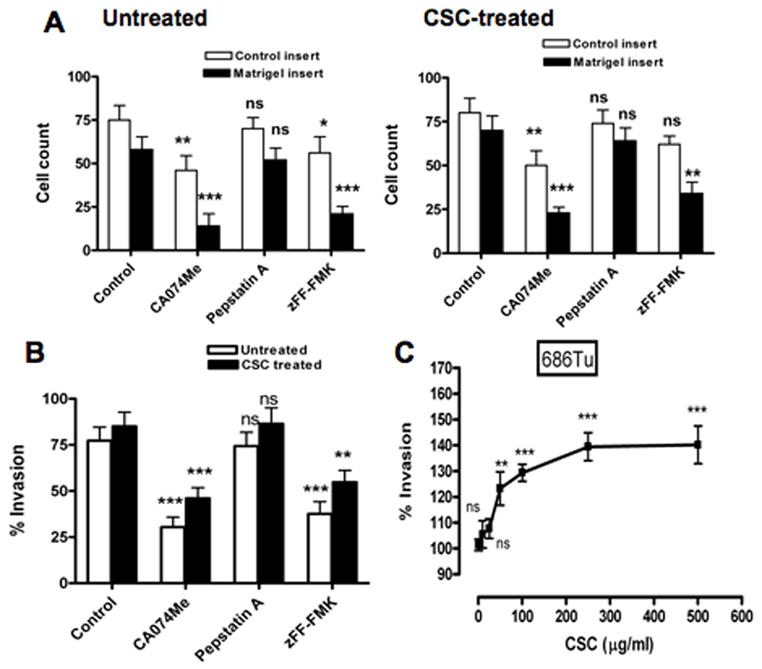
Effect of cathepsin inhibitors on invasiveness of CSC-treated 686Tu cells. Invasion was measured after 24 hours with Matrigel-coated membrane inserts and uncoated control inserts for CSC-treated (25 μg/ml or 1–500 μg/ml) 686Tu cells. Assays were done with or without inhibitors for cathepsin B (CA-074Me), cathepsin D (pepstatin A) or cathepsin L (zFF-FMK), and were performed twice, each in triplicate wells. A. Cell counts (open bars, control inserts; closed bars, Matrigel inserts) represented as the number of cells/area (means ± SD) from 5 to 7 photographs taken from triplicate wells. B. Invasion index, calculated as percentage of cells migrating through Matrigel-coated compared to uncoated membranes (set to 100%), for treated (25 μg/ml; closed bars) versus untreated (solvent only; open bars) 686Tu cells. C. Invasion index as a function of increasing concentration of CSC (1–500 μg/ml). Significance was determined by ANOVA: * p<0.05; ** p<0.01; *** p<0.001; ns, non-significant.
Fig. 5.
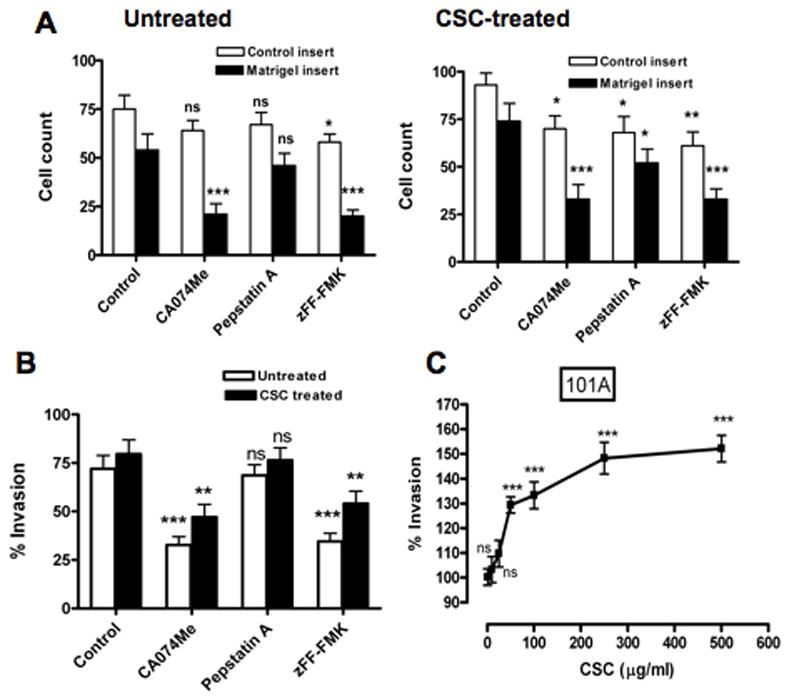
Effect of cathepsin inhibitors on invasiveness of CSC-treated 101A cells. Invasion was measured after 24 hours with Matrigel-coated membrane inserts and uncoated control inserts for CSC-treated (25 μg/ml or 1–500 μg/ml) 101A cells. Assays were done with or without inhibitors for cathepsin B (CA-074Me), cathepsin D (pepstatin A) or cathepsin L (zFF-FMK), and were performed twice, each in triplicate wells. A. Cell counts (open bars, control inserts; closed bars, Matrigel inserts) represented as the number of cells/area (means ± SD) from of 5 to 7 photographs taken from triplicate wells. B. Invasion index, calculated as percentage of cells migrating through Matrigel-coated compared to uncoated membranes (set to 100%), for treated (25 μg/ml; closed bars) versus untreated (solvent only; open bars) 101A cells. C. Invasion index as a function of increasing concentration of CSC (1–500 μg/ml). Significance was determined by ANOVA: * p<0.05; ** p<0.01; *** p<0.001; ns, non-significant.
