

Abstract
Background
Cardiac ion channelopathies are responsible for an ever-increasing number and diversity of familial cardiac arrhythmia syndromes. We describe a new clinical entity that consists of an ST-segment elevation in the right precordial ECG leads, a shorter-than-normal QT interval, and a history of sudden cardiac death.
Methods and Results
Eighty-two consecutive probands with Brugada syndrome were screened for ion channel gene mutations with direct sequencing. Site-directed mutagenesis was performed, and CHO-K1 cells were cotransfected with cDNAs encoding wild-type or mutant CACNB2b (Cavβ2b), CACNA2D1 (Cavα2δ1), and CACNA1C tagged with enhanced yellow fluorescent protein (Cav1.2). Whole-cell patch-clamp studies were performed after 48 to 72 hours. Three probands displaying ST-segment elevation and corrected QT intervals ≤360 ms had mutations in genes encoding the cardiac L-type calcium channel. Corrected QT ranged from 330 to 370 ms among probands and clinically affected family members. Rate adaptation of QT interval was reduced. Quinidine normalized the QT interval and prevented stimulation-induced ventricular tachycardia. Genetic and heterologous expression studies revealed loss-of-function missense mutations in CACNA1C (A39V and G490R) and CACNB2 (S481L) encoding the α1- and β2b-subunits of the L-type calcium channel. Confocal microscopy revealed a defect in trafficking of A39V Cav1.2 channels but normal trafficking of channels containing G490R Cav1.2 or S481L Cavβ2b-subunits.
Conclusions
This is the first report of loss-of-function mutations in genes encoding the cardiac L-type calcium channel to be associated with a familial sudden cardiac death syndrome in which a Brugada syndrome phenotype is combined with shorter-than-normal QT intervals.
Keywords: arrhythmia, genetics, electrophysiology, tachycardia, fibrillation
Cardiac arrhythmias are responsible for an estimated1 million cases of syncope and sudden cardiac death (SCD) among Europeans and Americans each year.1 Cardiac arrhythmias can be acquired as a consequence of coronary heart disease or may be secondary to familial or inherited syndromes. The past decade has witnessed an explosion of information linking cardiac ion channel mutations with a wide variety of inherited arrhythmia syndromes.2 The long-QT syndrome has been associated with 10 different genes, in large part owing to the pioneering studies of Keating and coworkers. The LQT8 form of long-QT syndrome, also known as Timothy syndrome, is associated with gain-of-function mutations in cardiac calcium channel activity.3,4 The cardiac L-type calcium channel is a protein complex formed by at least 3 subunits, α1, β, and α2δ. The pore-forming Cav1.2 α1-subunit is encoded by CACNA1C. The β- or Cavβ2b-subunit, encoded by CACNB2b, modulates calcium channel activity in the human heart and enables trafficking by suppressing an endoplasmic reticulum retention signal in the I-II loop of the α1-subunit.5
The short-QT syndrome (SQTS), a clinical entity first described in 2000,6 has been associated with a gain of function in 3 distinct potassium channels (KCNH2, KCNQ1, and KCNJ2) that leads to abbreviation of the ECG QT interval and the development of malignant arrhythmias.7–9 Loss-of-function mutations in SCN5A, the gene that encodes the α-subunit of the cardiac sodium channel, have been linked to Brugada syndrome, which is characterized by an ST-segment elevation in the right precordial leads (V1 through V3) of the ECG and the development of polymorphic ventricular tachycardia that can result in SCD.10 Here, we report for the first time a new clinical entity that consists of an ST-segment elevation in V1 through V3 and a shorter-than-normal QT interval caused by loss-of-function mutations in the α1- and β-subunits of the L-type cardiac calcium channel.
Methods
Patients were diagnosed with Brugada syndrome on the basis of established criteria.11,12 We define short QT as corrected QT (QTc) intervals ≤ 360 ms for males and ≤ 370 ms for females, on the basis of published reports.13,14 At a heart rate of 60 bpm, the predicted QT interval (QTp) is 410 ms and the lower limit of normal is defined as 2 SDs below QTp, or 360 ms. Clinical and genetic studies were performed in accordance with human subject guidelines after written informed consent was obtained according to protocols approved by the local institutional review boards.
ECG Measurement
The ECG was digitally scanned, magnified 4 to 8 times, and measured with digital calipers. The end of the T wave was defined as the intersection of a tangent, drawn to the descending portion of the T wave, with the isoelectric line. QT intervals were measured in lead II whenever possible.
Mutation Analysis
Genomic DNA was prepared from peripheral blood lymphocytes of patients 1, 2, and 3 and available family members. All known exons of the principal long-QT syndrome genes were amplified with intronic primers and sequenced in both directions to probe for mutations. The following genes were screened: SCN5A, SCN1B, SCN3B, KCNH2, KCNQ1, KCNJ2, KCNE1, KCNE2, KCNE3, KCND3 (Kv4.3), KCNIP2 (KCHiP2), KCNJ11, CACNA1C (Cav1.2), CACNB2b (Cavβ2b), and CACNA2D1 (Cavα2δ1). In addition, IRX5 was probed because of association of this transcriptional factor gene with Kv4 transient outward potassium channels.15
All individuals studied in the control groups for the different mutations, matched by race and ethnic background, were healthy and had no family history of cardiac arrhythmias based on written clinical history. ECGs of control individuals were not available.
Linkage Analysis
Two-point linkage analysis was performed with LINKAGE software (version 5.2). An autosomal-dominant mode of inheritance with complete penetrance and a disease allele frequency of 0.001 were used for the analysis. The allele frequencies for the calculated mutation were given as 0.001 for the T allele.
