

Abstract
The thalamocortical (TC) input to layer IV provides the major pathway for ascending sensory information to the mammalian sensory cortex. During development there is a dramatic refinement of this input that underlies the maturation of the topographical map in layer IV. Over the last ten years our understanding of the mechanisms of the developmental and experience-driven changes in synaptic function at TC synapses has been greatly advanced. Here we describe these studies that point to a key role for NMDA receptor-dependent synaptic plasticity, a role for kainate receptors and for a rapid maturation in GABAergic inhibition. The expression mechanisms of some of the forms of neonatal synaptic plasticity are novel and, in combination with other mechanisms, produce a layer IV circuit that exhibits functional properties necessary for mature sensory processing.
Introduction
The facial vibrissae (whiskers) of rodents are somatotopically represented in the primary somatosensory cortex by cylindrical arrangements of neurons known as barrels (Woolsey and Van der Loos, 1970). Sensory information from the whiskers passes via the brain stem and thalamus to layer IV neurons in “barrel cortex” (Fig. 1 A). Anatomical (Woolsey and Van der Loos, 1970) and electrophysiological (Welker, 1971) studies show that each whisker projects preferentially to a single barrel. In adult animals this preference is seen in electrophysiological recordings from layer IV as a short latency (5–10 ms) response to deflection of only the corresponding principle whisker mediated by activation of the thalamocortical (TC) input. Layer IV neurons also show responses to stimulation of non-principal whisker stimulation, so-called surround receptive field (SRF) responses, but unlike the principle whisker responses, these are thought to involve cortico-cortical connections (Armstrong-James and Callahan, 1991; Armstrong-James et al., 1991). This receptive field is, however, subject to experience-dependent plasticity: if all but one whisker is trimmed, short latency responses to deflection of the remaining whisker appear in other barrels. This plasticity in layer IV is most prominent if the whiskers are trimmed within the first postnatal week, although other forms of intracortical plasticity persist later in development (Diamond et al., 1993; Diamond et al., 1994; Fox, 1992, 2002; Wallace and Fox, 1999). Thus the first postnatal week represents a period of enhanced plasticity in layer IV, similar to critical periods described for other forms of experience-dependent plasticity, for example in the visual system.
Fig. 1.
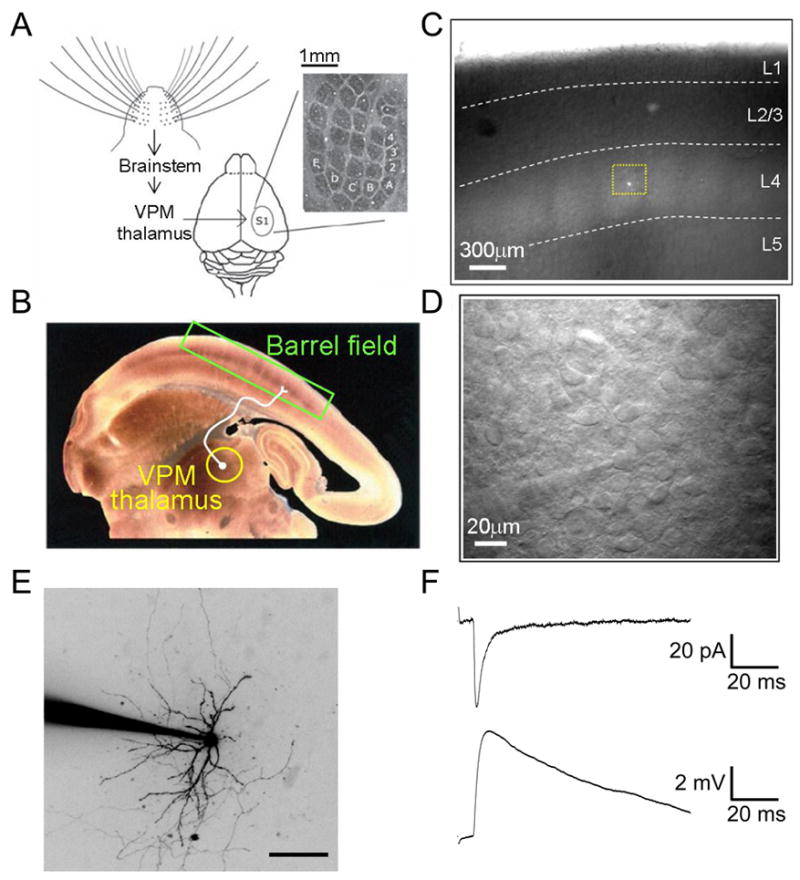
Electrophysiological recordings from neurons in layer IV barrel cortex in the thalamocortical (TC) slice preparation. (A) Schematic of the afferent pathway from the whiskers to barrel cortex. Inset: cytochrome oxidase C-stained tangential section through layer IV of barrel cortex showing the distinctive cytoarchitechnure. Reproduced from (Feldman and Brecht, 2005). (B) Cytochrome oxidase C-stained TC slice from 2 week old rat. The ventroposteriomedial nucleus of thalamus (VPM) that receives ascending sensory input from the whiskers, and the barrel cortex are indicated. Reproduced from (Petersen and Sakmann, 2000) (C) Unstained TC slice from P5 mouse as it appears in recording chamber during an electrophysiology experiment. Highlighted by the yellow box is a stellate cell (SC) filled with the fluorescent dye Alexa-594 during a whole-cell patch-clamp recording (Daw, Ashby and Isaac, unpublished). (D) High power image of the region highlighted in C, showing a whole-cell recording from an SC (patch electrode can be seen as the out of focus shadow in bottom left hand corner). (E) 2-photon image (flattened projection of Z-stack, contrast inverted for display) of an SC filled with Alexa-594 during patch clamp recording. (F) TC synaptic responses recorded during a whole-cell patch-clamp recording from an SC evoked by electrical stimulation of VPM thalamus. Top is an EPSC (voltage-clamp recording) and bottom EPSP (current clamp recording); responses collected in the same cell in response to the same stimulus (Daw, Ashby and Isaac, unpublished).