To demonstrate involvement of cathepsins in these phenotypic alterations, cell-permeable cathepsin inhibitors were used in invasion assay. It was shown previously that CA-074Me and Z-FF-FMK are irreversible and selective inhibitors for cathepsin B (Buttle et al., 1992) and cathepsin L (Shaw et al., 1993), respectively. Pepstatin A has been used for selective inhibition of aspartic proteases including cathepsin D (Chen et al., 2000). Cell motilities were decreased by 52.9–54% for CA-074Me, 13.5–23.5% for pepstatin A and 45.2–45.9% for zFF-FMK in CSC-treated cells (Figs. 4A and 5A). More importantly, substantial and significant decreases in invasion indices for CSC-treated 686Tu (Fig. 4B) and 101A cells (Fig. 5B) in the presence of cathepsin inhibitors were observed compared to assays without cathepsin inhibitors (62.5% and 54.4% decrease for CA-074Me, p ≤ 0.01; 53.8% and 52.1% decrease for zFF-FMK, p < 0.01; for 686Tu and 101A cells, respectively). Pepstatin A showed only small and non-significant decreases for both cells (8.6% and 4.7%, respectively). Apparently, the decreased cathepsin B and L activity resulted in the decreased ability of the OSCC cells to degrade the Matrigel barrier, whereas inhibition of cathepsin D activity had little effect. The invasion indices and their changes were very similar for 686Tu and 101A cells, indicating that the effect of the CSC treatment was the same in both OSCC cells. This differential functional increase of invasiveness after CSC treatment, and decrease with cathepsin B and L inhibitors, parallels the above molecular data which also show overall strong CSC-mediated increases in cathepsin mRNA, protein and activity.
3.5. Effects of CSC treatment on apoptosis markers
In order to test for apoptosis induction as a result of CSC exposure, we investigated possible activation of apoptosis marker proteins (Fig. 6). It is known that after initiation of the apoptotic cascade, the 23 kD Bid protein is cleaved into its 15 kD truncated form, the 32 kD pro-caspase-3 to its 20 kD active form, and the 113 kD poly-ADP-ribose-polymerase (PARP) is processed to the 85 kD active protein (Nagaraj et al, 2006b; Nagaraj et al, 2007). We observed that exposure to CSC at doses used for invasion assays did not cause detectable cleavage of pro-apoptotic Bid or the late apoptosis factors caspase-3 and PARP into their processed forms (Fig. 6A). Also, treatment for 24 hours with up to 500 μg/ml CSC resulted in only very low caspase-3 enzymatic activity (0.3 to 1.7 nMol/h/μg) (Fig. 6B), levels that were marginal compared to activation levels observed in OSCC cells after apoptosis induction by TRAIL (11–15 nMol/h/μg) (Nagaraj et al, 2006b; Nagaraj et al, 2007). These data document that apoptosis in exposed cells is minimal and does not contribute to the observed effects on migration and invasion.
Fig. 6.