Mutagenesis and Transfection
Site-directed mutagenesis was performed with QuikChange (Stratagene, La Jolla, Calif) on full-length human wild-type (WT) CACNA1C cDNA cloned in pcDNA3 containing exon 8 (accession No. AJ224873), the CACNA1C clone (EYFP)Nα1C,77 containing exon 8A (accession No. Z34815), and CACNB2b cloned in pcDNA3 (accession No. AF285239), which were a kind gift from Dr Nikolai Soldatov. Chinese hamster ovary (CHO-K1) cells were grown in GIBCO F12 nutrient mixture (Gibco, Invitrogen cell culture, Carlsbad, Calif) in 35-mm culture dishes and placed in a 5% CO2 incubator at 37°C. The cells were cotransfected with lipofectamine or FuGene6 (Roche Diagnostics, Indianapolis, Ind) with a 1:1:1 molar ratio of WT or mutant human CACNA1C, CACNB2b, and WT CACNA2D1.3 To assess the influence of WT on expression of the mutant channels, CHO-K1 cells were cotransfected with a combination of mutant and WT CACNA1C or mutant and WT CACNB2b with the same total molar ratio. Electrophysiological studies were performed after 48 to 72 hours of incubation. CACNA1C was transfected as either (EYFP)Nα1C,77 or pcDNA3-CACNA1C. In the latter case, 0.86 μg of enhanced green fluorescent protein cDNA was added to the transfection mixture. The 2 approaches yielded similar electrophysiological results. It is noteworthy that previous studies have demonstrated that the fusion yellow fluorescent protein [(EYFP)Nα1C,77] did not influence CaV1.2 channel expression.16
Previous studies have shown that transmembrane segment 6 in domain I of Cav1.2 can be encoded by 2 mutually exclusive exons, 8 and 8A.17 Exon 8 is highly expressed in the heart and to a much lesser extent in other tissues.4 By comparison, exon 8A expression is less prominent in heart but more impressive in organs with smooth muscle, including the aorta, bladder, and uterus.3 Specific mutations in either splice variant cause a gain of function in Cav1.2 responsible for 2 forms of Timothy syndrome, a multiorgan disease with severe QT prolongation, arrhythmia, and sudden death.3,4
Electrophysiology
Voltage-clamp recordings from transfected CHO-K1 cells were made with patch pipettes, fabricated from 1.5-mm OD borosilicate glass capillaries, filled with a solution containing (in mmol/L) 120 CsCl2, 2.0 MgCl2, 10 HEPES, 5 CaCl2, 2 MgATP, and 10 EGTA, (pH 7.25 with CsOH) that had a resistance of 2 to 4 MΩ. Extracellular solution contained (in mmol/L) 130 NMDG, 5 KCl, 15 CaCl2, 1 MgCl2, 10 HEPES, pH 7.35 with HCl. Current signals were recorded with an Axopatch 200A or MultiClamp 700A amplifier (Axon Instruments Inc, Foster City, Calif), and series resistance errors were reduced by 60% to 70% with electronic compensation. All recordings were made at room temperature.
Data Acquisition and Analysis
All signals were acquired at 20 to 50 kHz and analyzed with a personal computer running pCLAMP 9 software (Axon Instruments Inc, Foster City, Calif). Results from pooled data are presented as mean±SEM, and n represents the number of cells in each experiment. Statistical analysis was performed with ANOVA, followed by a Student-Newman-Keuls test with SigmaStat software. P< 0.05 was considered statistically significant.
Localization of Ca2+ Channels
Confocal microscopy was used to assess trafficking of Ca2+ channels tagged with enhanced yellow fluorescent protein (EYFP). Cells were grown on polylysine-coated 35-mm glass culture dishes and studied 3 days after transfection. Experiments were performed on an Olympus FluoView laser-scanning confocal microscope (Olympus America, Center Valley, Pa), and images were acquired with FluoView acquisition software. EYFP-labeled cells were analyzed in the XYZ configuration. A region of interest measurement confined to within 2 μm of the plasma membrane was made, and the average pixel intensity within this region of interest was defined as peripheral staining. The average pixel intensity for the remaining portion of the cell was also determined and defined as central staining. The ratio of peripheral to central fluorescence was calculated. Measurements were not normalized to cell area.
Rate Dependence of the QT Interval
The rate dependence of the QT interval was evaluated during a standard exercise stress test with a bicycle ergometer or treadmill.
The authors had access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Eighty-two consecutive probands with a clinical diagnosis of Brugada syndrome enrolled in our registry were systematically screened for ion channel gene mutations. Seven probands (8.5%) were found to have mutations in genes encoding the α1- and β2b-subunits of the cardiac L-type calcium channel. In addition to ST-segment elevation and a family history of SCD, 3 of the 7 probands exhibited QTc intervals ≤ 360 ms. The present study is focused on delineation of the clinical characteristics and genetic basis for this distinct clinical entity.
The first proband (III-6), a 25-year-old white male of European descent, presented with aborted SCD. QTc was 330 ms, and a coved-type ST-segment elevation was observed in V1 and V2 after an ajmaline challenge (Figure 1A). His 23-year-old brother (III-5) was also symptomatic, with syncope since age 21 years. Programmed atrial stimulation induced atrial fibrillation (AF) in both individuals and AV nodal reentrant tachycardia in the brother. The rest of the family was asymptomatic. The proband received an implantable cardioverter defibrillator, and over a 3-year follow-up period, he received only inappropriate shocks, which ceased after a cavotricuspid isthmus ablation. A total of 10 family members were evaluated clinically and genetically, and 6 were characterized as phenotype positive on the basis of the presence of ST-segment elevation ≥ 2 mm at baseline or after ajmaline and a QTc ≤ 360 ms in males and ≤ 370 ms in females (Figure 1B). III-3 showed a prominent r′ in V2 at baseline and an ST-segment elevation >2 mm in response to ajmaline that resulted in neither a coved nor saddleback morphology; therefore, the designation was +/-A. III-7, although genotype negative, showed a positive response to ajmaline; QTc was 414 ms. Tall, peaked T waves were observed in some family members presenting with shorter-than-normal QT intervals. QT/heart rate slope was −0.639 ms/bpm for patient 1 (III-6) and −0.869 ms/bpm for the symptomatic brother of patient 1 (III-5).
Figure 1.