A slice preparation has been developed in which the TC afferents are preserved for in vitro electrophysiological studies (Fig. 1 B–F). In this preparation electrical stimulation in the ventrobasal complex (VB) of the thalamus allows the study of monosynatically activated TC synapses (Agmon and Connors, 1991), something which has been possible for auditory (Cruikshank et al., 2002) and visual cortex (MacLean et al., 2006) only relatively recently. This preparation together with the ease of anatomically identifying the barrel field both histochemically (Woolsey and Van der Loos, 1970) and also in unstained slices (Agmon and Connors, 1991), and the well-defined mapping of whiskers to barrels, makes the barrel cortex a uniquely well-placed model for studying the synaptic mechanisms underling the development of the sensory map. Such studies have shown that a number of mechanisms are involved in mediating this development and refinement of the cortical map. NMDA receptor-dependent long-term plasticity occurs at the TC synapse only during the critical period for layer IV experience-dependent plasticity (Crair and Malenka, 1995) and results in a switch to fast AMPA receptor-mediated synaptic transmission from slow kainate receptor-mediated transmission (Bannister et al., 2005; Daw et al., 2006; Kidd and Isaac, 1999). At the same time TC afferents projections refine to within individual barrels in a manner dependent on synaptic activity (Lu et al., 2006; Persico et al., 2001; Schlaggar and O'Leary, 1991) and cortical neurons migrate to the edge of the barrels whilst their dendrites become orientated towards the centre, a process that may also require activity (Inan et al., 2006; Iwasato et al., 2000).
Long-term Potentiation (LTP) at TC Synapses
LTP is a long-lasting and activity-dependent increase in synaptic strength (Bliss and Collingridge, 1993). A variety of stimulation protocols both artificial and pseudo-natural have been used that result in an increase in synaptic transmission in many brain areas (Malenka and Bear, 2004). Although the mechanisms mediating this increase in synaptic transmission may vary depending on the synapse being studied, the majority of these mechanisms require activation of the NMDA subtype of glutamate receptors. The potential of such mechanisms to produce long-term changes in the weight of transmission at a particular synapse make LTP a likely candidate as the synaptic mechanism underlying certain forms of learning and memory (Eichenbaum, 1996; Malenka and Bear, 2004).
There is considerable evidence that NMDA receptor-dependent long-term synaptic plasticity is necessary for the correct formation and refinement of receptive fields in the barrel cortex. Animals in which the barrel cortex is chronically treated with the NMDA receptor antagonist, AP5, during the first postnatal week fail to develop the precise topographical map and exhibit deficient experience-dependent receptive field plasticity (Fox et al., 1996; Schlaggar et al., 1993). Additionally, NMDA receptor-dependent LTP can be induced in vitro at TC synapses by pairing presynaptic stimulation with postsynaptic depolarisation. LTP at these synapses can only be induced between postnatal days 3 and 7 (P3–7) (Barth and Malenka, 2001; Crair and Malenka, 1995), a time period very similar to the critical period for layer IV experience-dependent receptive field plasticity (Foeller and Feldman, 2004; Fox, 1992, 2002) (Fig. 2). In visual cortex, neuronal activity and synaptic plasticity are also thought to be important for the development of the cortical representation of sensory information (Malenka and Bear, 2004). In particular ocular dominance plasticity (ODP), in which the representation of a spared eye increases whilst the representation of a closed eye decreases (LeVay et al., 1980), has been studied as a model for experience-dependent visual cortex plasticity during development. Rearing animals in the dark extends both the critical period for ODP and LTP in visual cortex (Kirkwood et al., 1995) and blocking retinal activity by injecting tetrodotoxin in the open eye prevents ODP showing the requirement for activity in this form of plasticity (Antonini and Stryker, 1993). Again a role for NMDA receptor-dependent LTP is supported by the finding that NMDA receptor antagonists also prevent ODP (Bear et al., 1990; Daw et al., 1999). Blockade of NMDA receptors also reduces responses to visual stimulation complicating the conclusions of these studies. Subsequently, however, the same effect has been shown by knocking down activity of NR1 subunits using antisense DNA, a manipulation that does not affect visual responses (Roberts et al., 1998). These findings lend further support to the idea that experience-dependent plasticity in primary sensory systems involves NMDA receptor-dependent synaptic plasticity. A number of genetic and pharmacological manipulations have been studied for their affect on LTP and LTD in visual cortex and on ODP (Daw et al., 2004). Many of these studies have found that manipulations affecting plasticity, especially LTD, also prevent ODP (Beaver et al., 2001b; Choi et al., 2002; Fischer et al., 2004b; Hensch et al., 1998a; Liu et al., 2003a); however mGluR2 (Renger et al., 2002) and PKA R1β (Hensch et al., 1998b) knock out mice lack LTD but show normal ODP. Thus, there are examples of dissociation between long-term synaptic plasticity mechanisms and ODP, indicating that the precise connection between the two phenomena is not fully understood.
Fig. 2.