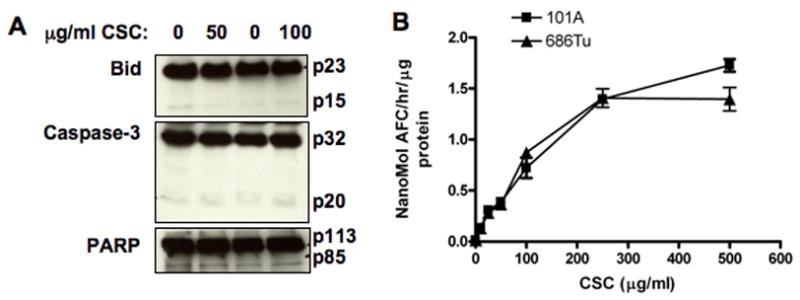
Effects of CSC treatment on apoptosis markers. The 686Tu and 101A cells were treated for 24 hours with different concentrations of CSC, and cellular proteins extracted for analysis. A: Western blots to test for cleavage of Bid, caspase-3, or PARP in 101A cells treated with 50 or 100 μg/ml CSC, or with DMSO solvent only as control. B: Caspase-3 enzyme activity in 686Tu (triangles) and 101A (squares) cell extracts after treatment with increasing concentrations of CSC for 24 hours. Results are the mean ± SD of three independent measurements for each point.
4. Discussion
The present study demonstrates that CSC treatment of oral cancer cells increases cathepsin expression and enhances the invasive phenotype of the exposed OSCC cells. This is the first report on the effects of CSC on malignant cells of the oral cavity, a site that is directly exposed to tobacco-borne carcinogens in smokers. We propose that the mechanism by which CSC stimulates invasion in OSCC cells is due to a dose-dependent induction of cathepsin expression and activity, leading to increased cell motility and invasion-promoting proteolytic activity. In order to define a biologically relevant range of CSC concentrations, our main objective has been to investigate the response of live cell populations without substantial loss due to cell death. Thus, a range of CSC concentrations was chosen for which we observed molecular responses on gene expression profiles without substantial cell death or toxicity (Zacharias et al., to be published). The highest dose applied (500 μg/ml CSC for 24 hours in 10 ml media, equivalent to a total of 5 mg CSC) corresponds to less than half a cigarette per 24-hour period, using the yield of 18.1 mg CSC/cigarette provided by the supplier company. Thus, our range of CSC doses is clearly very low compared to the uptake by any average smoker of several cigarettes per day.
Our data suggests that cathepsins B and L are the major determinants of the invasive capacity of CSC-treated OSCC cells. This mechanism is supported by the observations that: 1) CSC treatment at doses that stimulated invasion also increased cathepsins B and L expression and activity; 2) CSC-enhanced invasion was blocked by co-treatment with cathepsin inhibitors CA-074Me or zFF-FMK, indicating that such CSC-induced changes were dependent on cathepsin activity; 3) cathepsin B and L inhibitors strongly reduced cell motilities, whereas the cathepsin D inhibitor caused only a weak reduction; 4) CSC stimulated invasion and motility to the same degree in two different OSCC cells. In addition, since upregulation of cathepsins was observed at both the RNA and protein level, certain metabolites of cigarette smoke carcinogens may also induce cathepsin transcriptional gene activity. These effects may be due to the conversion of carcinogens by the cytochrome P450 enzymes CYP1a1 and CYP1b1 to metabolites that can bind to TNF-α receptors (Nagaraj et al., 2006a). Our findings are in agreement with prior studies which showed that cathepsin expression and activity were up-regulated by cigarette smoke treatment of pulmonary macrophages, dendritic cells, or alveolar macrophages (Chang et al., 1989; Gairola et al., 1989; Bracke et al., 2005).
Interestingly, although cathepsin D is also up-regulated in CSC-treated cells, the cathepsin D inhibitor only caused a small (5–9%) decrease of invasion, compared to much stronger effects (53–63%) by cathepsin B or L inhibitors. Also, it cannot be excluded that cathepsin B or L inhibitors may inhibit some other yet uncharacterized cysteine cathepsins as observed in the mouse genome (Turk et al., 2004).
It is important to note that cell proliferation, defined here as change in the number of viable cells over time, was also affected by CSC treatment. Under our invasion assay conditions, CSC exposure at any dose for short time periods (1 to 5 hours) did not affect proliferation for 101A cells and actually increased proliferation for the 686Tu cells. Yet, CSC-induced increases in invasion occurred although the cells started to decrease in number after 24 h treatment even at low dose (25 μg/ml) of CSC, presumably due to a decrease in proliferation rates. Clearly, elevated cathepsin activity did not lead to apoptosis in CSC-treated cells, and significant increases in invasive capacity occurred without initiation of apoptosis. Previously, we observed cathepsin B-mediated increased apoptosis susceptibility in OSCC cells via the receptor- or mitochondria-dependent pathways after exposure to TRAIL or hypoxia, respectively (Nagaraj et al., 2006b; Nagaraj et al., 2007). Apparently, CSC exposure leads to a different cellular response than apoptosis induction by those pathways, and the net response to CSC exposure in these cells favors invasiveness over apoptosis induction.