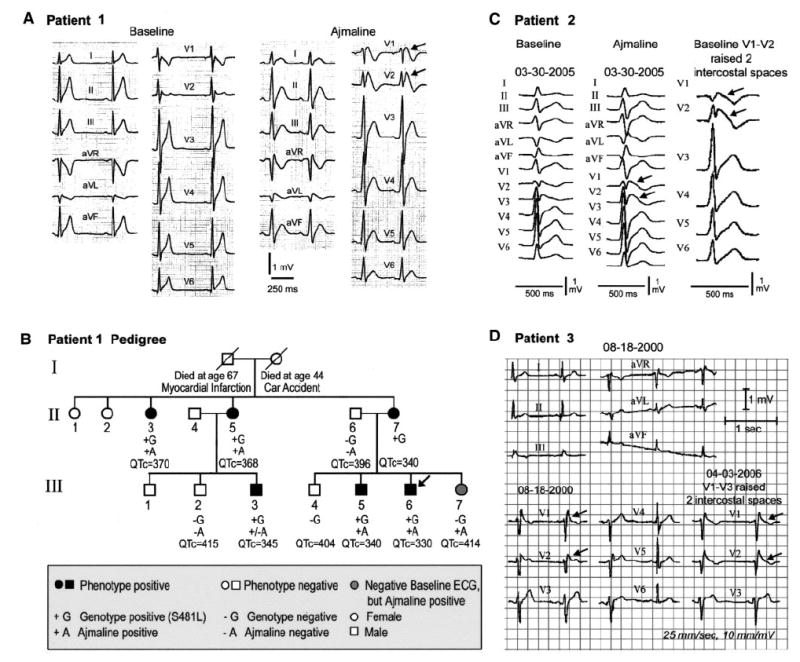
A, Twelve-lead ECG of patient 1 before and after ajmaline recorded with V1 and V2 displaced superiorly 2 intercostal spaces. B, Pedigree of family of patient 1 (III-6, arrow: proband). Arrows in ECGs depict prominent (type I) ST-segment elevation. C, Twelve-lead ECG of patient 2 before and after ajmaline (1 mg/kg) and at baseline with V1 and V2 displaced superiorly 2 intercostal spaces to unmask a type 1 ST-segment elevation. D, Twelve-lead ECG of patient 3 with V1 through V3 in normal position and displaced superiorly 2 intercostal spaces.
Patient 2, a 41-year-old white male of Turkish descent, presented with AF and an abbreviated QT interval of 300 ms (QTc=346 ms). His brother died of sudden cardiac arrest at age 45 years. Ajmaline administration (1 mg/kg) led to a further elevation of the ST segment in leads V1 to V2 (Figure 1C). The QT interval showed little rate dependence, with a QT/heart rate slope of −0.54 ms/bpm (Figure 2). Monomorphic ventricular tachycardia was inducible with 2 extrastimuli. Atrial and ventricular effective refractory periods were 150 and 170 ms at a 430-ms pacing cycle length, respectively. AH and HV intervals and sinus node recovery time were within normal limits. Structural heart disease was ruled out by coronary angiography and right ventricular angiography. Quinidine (750 mg/d for 5 days), administered for control of AF, prolonged QTc to 390 ms. An implantable cardioverter defibrillator was implanted for primary prevention. Programmed ventricular stimulation via the implantable cardioverter defibrillator lead was not able to induce ventricular tachycardia in the presence of quinidine. Patient 2 had several episodes of AF but no implantable cardioverter defibrillator discharges during a 1-year-follow-up.
Figure 2.
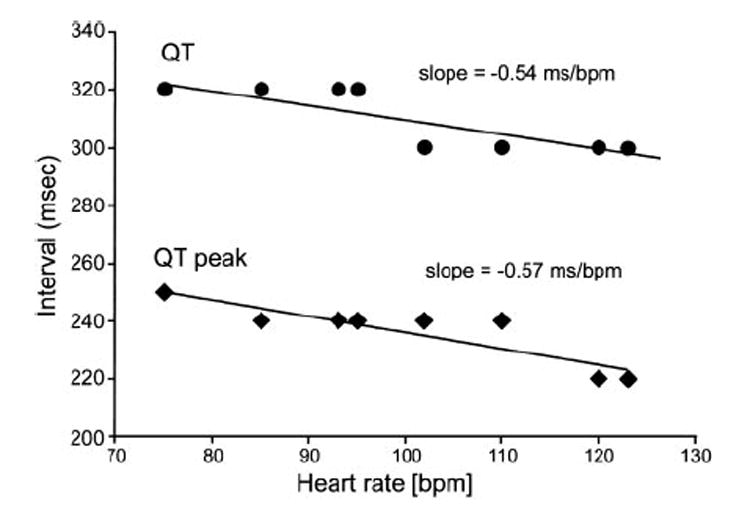
Heart rate dependence of QT interval of patient 2. Plotted are the QTpeak and QTend (QT) intervals as a function of heart rate recorded during bicycle ergometry exercise test.
Patient 3, a 44-year-old white male of European descent, presented with a prominent ST-segment elevation in V1, a saddleback ST-segment elevation in V2, and a prominent J wave in lead III (Figure 1D). His mother had 2 syncopal episodes at age 48 years that resulted in SCD. His father is 75 years old with no known medical problems; his brother (47 years old), sister (44 years old), and 3 children (8, 10, and 12 years old) declined examination but reportedly do not exhibit the Brugada phenotype. Patient 3 was recently diagnosed with facioscapulohumeral muscular dystrophy. His QTc was 360 ms, and his QT/heart rate slope was −0.991 ms/bpm.
In both patients 2 and 3, raising the position of the right precordial leads (V1 through V3) 2 intercostal spaces unmasked or accentuated the type I ST-segment elevation in V1 through V3 (Figures 1C and D). All 3 probands displayed ejection fractions, sinus node recovery times, and AV conduction values within normal limits. Genetic analysis revealed no mutations in SCN5A, the gene traditionally associated with the Brugada syndrome (20% of cases), or KCNH2, KCNQ1, or KCNJ2, the genes previously linked to the SQTS.