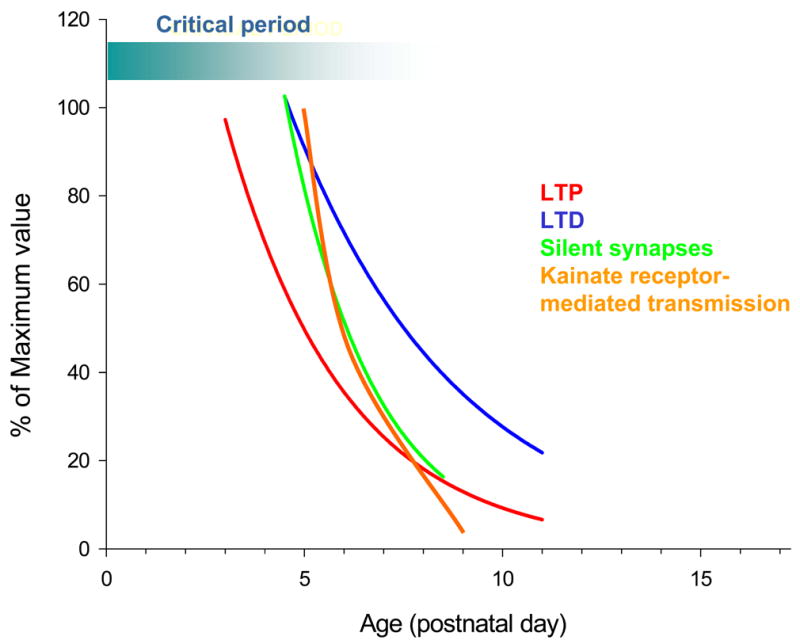
Developmental profile for synaptic properties and long-term synaptic plasticity at TC synapses in layer IV barrel cortex. Each parameter is plotted as a % of the maximum value. The critical period for experience dependent plasticity in layer IV is also shown for comparison.
Although it is generally agreed that Ca2+ influx through NMDA receptors is necessary for LTP induction at most synapses, exactly how this leads to an increase in postsynaptic response remains a matter of much debate (Bliss and Collingridge, 1993; Malenka and Bear, 2004; Malenka and Nicoll, 1999; Malinow and Malenka, 2002). There are a number of possible expression mechanisms for LTP: a presynaptic mechanism could involve an increase in the probability of neurotransmitter release or an increase in the amount of neurotransmitter released per quantum, and postsynaptically an increase in the number or conductance of AMPA receptors may mediate the increase in synaptic strength. LTP is associated with an increase in response to exogenously applied AMPA e.g.(Davies et al., 1989; Montgomery et al., 2001), interpreted as a postsynaptic change in receptors, but there is also a decrease in the number of failures of synaptic transmission that is commonly observed e.g.(Isaac et al., 1996; Malinow and Tsien, 1990),and classically interpreted as a presynaptic increase in probability of release. The existence of “silent” synapses, which express only NMDA receptors but can express AMPA receptors after LTP induction (Isaac, 2003), is a postsynaptic mechanism consistent with both the above findings. Indirect evidence for the existence of such silent synapses was provided using coefficient of variance analysis of synaptic responses at hippocampal CA1 inputs (Kullmann, 1994) and subsequently confirmed with direct experiments (Isaac et al., 1995; Liao et al., 1995). Other interpretations of the electrophysiological phenomenon of the silent synapse have been proposed, based on the higher affinity for glutamate that NMDA receptors have compared with AMPA receptors. The ‘spillover’ hypothesis is based on glutamate spilling over from one synapse and activating NMDA receptors in neighbouring synapses (Kullmann and Asztely, 1998). In the ‘whispering synapse’ hypothesis a low concentration of synaptically released glutamate due to incomplete synaptic vesicle fusion produces only NMDA receptor-mediated responses at synapses containing both AMPA and NMDA receptors (Choi et al., 2003). Although these mechanisms may contribute to silent synapses, there is now a large body of evidence supporting the idea that a fraction of glutamatergic synapses contain postsynaptic NMDA receptors but no postsynaptic AMPA receptors, and that one LTP expression mechanism is the rapid incorporation of AMPA receptors at silent synapses (Isaac, 2003).
The role of silent synapses in the expression of LTP at TC synapses was investigated using the TC slice (Isaac et al., 1997). The existence of silent synapses was directly demonstrated using low intensity stimulation in thalamus to activate one or a few TC axons. Under these conditions synaptic responses were evoked that were mediated by NMDA receptors but not by AMPA receptors. Such synapses are silent at resting membrane potentials due to the voltage-dependent Mg2+ block of NMDA receptors. An LTP induction protocol resulted in the appearance of AMPA receptor-mediated EPSCs at these synapses, thus causing an ‘unsilencing’. In addition, it was shown that the occurrence of silent synapses decreases with development such that silent synapses are not detected by postnatal day 8–9 (Fig. 2). This developmental decrease closely matches the developmental loss of the ability to induce LTP at these synapses (Crair and Malenka, 1995) supporting the idea that unsilencing is an important mechanism for TC LTP. Subsequent evidence for the presence of silent TC synapses has come in the form of differing short-term plasticity of AMPA and NMDA receptor-mediated synaptic currents (Yanagisawa et al., 2004) although these data appear to contradict finding of other groups at the same synapse (Gil et al., 1997; Gil et al., 1999; Kidd et al., 2002).
Long-term Depression (LTD) at TC Synapses
In addition to LTP, a synapse-specific long-term depression (LTD) of synaptic transmission can be induced by a period of low-frequency stimulation (LFS, typically 900 stimuli at 1 Hz) (Dudek and Bear, 1992; Mulkey and Malenka, 1992) or a pairing protocol (Goda and Stevens, 1996), which, like LTP, is NMDA receptor dependent (Dudek and Bear, 1992). LTD at TC synapses in visual cortex could explain aspects of plasticity in the visual system (Blakemore and Van Sluyters, 1974), and models reproducing many elements of plasticity in visual cortex depend on the existence of LTD (Bear et al., 1987; Bienenstock et al., 1982; Malenka and Bear, 2004). The existence of LTD had been demonstrated at layer IV cells in visual cortex (Dudek and Friedlander, 1996); however subsequent work on barrel cortex using the TC slice preparation studied LTD specifically at TC synapses. Since the LFS protocol typically used to study hippocampal LTD may damage axons in the immature TC slices, a pairing protocol was chosen to test for the presence of LTD at TC synapses (Feldman et al., 1998). This protocol reliably induced LTD in very young animals but, similar to LTP, the amount of plasticity induced exhibited a developmental reduction with little or no depression remaining by P10–12 (Fig. 2). This form of LTD is NMDA receptor-dependent and pathway specific, similar to the prototypic form of LTD originally described in the hippocampus (Dudek and Bear, 1992; Mulkey and Malenka, 1992). In addition, minimal stimulation experiments were used to study the expression mechanisms of LTD at TC synapses. This form of LTD caused a decrease in potency (amplitude of the synaptic response excluding failures) that completely accounted for the magnitude of LTD (Feldman et al., 1998). This finding indicates that LTD does not reverse LTP at this input by resilencing synapses and argues against the idea that LTD leads to the elimination of inappropriately targeted synapses for the TC input to layer IV barrel cortex, at least (Goodman and Shatz, 1993). Rather it suggests that LTD modulates the efficacy of synapses previously unsilenced by LTP. Alternatively, it is possible that LTD could cause the complete silencing of synapses but that this may take longer to develop or require a stronger LTD induction protocol.