The enhanced invasion of the CSC-treated cells may not be solely attributable to cathepsin B or L alone, but may also involve activation of matrix metalloproteinases. Cigarette smoking increased MMP-1 and MMP-12 expression in human lung fibroblasts and airway-like epithelia (Kim et al., 2004; Lavigne and Eppihimer, 2005). Inhibition of cathepsin B and MMP-9 expression in glioblastoma cells reduced tumor cell invasion (Lakka et al., 2004). These and our data indicate that smoking may increase MMP expression and/or activity indirectly following activation by cathepsins. However, the role of CSC on MMP expression and activity in oral cancer cells has yet to be determined. Also, the mechanisms responsible for the influence of cathepsin B and L on tumor cell motility per se are still unclear.
In conclusion, we observed several-fold increases in cathepsin B and L expression and activity in OSCC cells exposed to CSC, leading to elevated invasiveness in these cells. Inhibition of cathepsin activity by selective peptide inhibitors blocked the invasive capacity of the cells through a Matrigel matrix. These results indicate that cathepsins B and L, possibly in addition to some other yet uncharacterized cysteine proteases, play an important role in matrix degradation and cell invasion of the CSC-treated cells. These results provide mechanism to explain, in part, the increased risk for smokers to develop metastatic oral cancers, and suggest that administration of cathepsin inhibitors may prevent the spread and metastasis of oral cancer. Since cathepsins B and L are key contributors to tumor invasiveness, they may be valuable potential targets for future cancer therapies to prevent smoking-related oral cancer progression and metastatic spread.
Acknowledgments
Supported by NIH grant DE13150 (W.Z.) and by Philip Morris USA Inc. and Philip Morris International (W.Z.). Cell lines were generous gifts from T. E. Carey, University of Michigan (101A) and P. G. Sacks, New York University (686Tu).
Footnotes
Conflict of Interest Statement: None of the authors have any conflicts of interest with respect to the work described here.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bervar A, Zajc I, Sever N, Katunuma N, Sloane BF, Lah TT. Invasiveness of transformed human breast epithelial cell lines is related to cathepsin B and inhibited by cysteine proteinase inhibitors. Biol Chem. 2003;384:447–455. doi: 10.1515/BC.2003.050. [DOI] [PubMed] [Google Scholar]
- Besaratinia A, Van Straaten HW, Godschalk RW, Van ZN, Balm AJ, Kleinjans JC, Van Schooten FJ. Immunoperoxidase detection of polycyclic aromatic hydrocarbon-DNA adducts in mouth floor and buccal mucosa cells of smokers and nonsmokers. Environ Mol Mutagen. 2000;36:127–133. doi: 10.1002/1098-2280(2000)36:2<127::aid-em7>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Bracke K, Cataldo D, Maes T, Gueders M, Noel A, Foidart JM, Brusselle G, Pauwels RA. Matrix metalloproteinase-12 and cathepsin D expression in pulmonary macrophages and dendritic cells of cigarette smoke-exposed mice. Int Arch Allergy Immunol. 2005;138:169–179. doi: 10.1159/000088439. [DOI] [PubMed] [Google Scholar]
- Buttle DJ, Murata M, Knight CG, Barrett AJ. CA074 methyl ester: A proinhibitor for intracellular cathepsin B. Arch Biochem Biophys. 1992;299:377–380. doi: 10.1016/0003-9861(92)90290-d. [DOI] [PubMed] [Google Scholar]
- Chen CS, Chen WN, Zhou M, Arttamangkul S, Haugland RP. Probing the cathepsin D using a BODIPY FL-pepstatin A: applications in fluorescence polarization and microscopy. J Biochem & Biophys Methods. 2000;42:137–151. doi: 10.1016/s0165-022x(00)00048-8. [DOI] [PubMed] [Google Scholar]