Patient 1 carried a heterozygous C1442T transition in exon 13 that predicted a substitution of leucine for serine at position 481 (S481L) of CACNB2b, which was not present in 400 ethnically matched control alleles (Figure 3A). The mutation is located downstream of the β-subunit interaction domain segment (Figure 3B). The S481L mutation was present in all 6 phenotype-positive and absent in all 4 phenotype-negative family members (Figure 1B). Linkage analysis with the identified C1442T mutation was performed that included genotypes of available family members (Figure 1B) with LINKAGE software under the dominant model of inheritance, and we obtained a maximum logarithm of the odds (LOD) score of 2.10.
Figure 3.
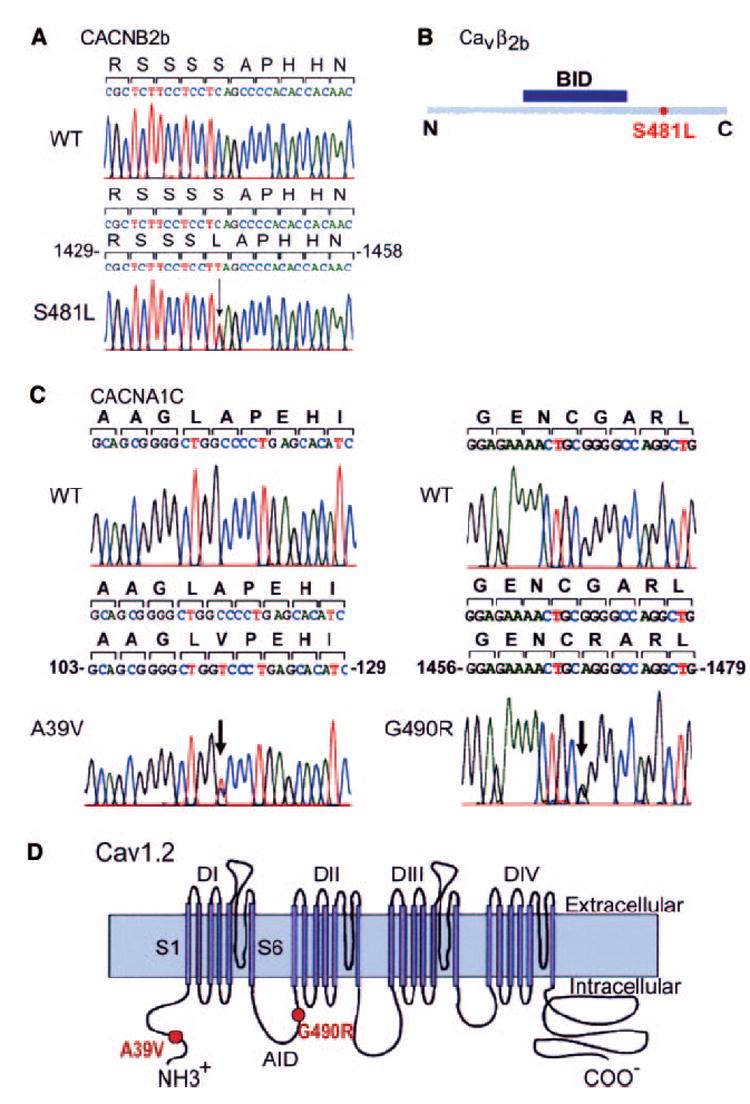
DNA sequence analysis. A, Heterozygous C1442T transition in exon 13 of CACNB2b predicting replacement of serine by leucine at position 481. B, Cartoon of Cavβ2b (cytoplasmic) showing the location of the mutation and the position of the β-subunit interaction domain (BID) segment. C, Left, Heterozygous C to T transition (arrow) at position 116 in exon 2 of CACNA1C allele in patient 2 predicting a substitution of a valine for alanine at position 39; Right, heterozygous A to G transition (arrow) at position 1468 in exon 10 of CACNA1C allele in patient 1 predicting replacement of glycine by arginine at position 490. D, Predicted topology of Cav1.2 showing the location of the mutations. Sschematic modified from Splawski et al,3 with permission from Elsevier. Copyright 2004. The loop between domains I and II contains a conserved motif named “AID” (α-subunit interaction domain) that binds to the β-subunit segment called BID.
Patient 2 showed a heterozygous substitution of an adenine for a guanine at position 1468 in exon 10 of CACNA1C, which predicted substitution of an arginine for a glycine at position 490 (G490R; Figure 3C), which was not present in 640 ethnically matched control alleles. Patient 2 also had 2 polymorphisms in CACNA1C, P1820L and V1821M, which were found in 31 and 27 of 114 healthy control subjects, respectively. The G490R mutation was also found in his 2 daughters (QTc=360 and 373 ms, respectively). The daughter with the longer QTc interval (373 ms) also displayed a known polymorphism in KCNH2 (K897T).
Patient 3 showed a heterozygous C116T transition in exon 2 of CACNA1C, which predicted a substitution of a valine for an alanine at position 39, A39V (Figure 3C), which was not present in 404 ethnically matched control alleles. In the proposed topology of the Cav1.2 channel subunit, the G490 is located in the cytoplasmic linker between domains I and II. A39 is located near the N-terminus of the protein (Figure 3D). Both mutations are located within a highly conserved region of the Cav1.2 protein.
To determine the contribution of each mutation to the clinical phenotype, we expressed each of the WT and mutated CACNA1C and CACNB2b constructs in CHO cells and performed patch-clamp experiments. The WT of the other 2 calcium channel subunits were cotransfected. We first compared the current-voltage (I-V) relationship between WT, A39V, and G490R channels in the EYFP-tagged, exon 8A variant of Cav1.2. A set of depolarizing pulses applied in 10-mV increments from a holding potential of −90 mV elicited robust WT currents (Figure 4A). In contrast, the amplitudes of A39V and G490R currents were drastically reduced, although the voltage at peak current remained unchanged (Figures 4B, 4C, and 4E). Similar results were obtained when the exon 8 variant of Cav1.2 was used (Figure 4F). CACNB2b WT and the S481L mutant were studied only with the EYFP-tagged, exon 8A variant of Cav1.2 (Figures 4D and 4G). The results indicate that the 2 mutations in CACNA1C and the mutation in CACNB2b all cause a major loss of function in calcium channel activity.