Recent work has demonstrated that activity patterns much more akin to those observed in vivo in primary sensory cortical areas can induce LTP and LTD (Dan and Poo, 2006). Such spike timing dependent synaptic plasticity can be induced in layer II/III of barrel cortex, both LTP and LTD (Feldman, 2000) and evidence from in vivo studies suggest that LTD, in particular, may be an important mechanism contributing to experience-dependent plasticity in barrel cortex (Celikel et al., 2004; Glazewski et al., 1998).
Role of Protein Kinases in Barrel Cortex Plasticity
Post-translational modifications in the form of protein kinase-dependent phosphorylation involving a wide variety of kinases and postsynaptic proteins has been shown to be important in a number of forms of synaptic plasticity (Roche et al., 1994; Soderling and Derkach, 2000; Song and Huganir, 2002). Kinase activity has also been implicated in plasticity in the barrel cortex. Knock out (Glazewski et al., 1996) or mutation (Glazewski et al., 2000; Hardingham et al., 2003) of calcium/calmodulin-dependent kinase II (CaMKII), an enzyme necessary for LTP (Malenka and Nicoll, 1999; Silva et al., 1992), blocks both receptive field plasticity and NMDA receptor-dependent LTP in barrel cortex. In addition, a recent study has identified a role for PKC in regulating synaptic strength at TC synapses in barrel cortex (Scott et al., 2006). In this study it was shown that postsynaptic PKC activation increases synaptic strength and that PKC activity is required for LTP at TC inputs in neonatal barrel cortex.
There is considerable evidence for a role of PKA signalling in barrel cortex development and plasticity. The first identified mutant mouse strain that lacks barrels, barrelless, has a loss of function mutation in the gene encoding adenylate cyclase I (Abdel-Majid et al., 1998; Welker et al., 1996), which activates PKA via production of cAMP The barrelless mouse exhibits a deficit in TC afferent clustering, suggesting a presynaptic role for AC1; however this mutant also exhibits a prominent disruption of AMPA receptor surface expression and LTP and LTD in layer IV neurons, also demonstrating a postsynaptic phenotype (Lu et al., 2003). Moreover, blockade of PKA prevents TC LTP and knockout of the PKA regulatory subunit IIβ (PKARIIβ) results in defective barrel formation and lack of LTP through a postsynaptic mechanism, without affecting TC afferent clustering (Inan et al., 2006). Interestingly, a recent study shows that PKA knock out mice do not exhibit disrupted barrel cortex development, although the PKARIIβ knock out does exhibit a barrel cortex phenotype and reduced synaptic GluR1 levels in layer IV (Watson et al., 2006). A presynaptic role of AC1 is also implicated by the finding that barrelless mice exhibit defective short-term plasticity (Lu et al., 2006). This effect on short-term plasticity may be via the presynaptic active zone protein Rab3-interacting molecule 1α (RIM 1α), which is involved in the regulation of transmitter release. RIM1α is a phosphorylation target of AC1/PKA and a RIM1α knockout mouse exhibits similar barrel pattern phenotype to the barrelless mouse (Lu et al., 2006). Thus, there is evidence for a role of AC1/PKA in barrel cortex development and synaptic plasticity, acting both pre- and postsynaptically, although the role and mechanisms appears to be complex suggesting a disconnect between structural and synaptic plasticity. The critical role of PKA in sensory cortex development and plasticity is further supported by studies in visual cortex in which knock-outs of PKARIIβ exhibit a lack of ODP and LTD, although LTP is unaffected (Fischer et al., 2004a). In addition, direct blockade of PKA in visual cortex also blocks ODP (Beaver et al., 2001a) and LTP and LTD (Liu et al., 2003b). However, in this study knockout of AC1 and AC8, the two cortically expressed Ca2+-stimulated adenylate cyclases, did not affect ODP or synaptic plasticity, indicating the PKA activation via the canonical adenylyl signalling pathway is not necessary for visual cortex plasticity. Thus AC/PKARIIβ/PKA signalling clearly plays a role in barrel cortex and visual cortex development and plasticity; however, the exact mechanisms remain to be fully elucidated.