- Chang JC, Yoo OH, Lesser M. Cathepsin D activity is increased in alveolar macrophages and bronchoalveolar lavage fluid of smokers. Am Rev Respir Dis. 1989;140:958–960. doi: 10.1164/ajrccm/140.4.958. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Bray F, Pisani P, Parkin DM. Mortality and Prevalence Worldwide, IARC CancerBase No. 5, version 2.0. IARC Press; Lyon: 2004. GLOBOCAN 2002: Cancer Incidence. [Google Scholar]
- Gairola CG, Galicki NI, Cardozo C, Lai YL, Lesser M. Cigarette smoke stimulates cathepsin B activity in alveolar macrophages of rats. J Lab Clin Med. 1989;114:419–425. [PubMed] [Google Scholar]
- Hashimoto Y, Kondo C, Kojima T, Nagata H, Moriyama A, Hayakawa T, Katunuma N. Significance of 32-kDa cathepsin L secreted from cancer cells. Cancer Biother Radiopharm. 2006;21:217–224. doi: 10.1089/cbr.2006.21.217. [DOI] [PubMed] [Google Scholar]
- Hoffmann D, Hoffmann I, El-Bayoumy K. The less harmful cigarette: a controversial issue. A tribute to Ernst L Wynder. Chem Res Toxicol. 2001;14:767–790. doi: 10.1021/tx000260u. [DOI] [PubMed] [Google Scholar]
- Joyce JA, Baruch A, Chehade K, Meyer-Morse N, Giraudo E, Tsai FY, Greenbaum DC, Hager JH, Bogyo M, Hanahan D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5:443–453. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]
- Kawamata H, Nakashiro K, Uchida D, Harada K, Yoshida H, Sato M. Possible contribution of active MMP2 to lymph-node metastasis and secreted cathepsin L to bone invasion of newly established human oral-squamous-cancer cell lines. Int J Cancer. 1997;70:120–127. doi: 10.1002/(sici)1097-0215(19970106)70:1<120::aid-ijc18>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Kawasaki G, Kato Y, Mizuno A. Cathepsin expression in oral squamous cell carcinoma: relationship with clinicopathologic factors. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;93:446–454. doi: 10.1067/moe.2002.122834. [DOI] [PubMed] [Google Scholar]
- Kim H, Liu X, Kohyama T, Kobayashi T, Conner H, Abe S, Fang Q, Wen FQ, Rennard SI. Cigarette smoke stimulates MMP-1 production by human lung fbroblasts through the ERK1/2 pathway. COPD. 2004;1:13–23. doi: 10.1081/COPD-120030164. [DOI] [PubMed] [Google Scholar]
- Koblinski JE, Ahram M, Sloane BF. Unraveling the role of proteases in cancer. Clin Chim Acta. 2000;291:113–135. doi: 10.1016/s0009-8981(99)00224-7. [DOI] [PubMed] [Google Scholar]
- Lah TT, Kalman E, Najjar D, Gorodetsky E, Brennan P, Somers R, Daskal I. Cells producing cathepsins D, B, and L in human breast carcinoma and their association with prognosis. Hum Pathol. 2000;31:149–160. doi: 10.1016/s0046-8177(00)80214-2. [DOI] [PubMed] [Google Scholar]
- Lakka SS, Gondi CS, Yanamandra N, Olivero WC, Dinh DH, Gujrati M, Rao JS. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene. 2004;23:4681–4689. doi: 10.1038/sj.onc.1207616. [DOI] [PubMed] [Google Scholar]
- Lavigne MC, Eppihimer MJ. Cigarette smoke condensate induces MMP-12 gene expression in airway-like epithelia. Biochem Biophys Res Commun. 2005;330:194–203. doi: 10.1016/j.bbrc.2005.02.144. [DOI] [PubMed] [Google Scholar]
- Leto G, Tumminello FM, Crescimanno M, Flandina C, Gebbia N. Cathepsin D expression levels in nongynecological solid tumors: clinical and therapeutic implications. Clin Exp Metastasis. 2004;21:91–106. doi: 10.1023/b:clin.0000024740.44602.b7. [DOI] [PubMed] [Google Scholar]
- Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- Nagaraj NS, Beckers S, Mensah JK, Waigel S, Vigneswaran N, Zacharias W. Cigarette smoke condensate induces cytochromes P450 and aldo-keto reductases in oral cancer cells. Toxicol Lett. 2006a;165:182–194. doi: 10.1016/j.toxlet.2006.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj NS, Vigneswaran N, Zacharias W. Cathepsin B promotes TRAIL-induced apoptosis in oral cancer cells. J Cancer Res Clin Oncol. 2006b;132:171–183. doi: 10.1007/s00432-005-0053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj NS, Vigneswaran N, Zacharias W. Hypoxia inhibits TRAIL-induced tumor cell apoptosis: Involvement of lysosomal cathepsins. Apoptosis. 2007;12:125–139. doi: 10.1007/s10495-006-0490-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Katunuma N. Involvement of cathepsins in the invasion, metastasis and proliferation of cancer cells. J Med Invest. 2005;52:1–9. doi: 10.2152/jmi.52.1. [DOI] [PubMed] [Google Scholar]
- Sacks PG. Cell, tissue and organ culture as in vitro models to study the biology of squamous cell carcinomas of the head and neck. Cancer Metast Rev. 1996;15:27–51. doi: 10.1007/BF00049486. [DOI] [PubMed] [Google Scholar]
- Schedel J, Seemayer CA, Pap T, Neidhart M, Kuchen S, Michel BA, Gay RE, Muller-Ladner U, Gay S, Zacharias W. Targeting cathepsin L (CL) by specific ribozymes decreases CL protein synthesis and cartilage destruction in rheumatoid arthritis. Gene Ther. 2004;11:1040–1047. doi: 10.1038/sj.gt.3302265. [DOI] [PubMed] [Google Scholar]
- Sebzda T, Saleh Y, Gburek J, Andrzejak R, Gnus J, Siewinski M, Grzebieniak Z. Cathepsin D expression in human colorectal cancer: relationship with tumour type and tissue differentiation grade. J Exp Ther Oncol. 2005;5:145–150. [PubMed] [Google Scholar]
- Shaw E, Mohanty S, Colic A, Stoka V, Turk V. The affinity-labelling of cathepsin S with peptidyl diazomethyl ketones : Comparison with the inhibition of cathepsin L and calpain. FEBS Letters. 1993;334:340–342. doi: 10.1016/0014-5793(93)80707-2. [DOI] [PubMed] [Google Scholar]
- Takebayashi S, Ogawa T, Jung KY, Muallem A, Mineta H, Fisher SG, Grenman R, Carey TE. Identification of new minimally lost regions on 18q in head and neck squamous cell carcinoma. Cancer Res. 2000;60:3397–3403. [PubMed] [Google Scholar]
- Troy AM, Sheahan K, Mulcahy HE, Duffy MJ, Hyland JM, O’Donoghue DP. Expression of Cathepsin B and L antigen and activity is associated with early colorectal cancer progression. Eur J Cancer. 2004;40:1610–1616. doi: 10.1016/j.ejca.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Turk V, Kos J, Turk B. Cysteine cathepsins (proteases) - on the main stage of cancer? Cancer Cell. 2004;5:409–410. doi: 10.1016/s1535-6108(04)00117-5. [DOI] [PubMed] [Google Scholar]
- Vasiljeva O, Papazoglou A, Kruger A, Brodoefel H, Korovin M, Deussing J, Augustin N, Nielsen BS, Almholt K, Bogyo M, Peters C, Reinheckel T. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. 2006;66:5242–5250. doi: 10.1158/0008-5472.CAN-05-4463. [DOI] [PubMed] [Google Scholar]
- Vigneswaran N, Zhao W, Dassanayake A, Muller S, Miller DM, Zacharias W. Variable expression of cathepsin B and D correlates with highly invasive and metastatic phenotype of oral cancer. Hum Pathol. 2000;31:931–937. doi: 10.1053/hupa.2000.9035. [DOI] [PubMed] [Google Scholar]
- Vigneswaran N, Wu J, Sacks P, Gilcrease M, Zacharias W. Microarray gene expression profiling of cell lines from primary and metastatic tongue squamous cell carcinoma: Possible insights from emerging technology. J Oral Pathol & Med. 2005;34:77–86. doi: 10.1111/j.1600-0714.2004.00258.x. [DOI] [PubMed] [Google Scholar]
- Warnakulasuriya S, Sutherland G, Scully C. Tobacco, oral cancer, and treatment of dependence. Oral Oncol. 2005;41:244–260. doi: 10.1016/j.oraloncology.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe NS, Nagaraj NS, Vigneswaran N, Zacharias W. Cathepsin B promotes both motility and invasiveness of oral carcinoma cells. Arch Biochem Biophys. 2005;436:187–195. doi: 10.1016/j.abb.2005.01.023. [DOI] [PubMed] [Google Scholar]