Figure 4.
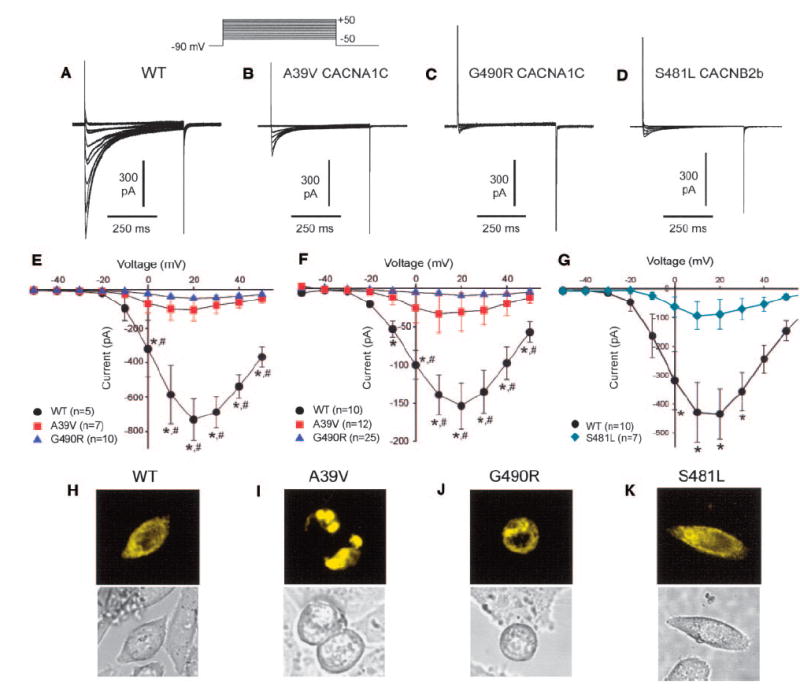
Representative whole-cell Ca2+ currents recorded from CHO cells transfected with WT CACNA1C (A) or A39V (B) and G490R (C) mutant CACNA1C and S481L mutant CACNB2b (D). Currents were elicited with the pulse protocol illustrated in the inset above panel B. E, Current-voltage (I-V) relationship for WT (n=5), A39V (n=7), and G490R (n=10) Cav1.2 channels (exon 8A variant). F, I-V relationship for WT (n=10), A39V (n=12), and G490R (n=25) Cav1.2 channels (exon 8 variant). *P< 0.05 compared with G490R. #P< 0.05 compared with A39V. G, I-V relationship for WT (n=10), S481L (n37) Cavβ2b channels. *P< 0.05 compared with S481L. H through K, Representative confocal XYZ scans showing localization of EYFP-tagged Cav1.2 channels in CHO cells. H, Cell expressing WT Cav1.2 channels showed marked peripheral and cytoplasmic fluorescence. I, Cells expressing A39V Cav1.2 channels showed fluorescence localized in the perinuclear region of the cell. J, Cell expressing G490R Cav1.2 channels exhibit a fluorescence pattern similar to WT, which suggests that trafficking is not impaired. K, Cells expressing S481L Cavβ2b channels exhibit a fluorescence pattern similar to WT, which suggests that trafficking is not impaired.
To determine whether the mutation-induced loss of function was related to a trafficking defect, we used confocal microscopy to determine the intracellular expression pattern of WT or mutant channels (S481L Cavβ2b, or A39V and G490R Cav1.2) tagged with EYFP (Figures 4H through 4K). XYZ scans of WT and G490R channels revealed both a central and peripheral pattern of fluorescence, which suggests that a pool of these channels exists in intracellular organelles and that the proteins translocate normally to the cell membrane. In contrast, the fluorescence pattern of A39V channels was almost exclusively localized to intracellular organelles. The peripheral:central fluorescence ratio was similar for WT, G490R, and S481L (1.34±0.17, 1.58±0.31, and 0.94±0.1; n=6) but much smaller for A39V (0.41±0.26; P< 0.05 compared with WT and G490R, n=6 cells), which indicates that very few A39V channels were localized at the sarcolemma. Coexpression of WT and A39V channels was associated with more peripheral fluorescence (ratio=1.16±0.10) than A39V alone, which suggests that the mutant channel does not interfere with trafficking of WT channels. Coexpression of WT and S481L Cavβ2b channels yielded a ratio (1.04±0.10) similar to that of WT or S481L alone. These findings suggest that the loss of current observed with A39V is due to a defect in trafficking of mature Cav1.2 channels from the endoplasmic reticulum/Golgi complex to the cell membrane, whereas channels formed from G490R Cav1.2 or S481L Cavβ2b subunits traffic normally.
Discussion
Although the Brugada syndrome has thus far been linked to mutations that impede sodium channel expression or function, experimental studies have shown that the electrocardiographic and arrhythmic manifestations of the disease phenotype can be recapitulated in the coronary-perfused canine right ventricular wedge preparation with calcium channel blockers,18 which points to genes encoding the various subunits of the calcium channel as candidates to explain the disease phenotype. Moreover, calcium channel blockers have been reported to produce an acquired form of the Brugada syndrome in humans.19 Consistent with these findings, the present study demonstrates an association between loss-of-function mutations in the α1- and β2b-subunits of the cardiac L-type calcium channel and the Brugada syndrome phenotype.