BDNF in Barrel Cortex Development and Plasticity
Expression of the mRNA for the neurotrophin, brain-derived neurotrophic factor (BDNF), is transiently increased following sensory stimulation (Rocamora et al., 1996) and optical mapping of the NMDA and AMPA receptor-mediated responses to thalamic stimulation in BDNF knock-out mice reveal a pattern similar to that seen in mice that had previously undergone whisker deprivation (Itami et al., 2000). These findings suggest that BDNF is important for the experience-dependent development of barrel cortex. Barrel formation, however, is unaffected in the BDNF knock-out although ultra-structural studies show that whisker stimulation does not result in new synapse formation that occurs in wild-type animals (Genoud et al., 2004). Consistent with this, BDNF knock-out mice have a higher proportion of silent synapses at TC inputs than do wild types, and an LTP pairing protocol will not cause unsilencing in these knock outs unless BDNF is exogenously applied at the same time (Itami et al., 2003). Moreover, unsilencing of silent synapses during LTP is prevented by inhibition of the TrkB tyrosine kinase activity, the receptor for BDNF. Both BDNF and NGF have also been implicated in visual cortex experience-dependent plasticity (Cabelli et al., 1995; Maffei et al., 1992; Prakash et al., 1996; 2004), and this has led to the hypothesis that thalamic afferents may compete for a limited supply of neurotrophin required for their development (Katz and Shatz, 1996). The recent results in barrel cortex provide additional evidence for a direct role of BDNF in synaptic plasticity underlying cortical map plasticity. TC afferents may require both activity and BDNF to cause an unsilencing of silent synapses. TC afferents may compete for a limited supply of BDNF during early development and thus BDNF could limit the number of functional synapses generated in the map.
Kainate Receptors at TC Synapses
The glutamate ionotropic receptors are subdivided into the NMDA, AMPA and kainate classes (Collingridge and Lester, 1989; Hollmann and Heinemann, 1994). Although NMDA and AMPA receptors have been extensively studied, the lack, until recently, of selective ligands that allow kainate receptor-mediated responses to be distinguished from AMPA receptor-mediated currents, initially made the study of kainate receptors difficult. This changed with the development of the selective AMPA receptor antagonist GYKI 53655 (Wilding and Huettner, 1995) and the subsequent development of selective kainate receptor antagonists and knock out mice (Bleakman and Lodge, 1998; Lerma, 2003). Kainate receptor-mediated EPSCs were first identified at the CA3 mossy fibre synapse (Castillo et al., 1997; Vignes and Collingridge, 1997). Repetitive stimulation of the mossy fibre pathway resulted in a dual component EPSC, the slow component of which was sensitive to the non-selective AMPA/kainate antagonist CNQX, but resistant to GYKI 53655, and thus mediated by kainate receptors. Similarly, a dual component EPSC can be evoked in layer IV cells of the barrel cortex by single shock thalamic stimulation in the presence of the NMDA receptor antagonist APV (Kidd and Isaac, 1999). The GYKI-resistant, CNQX-sensitive slow component at the TC input has a similar decay time constant (~150 ms) to the kainate receptor-mediated current in CA3 neurons, but unlike the mossy fibre-CA3 synapse the I-V relationship for the kainate receptor-mediated current is strongly rectifying. This type of rectification has previously been shown for heterologously expressed kainate receptors containing unedited Ca2+-permeable GluR5 or 6 subunits and is produced by a voltage-dependent block of the channel by intracellular polyamines (Bowie and Mayer, 1995; Kamboj et al., 1995; Ruano et al., 1995). In some experiments, TC EPSCs were evoked that contained a fast AMPA component but no slow kainate component or vice versa (Kidd and Isaac, 1999). Spontaneous EPSCs (sEPSCs), however, only ever showed a single component, fast or slow. This suggests that the evoked dual component EPSCs at developing TC inputs to layer IV barrel cortex neurons are the result of the activation of two separate types of synapse: those containing only AMPA receptors and those containing only kainate receptors.
The quantal properties of these AMPA receptor and kainate receptor-mediated EPSCs were investigated in more detail in experiments in which extracellular Ca2+ was replaced with Sr2+(Bannister et al., 2005). Like Ca2+, Sr2+ supports action potential dependent release of neurotransmitter, but is much less efficient. This results in a smaller evoked synchronous EPSC that is followed by a barrage of quantal miniature EPSCs (mEPSCs) lasting up to 1s after axon stimulation (Goda and Stevens, 1994). The two main advantages of this technique are that the quantal properties of transmission can be studied in systems where mEPSC frequency is ordinarily prohibitively low (as is the case in neonatal layer IV barrel cortex), and that the source of the mEPSCs is known. Thus the use of Sr2+ allows mEPSCs to be evoked at TC synapses thus enabling quantal transmission to be studied at this input. In these experiments fast-rising AMPA receptor-mediated mEPSCs were evoked in the presence of Sr2+ in cells which also exhibited a dual AMPA and kainate receptor-mediated evoked EPSC in extracellular [Ca2+] in the absence of Sr2+. In addition, slow-rising slow-decaying kainate receptor-mediated mEPSCs were also recorded in a subset of cells, with an amplitude ~20 % of that of the AMPA receptor-mediated events. Most importantly no dual component mEPSCs were observed providing further evidence that AMPA receptors and kainate receptors never co-localize at the same TC synapse. Despite the smaller peak amplitude of these slow kainate receptor-mediated mEPSCs the charge transfer is almost five times greater than that for the AMPA receptor-mediated mEPSCs; this is due to the slow kinetics of the kainate receptor-mediated currents and indicates that they may provide more postsynaptic drive than the fast AMPA receptor-mediated component of transmission.
When the effect of LTP induction on the kainate receptor-mediated component of TC EPSCs was investigated it was found that whilst LTP causes an increase in the AMPA receptor-mediated component combined with a reduction in the slow kainate receptor-mediated component (Kidd and Isaac, 1999). This, together with the segregation of AMPA and kainate receptors at separate synapses, suggests that LTP expression involves the rapid conversion of slow kainate receptor-mediated transmission to fast AMPA receptor-mediated transmission. The mechanism for this change could be a receptor switch at individual synapses, for example involving the rapid removal of kainate receptors combined with a rapid recruitment of AMPA receptors. Alternatively, LTP could cause a presynaptic inactivation of KA receptor containing synapses and a coincident activation of different AMPA receptor containing inputs onto the same cell. However, the former type of mechanism seems a simpler explanation for the data.
The relative contribution of kainate receptor-mediated transmission decreases with development during the first postnatal week (Kidd and Isaac, 1999).This decrease involves a reduction in quantal content independent of changes in quantal amplitude (Bannister et al., 2005), suggesting that this mechanism occurs in vivo during early postnatal development (Fig. 2). The consequences of the rapid switch from slow kainate receptor- to fast AMPA receptor-mediated transmission at the TC input was recently addressed using recordings made from layer IV cells whilst sequentially monitoring TC EPSCs in voltage clamp and EPSPs in current clamp (Daw et al., 2006). Consistent with the previous report (Kidd and Isaac, 1999) an LTP induction caused a robust potentiation of the fast AMPA receptor-mediated component of the EPSC and a reduction in the slow component of the EPSC. The consequence of this was a variable effect on EPSP amplitude but a very consistent speeding of EPSP kinetics. The change in EPSP kinetics caused by LTP had a profound influence on the timing of action potentials generated by suprathreshold synaptic responses (Daw et al., 2006). Prior to LTP induction, action potentials evoked by TC EPSPs showed a large and highly variable latency when compared to other cells (Fricker and Miles, 2000, 2001; Jonas et al., 2004). The induction of LTP caused a substantial and consistent decrease in both the latency and the variability of the latency. However, LTP caused only a variable change in the probability of an EPSP evoking an action potential; some cells showed a large increase in firing probability with LTP while others exhibited no change or a reduction. In addition, similar changes in EPSP kinetics and action potential timing to those produced acutely by LTP induction are observed during development in the first postnatal week, suggesting that a similar LTP-like process may occur in vivo during this developmental period (Daw et al., 2006).
Thus development or induction of LTP results in action potentials evoked in layer IV neurons by TC stimulation that exhibit the high timing precision. Such timing precision is thought to be necessary for information processing in mature somatosensory cortex (Celikel et al., 2004; Foffani et al., 2004; Ghazanfar et al., 2000; Panzeri et al., 2001). If such precisely timed action potentials are desirable, the role of the slow kainate receptor-mediated EPSP in neonatal animals is not immediately apparent. One possibility is that the slow EPSP may be allowing a wide time window for synaptic integration and thus promote neonatal synaptic plasticity. Daw et al also tested this idea, and found that the slow kainate receptor-mediated EPSP produced increased temporal summation and that the change in EPSP kinetics with LTP reduced the window for integration (Daw et al., 2006). This suggests that the switch from slow, kainate receptor-mediated to fast, AMPA receptor-mediated transmission results in a change from a state that favours long temporal integration of inputs, which may be important for developmental plasticity, to precise coincidence detection that is likely to be important for mature information processing.
Mechanisms Contributing to the Close of the Critical Period
The molecular mechanisms defining the duration of the critical period are not well understood. One hypothesis is been that a change in NMDA receptor subunit composition produces a reduction in the ability to induce synaptic plasticity that may contribute to the end of the critical period in barrel cortex and in visual cortex. Functional NMDA receptors are composed of heteromeric tetramers containing a combination of NR1 subunits and NR2 or NR3 subunits (Wenthold et al., 2003). In the neocortex NMDA receptors primarily contain NR1 and either NR2A or NR2B subunits with a switch from mostly NR1/NR2B heteromers shortly after birth to mixed NR1-NR2A&B-containing receptors at older ages (Monyer et al., 1994; Sheng et al., 1994; Watanabe et al., 1992). Pure NR1/NR2B channels exhibit slower channel kinetics than those containing NR2A subunits (Flint et al., 1997; Vicini et al., 1998) and this has led to the suggestion that an increase in NR2A content causes the shortening of NMDA receptor channel kinetics observed during development (Carmignoto and Vicini, 1992) and is responsible for the end of the critical period both for ocular dominance plasticity (ODP) in visual cortex and for barrel cortex plasticity. The basis for this theory is that a shorter NMDA receptor-mediated EPSC would not allow sufficient Ca2+ entry for LTP induction. A series of studies showed that manipulating the visual experience of the animal can affect the developmental progression from NR2B to NR2A expression in visual cortex. Specifically, dark rearing extends NR2B expression whilst subsequent exposure to light results in a rapid switch to NR2A (Philpot et al., 2001; Quinlan et al., 1999a; Quinlan et al., 1999b). Dark-rearing also extends the critical period for ODP, and the correlation with changes in the NR2B to NR2A switch supports the idea that this mechanism may contribute to the ending of the critical period for ODP. However, another study found that the change in both subtype and kinetics of NMDA receptors coincides with the onset rather than the end of the critical period (Roberts and Ramoa, 1999).
At TC inputs in barrel cortex there is also a change in subunit composition from NR2B- to NR2A-containing NMDA receptors, as determined by sensitivity to the NR2B-selective antagonist ifenprodil, and this correlates well with the end of the layer IV critical period for LTP and for experience-dependent plasticity (Barth and Malenka, 2001). Surprisingly, this switch in subunit composition does not produce a change in kinetics of the NMDA receptor-mediated EPSC, although a speeding of kinetics does occur later in development. Another study, however, reported a simultaneous change in NMDA receptor kinetics and subunit composition, and showed that ifenprodil blocks TC LTP (Lu et al., 2001). These findings indicate that NR2B is required for TC LTP and the reduction in expression of this subunit may be responsible for the loss of synaptic plasticity in layer IV barrel cortex at the end of the first postnatal week. However, Lu et al also found that NR2A knock-out mice exhibit exactly a similar critical period for TC LTP compared to wild type and show no deficit in experience-dependent plasticity of the anatomical barrel map (Lu et al., 2001). Thus, a simple switch from NR2B to NR2A alone is not responsible for the end of the critical period. Given the apparent correlation between the critical period and NMDA receptor subunit expression in two areas of sensory cortex (somatosensory and visual) that develop at different ages, it seems likely that there is a link between these two events. However, the role of the NMDA receptor subunit switch is still unclear and this issue appears to be complex.