Evidence in support of mutations in the calcium channel being causal include the following findings: (1) mutations in 2 different subunits of the calcium channel are associated with similar disease phenotypes in probands with a family history of SCD; (2) heterologous expression of mutant channels in CHO cells leads to a major loss of function consistent with the Brugada syndrome phenotype and with a shorter-than-normal QT interval; (3) the 3 mutations are not observed in ethnically matched healthy individuals; and (4) there is a clear genotype-phenotype correlation. The absence of an ST-segment elevation in the 2 daughters of patient 2 may be explained on the basis of the well-known low penetrance for clinical manifestation of the Brugada syndrome in young females.12 The apparently falsepositive response to ajmaline in III-7 has been reported previously in patients with Brugada syndrome.20 The LOD score of 2.1 does not reach statistical significance for linkage but represents the maximal theoretical LOD score for this relatively small family. This notwithstanding, cosegregation of the mutation with the disease in the family and the in vitro expression data provide proof of the causative nature of the mutation.
Arrhythmogenesis in both Brugada syndrome and SQTS is thought to be due to amplification of heterogeneities in action potential characteristics among the different transmural cell types.18 In Brugada syndrome, a decrease in INa or ICa or augmentation of any one of a number of outward currents, including IKr, IKs, ICl(Ca), and Ito, can cause preferential abbreviation of the right ventricular epicardial action potential secondary to all-or-none repolarization of the action potential at the end of phase 1. This leads to loss of the action potential dome and the development of spatial dispersion of repolarization and thus the substrate and trigger for ventricular tachycardia, which is usually polymorphic and less frequently monomorphic.12 In the short-QT syndromes, preferential abbreviation of either the epicardial or endocardial response amplifies spatial dispersion of repolarization and creates the substrate for reentrant arrhythmias.21 An increase in outward current7–9 or a decrease in inward current, including calcium current, may be responsible.
The present study is the first to associate a cardiac calcium channel mutation with the Brugada syndrome or short QT intervals. Although the QTc intervals in probands in the present study may be defined as “short” on the basis of published reports,13,14 they may not in all cases be considered as representing an SQTS, which thus far has been associated with QTc intervals ≤ 330 ms. Only 1 of the probands in the present study presented with a QTc ≤ 330 ms (patient 1). It is noteworthy that Brugada syndrome is generally associated with a slight prolongation of the QT interval, particularly in the right precordial leads, presumably due to an accentuation of the action potential notch (without loss of the dome), which prolongs the action potential in right ventricular epicardium.
Viskin and coworkers14 reported that short QT intervals (QTc of ≤ 360 ms for males and ≤ 370 ms for females) are commonly observed in patients with idiopathic ventricular fibrillation. A less-steep QT-RR relationship is also observed in these patients, similar to the abnormal rate dependence of QT reported in patients in the present study. The slope of the QT/heart rate relation was −0.639 ms/bpm for patient 1 (III-6) and −0.869 ms/bpm for the symptomatic brother of patient 1 (III-5), −0.540 ms/bpm for patient 2, and −0.991 ms/bpm for patient 3. These values are considerably less steep that those reported by Magnano et al22 for normal controls (−1.37 ms/bpm). These distinctions are similar to those reported between SQT1 patients (−0.54 ms/bpm) and noncarrier controls (−1.29 ms/bpm.23
AF is known to be associated with 20% to 30% of Brugada syndrome cases24 and with a similar or higher fraction of short-QT cases.25 The high incidence of AF in the present cohort is therefore not unexpected.
Quinidine has been proposed to be of therapeutic value in the Brugada syndrome26 and in the SQTS.27 In the setting of Brugada syndrome, it is the Ito blocking effect of the drug that is salutary, whereas in the SQTS, it is the effect of the drug to block IKr and IKs. Clinical evidence of the effectiveness of quinidine in inducible and spontaneous ventricular fibrillation was reported by Belhassen and coworkers28 in a prospective study of 25 Brugada syndrome patients. The ability of quinidine to prevent induction of ventricular tachycardia and ventricular fibrillation and its effect to prolong QTc in patient 1 is consistent with these earlier reports.
Among 82 probands with a clinically robust diagnosis of Brugada syndrome in the present registry, 6% (5) presented with a shorter-than-normal QT interval. Three (60%) of these 5 probands carried a calcium channel mutation, which points to genetic heterogeneity for this phenotype. Fifteen percent of probands harbored a putative pathogenic mutation in SCN5A, and 4.9% carried a mutation in calcium channel genes associated with Brugada syndrome and QT intervals >370 ms. Expression studies are under way to determine the nature of the defect, if any, in calcium channel function in these other mutations. The fraction of probands with SCN5A mutations (15%) is similar to that reported by Schulze-Bahr and coworkers (14%).29 Whereas a gain of function in calcium channel current secondary to mutations in CANCA1C produces a sudden death syndrome associated with prolongation of the QT interval,3,4 the present findings indicate that a loss of function in calcium channel activity secondary to mutations in CACNA1C or CACNB2b can contribute to a sudden death syndrome that consists of a shorter-than-normal QT interval and ST-segment elevation (Brugada syndrome phenotype). A similar mirror image of malignant syndromes has been demonstrated for a loss and gain of function in SCN5A (Brugada versus LQT3 syndromes),10,30 KCNH2 (LQT2 versus SQT1),7,31 KCNQ1 (LQT1 versus SQT2),8,32 and KCNJ2 (Andersen-Tawil syndrome, LQT7 versus SQT3).9
CACNA1C- and CACNB2b-mediated syndromes may be considered as Brugada syndrome types 3 and 4 (GPD1-L has been reported by London et al33 as type 2) or SQTS types 4 and 5, depending on which definition of short QT and which QT correction formula one chooses to employ. Because there is no clear basis on which to select Brugada syndrome over short QT or vice versa, we have chosen to present this as a distinct clinical entity in which these 2 sudden death syndromes are combined.
CLINICAL PERSPECTIVE.