In visual cortex there is considerable evidence for a maturation of GABAergic inhibition as an important mechanism regulating the timing of the critical period (Hensch, 2004; Rozas et al., 2001). Genetic or pharmacological disruption of inhibition alters the critical period for ODP suggesting that maturation of the inhibitory:excitatory balance is required for the opening of the critical period in visual cortex (Hensch, 2004; Hensch et al., 1998a) . However, other work indicates that inhibition may act as a ‘gate’ leading to closure of the critical period (Kirkwood and Bear, 1994; Rozas et al., 2001). The role of the developmental regulation of excitatory-inhibitory balance in plasticity and critical period timing has not been addressed in layer IV barrel cortex. The layer IV critical period occurs much earlier in development compared to visual cortex, at a time when GABAergic transmission may still be immature (Owens and Kriegstein, 2002). Indeed, GABAA receptor-mediated transmission is found to be depolarising in neonatal cortex and may contribute to excitation rather than inhibition (Ben-Ari et al., 1989; Owens and Kriegstein, 2002). In barrel cortex there is evidence for depolarizing GABAergic transmission in the first postnatal week (Agmon et al., 1996; Daw et al., 2006). However recent work (Daw, Ashby and Isaac, unpublished observations) demonstrates that GABAergic interneurons are not integrated into the layer IV network at this age, but are rapidly recruited in a feed forward circuit at the end of the first postnatal week (Fig. 3). This recruitment of feed forward GABAergic transmission also precisely coincides with a switch from depolarising to hyperpolarizing GABAA receptor-mediated responses, producing the rapid development of fast hyperpolarizing inhibition at the end of the first postnatal week. This correlates well with the end of the layer IV critical period, providing support for the idea that a switch in excitatory-inhibitory balance may also be important in defining the end of the layer IV barrel cortex critical period, as has been suggested in visual cortex. A critical test of this hypothesis, however, remains to be performed.
Fig. 3.
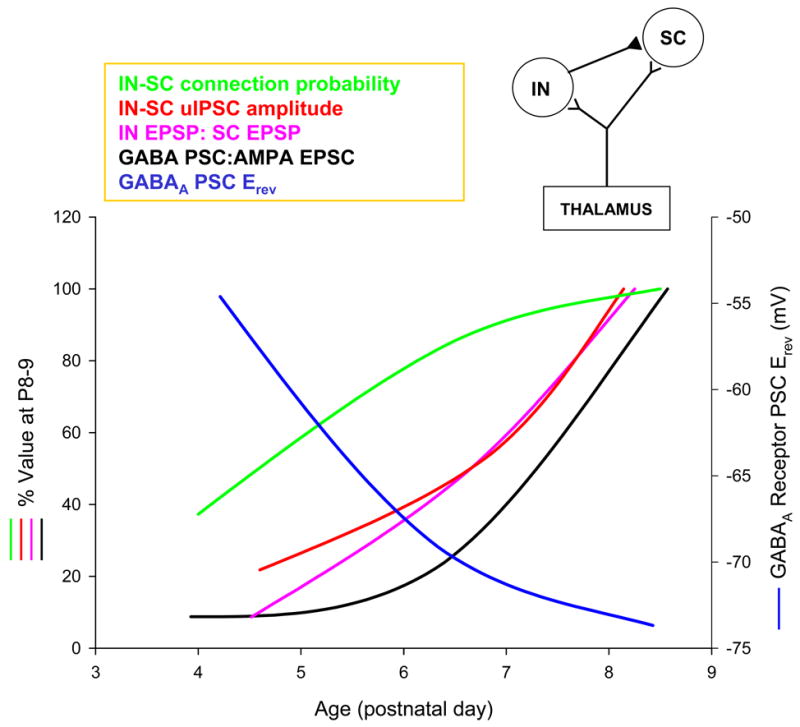
Developmental profile of GABAergic inhibition in layer IV barrel cortex. GABAA PSC Erev is the reversal potential of the pharmacologically isolated GABAA receptor-mediated PSC evoked in stellate cells (SCs) by local stimulation in layer IV/V border (measured using gramicidin perforated patch recordings). GABA PSC : AMPA EPSC ratio is the ratio of the TC EPSC and the disynaptic GABAergic PSC evoked in individual SCs in response to TC stimulation. IN-SC connection probability and IN-SC uIPSC (unitary IPSC) amplitude values are determined from paired recordings of fast spiking (FS) interneurons and SCs. IN EPSP : SC EPSP is the TC EPSP evoked in FS interneurons and SCs, and is determined from simultaneous recordings combined with thalamic stimulation. Inset top right: schematic showing the circuit for feed forward inhibition. All data from Daw, Ashby and Isaac, unpublished.
In summary, a combination of mechanisms likely produces the close of the critical period for experience-dependent plasticity in layer IV barrel cortex and in visual cortex. It is difficult to make direct comparisons between these two critical periods because they occur at different developmental stages and are mediated by different cortical layers. However, accumulating evidence suggests that appropriate regulation of the excitatory-inhibitory balance may be the common effector in producing the close of the critical period, and there appear to be a number of similarities in mechanisms producing this regulation of the circuit.