Brugada syndrome, characterized electrocardiographically as displaying a coved-type (type I) ST-segment elevation, was first identified as a new clinical entity associated with sudden death in 1992. The short-QT syndrome, characterized by corrected QT intervals ≤ 330 ms, was first identified as a distinct clinical entity associated with sudden cardiac death in 2000. Short QT intervals (corrected QT of ≤ 360 ms for males and ≤ 370 ms for females) are also commonly observed in patients with idiopathic ventricular fibrillation. Both short-QT syndrome and idiopathic ventricular fibrillation patients display an abnormal rate adaptation of the QT interval. The present study identifies a new clinical entity associated with sudden cardiac death that combines all of these characteristics. This syndrome is linked to loss-of-function mutations in genes encoding the α- and β-subunits of the cardiac L-type calcium channel. This is the first report of a calcium channel loss of function to be associated with a familial sudden cardiac death syndrome.
Acknowledgments
We are grateful to Tabitha Carrier for providing expert technical assistance and to Susan Bartkowiak for maintaining our genetics database. We thank Dr Nikolai Soldatov for expression constructs.
Sources of Funding: This study was supported by grant No. 20/2006 from the Koeln Fortune Program, Faculty of Medicine, University of Cologne (Dr Wollnik) and New York State and Florida Grand Lodge, Free and Accepted Masons.
Footnotes
Disclosures None.
Contributor Information
Charles Antzelevitch, Masonic Medical Research Laboratory, Utica, NY.
Guido D. Pollevick, Masonic Medical Research Laboratory, Utica, NY.
Jonathan M. Cordeiro, Masonic Medical Research Laboratory, Utica, NY.
Oscar Casis, Nora Eccles Harrison Cardiovascular Research and Training Institute, University of Utah, Salt Lake City, Utah.
Michael C. Sanguinetti, Nora Eccles Harrison Cardiovascular Research and Training Institute, University of Utah, Salt Lake City, Utah.
Alejandra Guerchicoff, Masonic Medical Research Laboratory, Utica, NY.
Ryan Pfeiffer, Masonic Medical Research Laboratory, Utica, NY.
Antonio Oliva, Masonic Medical Research Laboratory, Utica, NY.
Bernd Wollnik, Center for Molecular Medicine Cologne, Institute of Human Genetics, University of Cologne, Germany.
Philip Gelber, Cardiovascular Consultants of Long Island, New Hyde Park, NY.
Elias P. Bonaros, Jr, Cardiovascular Consultants of Long Island, New Hyde Park, NY.
Elena Burashnikov, Masonic Medical Research Laboratory, Utica, NY.
Yuesheng Wu, Masonic Medical Research Laboratory, Utica, NY.
John D. Sargent, Nora Eccles Harrison Cardiovascular Research and Training Institute, University of Utah, Salt Lake City, Utah.
Stefan Schickel, Department of Internal Medicine, Academic Hospital Oberhausen, Oberhausen, Germany.
Ralf Oberheiden, Department of Internal Medicine, Academic Hospital Oberhausen, Oberhausen, Germany.
Atul Bhatia, University of Wisconsin Medical School, Milwaukee, Wis.
Li-Fern Hsu, Hopital Cardiologique du Haut-Leveque, Bordeaux, France.
Michel Haïssaguerre, Hopital Cardiologique du Haut-Leveque, Bordeaux, France.
Rainer Schimpf, 1st Department of Medicine-Cardiology, University Hospital Mannheim, Faculty of Clinical Medicine of the University of Heidelberg, Mannheim, Germany.
Martin Borggrefe, 1st Department of Medicine-Cardiology, University Hospital Mannheim, Faculty of Clinical Medicine of the University of Heidelberg, Mannheim, Germany.
Christian Wolpert, 1st Department of Medicine-Cardiology, University Hospital Mannheim, Faculty of Clinical Medicine of the University of Heidelberg, Mannheim, Germany.
References
- 1.Priori SG, Aliot E, Blomstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, Camm JA, Cappato R, Cobbe SM, Di MC, Maron BJ, McKenna WJ, Pedersen AK, Ravens U, Schwartz PJ, Trusz-Gluza M, Vardas P, Wellens HJ, Zipes DP. Task Force on Sudden Cardiac Death, European Society of Cardiology. Europace. 2002;4:3–18. doi: 10.1053/eupc.2001.0214. [DOI] [PubMed] [Google Scholar]
- 2.Antzelevitch C. Molecular genetics of arrhythmias and cardiovascular conditions associated with arrhythmias. Heart Rhythm. 2004;1:42C–56C. doi: 10.1016/j.hrthm.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 3.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 119:2004. 19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 4.Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A. 2005;102:8089–8096. doi: 10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bichet D, Cornet V, Geib S, Carlier E, Volsen S, Hoshi T, Mori Y, De Waard M. The I-II loop of the Ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron. 25:2000. 177–190. doi: 10.1016/s0896-6273(00)80881-8. [DOI] [PubMed] [Google Scholar]
- 6.Gussak I, Antzelevitch C, Goodman D, Bjerregaard P. Short QT interval: ECG phenomenon and clinical syndrome. In: Gussak I, Antzelevitch C, editors. Cardiac Repolarization: Bridging Basic and Clinical Sciences. Totowa, NJ: Humana Press; 2003. pp. 497–506. [Google Scholar]
- 7.Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- 8.Bellocq C, Van Ginneken AC, Bezzina CR, Alders M, Escande D, Mannens MM, Baro I, Wilde AA. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109:2394–2397. doi: 10.1161/01.CIR.0000130409.72142.FE. [DOI] [PubMed] [Google Scholar]
- 9.Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, Napolitano C, Anumonwo J, Raffaele di BM, Gudapakkam S, Bosi G, Stramba-Badiale M, Jalife J. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96:800–807. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 10.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O’Brien RE, Schultze-Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanisms for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 11.Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, Corrado D, Hauer RN, Kass RS, Nademanee K, Priori SG, Towbin JA. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation. 2002;106:2514–2519. doi: 10.1161/01.cir.0000034169.45752.4a. [DOI] [PubMed] [Google Scholar]
- 12.Antzelevitch C, Brugada P, Borggrefe J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze-Bahr E, Tan H, Wilde A. Brugada syndrome: report of the Second Consensus Conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–670. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- 13.Rautaharju PM, Zhou SH, Wong S, Calhoun HP, Berenson GS, Prineas R, Davignon A. Sex differences in the evolution of the electrocardiographic QT interval with age. Can J Cardiol. 1992;8:690–695. [PubMed] [Google Scholar]
- 14.Viskin S, Zeltsner D, Ish-Shalom M, Katz A, Glikson M, Justo D, Tekes-Manova D, Belhassen B. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm. 2004;1:587–591. doi: 10.1016/j.hrthm.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 15.Costantini DL, Arruda EP, Agarwal P, Kim KH, Zhu Y, Zhu W, Lebel M, Cheng CW, Park CY, Pierce SA, Guerchicoff A, Pollevick GD, Chan TY, Kabir MG, Cheng SH, Husain M, Antzelevitch C, Srivastava D, Gross GJ, Hui CC, Backx PH, Bruneau BG. The homeodomain transcription factor Irx5 establishes the mouse cardiac ventricular repolarization gradient. Cell. 2005;123:347–358. doi: 10.1016/j.cell.2005.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobrinsky E, Tiwari S, Maltsev VA, Harry JB, Lakatta E, Abernethy DR, Soldatov NM. Differential role of the alpha1C subunit tails in regulation of the Cav1.2 channel by membrane potential, beta subunits, and Ca2+ ions. J Biol Chem. 2005;280:12474–12485. doi: 10.1074/jbc.M412140200. [DOI] [PubMed] [Google Scholar]
- 17.Zuhlke RD, Bouron A, Soldatov NM, Reuter H. Ca2+ channel sensitivity towards the blocker isradipine is affected by alternative splicing of the human alpha 1C subunit gene. FEBS Lett. 1998;427:220–224. doi: 10.1016/s0014-5793(98)00425-6. [DOI] [PubMed] [Google Scholar]
- 18.Fish JM, Antzelevitch C. Role of sodium and calcium channel block in unmasking the Brugada syndrome. Heart Rhythm. 2004;1:210–217. doi: 10.1016/j.hrthm.2004.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimizu W. Acquired forms of the Brugada syndrome. J Electrocardiol. 2005;38(suppl):22–25. doi: 10.1016/j.jelectrocard.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Hong K, Berruezo-Sanchez A, Poungvarin N, Oliva A, Vatta M, Brugada J, Brugada P, Towbin JA, Dumaine R, Pinero-Galvez C, Antzelevitch C, Brugada R. Phenotypic characterization of a large European family with Brugada syndrome displaying a sudden unexpected death syndrome mutation in SCN5A. J Cardiovasc Electrophysiol. 2004;15:64–69. doi: 10.1046/j.1540-8167.2004.03341.x. [DOI] [PubMed] [Google Scholar]
- 21.Extramiana F, Antzelevitch C. Amplified transmural dispersion of repolarization as the basis for arrhythmogenesis in a canine ventricular-wedge model of Short-QT syndrome. Circulation. 2004;110:3661–3666. doi: 10.1161/01.CIR.0000143078.48699.0C. [DOI] [PubMed] [Google Scholar]
- 22.Magnano AR, Holleran S, Ramakrishnan R, Reiffel JA, Bloomfield DM. Autonomic nervous system influences on QT interval in normal subjects. J Am Coll Cardiol. 2002;39:1820–1826. doi: 10.1016/s0735-1097(02)01852-1. [DOI] [PubMed] [Google Scholar]
- 23.Wolpert C, Schimpf R, Giustetto C, Antzelevitch C, Cordeiro J, Dumaine R, Brugada R, Hong K, Bauersfeld U, Gaita F, Borggrefe M. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol. 2005;16:54–58. doi: 10.1046/j.1540-8167.2005.04470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borggrefe M. Atrial tachyarrhythmias in Brugada syndrome. In: Antzelevitch C, Brugada P, Brugada J, Brugada R, editors. The Brugada Syndrome: From Bench to Bedside. Oxford, UK: Blackwell Futura; 2004. pp. 178–183. [Google Scholar]
- 25.Giustetto C, Di MF, Wolpert C, Borggrefe M, Schimpf R, Sbragia P, Leone G, Maury P, Anttonen O, Haissaguerre M, Gaita F. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J. 2006;27:2440–2447. doi: 10.1093/eurheartj/ehl185. [DOI] [PubMed] [Google Scholar]
- 26.Antzelevitch C, Brugada P, Brugada J, Brugada R, Nademanee K, Towbin JA. Clinical Approaches to Tachyarrhythmias The Brugada Syndrome. Armonk, NY: Futura; 1999. [Google Scholar]
- 27.Gaita F, Giustetto C, Bianchi F, Schimpf R, Haissaguerre M, Calo L, Brugada R, Antzelevitch C, Borggrefe M, Wolpert C. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43:1494–1499. doi: 10.1016/j.jacc.2004.02.034. [DOI] [PubMed] [Google Scholar]
- 28.Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004;110:1731–1737. doi: 10.1161/01.CIR.0000143159.30585.90. [DOI] [PubMed] [Google Scholar]
- 29.Schulze-Bahr E, Eckardt L, Breithardt G, Seidl K, Wichter T, Wolpert C, Borggrefe M, Haverkamp W. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum Mutat. 2003;21:651–652. doi: 10.1002/humu.9144. [DOI] [PubMed] [Google Scholar]
- 30.Wang Q, Shen J, Splawski I, Atkinson DL, Li ZZ, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 31.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, Van Raay TJ, Shen J, Timothy KW, Vincent GM, De Jager T, Schwartz PJ, Towbin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 33.London B, Sanyal S, Michalec M, Pfahnl AE, Shang LL, Kerchner BS, Lagana S, Aleong RG, Mehdi H, Gutmann R, Weiss R, Dudley SC. A mutation in the glycerol-3-phosphate dehydrogenase 1-like gene (GPD1L) causes Brugada syndrome. Heart Rhythm. 2006;3:S32. Abstract. [Google Scholar]