Conclusions
Primary sensory cortical areas contain topographical representations of their corresponding sensory input, and these representations refine during development in response to sensory experience. This experience-dependent plasticity occurs most prominently during a narrow postnatal time window, known as the critical period, the timing of which varies according to sensory modality (Fox, 1992; Hensch, 2004; Hubel and Wiesel, 1970). The development of a barrel cortex slice preparation in which the TC afferents are maintained (Agmon and Connors, 1991) has allowed great advances in our understanding of the synaptic and molecular mechanisms of developmental and experience-dependent plasticity in layer IV barrel cortex. This approach has also allowed comparisons to be made with plasticity mechanisms in other sensory cortical regions, most notably visual cortex. Such studies indicate that NMDA receptor-dependent forms of synaptic plasticity (LTP and LTD) are likely to be important synaptic mechanisms underlying the experience-dependent regulation of synaptic strength required for refinement of the map. In addition, some of these mechanisms are also required for formation of the distinct cytoarchitecture of the barrel cortex. However, in only a few cases has a direct causality been demonstrated between synaptic plasticity and map plasticity. Moreover, there are also studies showing a dissociation between LTP/LTD and receptive field plasticity. Thus it is clear that there is not a simple relationship between synaptic plasticity and map plasticity; more work will be required to determine the precise relationship between these synaptic and systems-level phenomena.
Several novel mechanisms and functional consequences of neonatal synaptic plasticity at the synapse level have been described. The switch from slow NR2B-containing NMDA and kainate receptor-mediated transmission at TC synapses to faster NR2A and AMPA receptor-mediated transmission has been characterized. This has profound consequences for the induction of synaptic plasticity, and the development of coincidence detection and information processing in barrel cortex. Finally, a rapid maturation of GABAergic transmission occurs towards the end of the first postnatal week that has similarities with the maturation of GABAergic circuits in visual cortex thought to contribute to the closing of the critical period for ODP.
Taken together, these studies show that the neonatal TC input to layer IV barrel cortex favours temporal summation and a long integration window at the expense of precise coincidence detection. This likely serves to enable a strong drive of layer IV by neonatal TC synapses and promote induction of long-term synaptic plasticity at this input at a time when excitatory circuitry in neocortex is poorly developed. The switch to fast excitation and strong feed forward inhibition at the end of the first postnatal week alters the mode of operation to produce a circuit capable of high precision coincidence detection that is required for mature information processing. This likely also makes a major contribution to the close of the critical period in layer IV by producing much less favourable conditions for the induction of long-term synaptic plasticity.
Acknowledgments
Supported by the NINDS Intramural Program (J.T.R.I.) and The Wellcome Trust (M.I.D., H.L.S., J.T.R.I.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdel-Majid RM, Leong WL, Schalkwyk LC, Smallman DS, Wong ST, Storm DR, Fine A, Dobson MJ, Guernsey DL, Neumann PE. Loss of adenylyl cyclase I activity disrupts patterning of mouse somatosensory cortex. Nat Genet. 1998;19:289–291. doi: 10.1038/980. [DOI] [PubMed] [Google Scholar]
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- Agmon A, Hollrigel G, O'Dowd DK. Functional GABAergic synaptic connection in neonatal mouse barrel cortex. J Neurosci. 1996;16:4684–4695. doi: 10.1523/JNEUROSCI.16-15-04684.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonini A, Stryker MP. Development of individual geniculocortical arbors in cat striate cortex and effects of binocular impulse blockade. J Neurosci. 1993;13:3549–3573. doi: 10.1523/JNEUROSCI.13-08-03549.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong-James M, Callahan CA. Thalamo-cortical processing of vibrissal information in the rat. II. spatiotemporal convergence in the thalamic ventroposterior medial nucleus (VPm) and its relevance to generation of receptive fields of S1 cortical "barrel" neurones. J Comp Neurol. 1991;303:211–224. doi: 10.1002/cne.903030204. [DOI] [PubMed] [Google Scholar]
- Armstrong-James M, Callahan CA, Friedman MA. Thalamo-cortical processing of vibrissal information in the rat. I. Intracortical origins of surround but not centre-receptive fields of layer IV neurones in the rat S1 barrel field cortex. J Comp Neurol. 1991;303:193–210. doi: 10.1002/cne.903030203. [DOI] [PubMed] [Google Scholar]
- Bannister NJ, Benke TA, Mellor J, Scott H, Gurdal E, Crabtree JW, Isaac JT. Developmental changes in AMPA and kainate receptor-mediated quantal transmission at thalamocortical synapses in the barrel cortex. J Neurosci. 2005;25:5259–5271. doi: 10.1523/JNEUROSCI.0827-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AL, Malenka RC. NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses. Nat Neurosci. 2001;4:235–236. doi: 10.1038/85070. [DOI] [PubMed] [Google Scholar]
- Bear MF, Cooper LN, Ebner FF. A physiological basis for a theory of synapse modification. Science. 1987;237:42–48. doi: 10.1126/science.3037696. [DOI] [PubMed] [Google Scholar]
- Bear MF, Kleinschmidt A, Gu QA, Singer W. Disruption of experience-dependent synaptic modifications in striate cortex by infusion of an NMDA receptor antagonist. J Neurosci. 1990;10:909–925. doi: 10.1523/JNEUROSCI.10-03-00909.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaver CJ, Ji Q, Fischer QS, Daw NW. Cyclic AMP-dependent protein kinase mediates ocular dominance shifts in cat visual cortex. Nat Neurosci. 2001a;4:159–163. doi: 10.1038/83985. [DOI] [PubMed] [Google Scholar]
- Beaver CJ, Ji Q, Fischer QS, Daw NW. Cyclic AMP-dependent protein kinase mediates ocular dominance shifts in cat visual cortex. Nat Neurosci. 2001b;4:159–163. doi: 10.1038/83985. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore C, Van Sluyters RC. Reversal of the physiological effects of monocular deprivation in kittens: further evidence for a sensitive period. J Physiol. 1974;237:195–216. doi: 10.1113/jphysiol.1974.sp010478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleakman D, Lodge D. Neuropharmacology of AMPA and kainate receptors. Neuropharmacology. 1998;37:1187–1204. doi: 10.1016/s0028-3908(98)00139-7. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bowie D, Mayer ML. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15:453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Vicini S. Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science. 1992;258:1007–1011. doi: 10.1126/science.1279803. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- Celikel T, Szostak VA, Feldman DE. Modulation of spike timing by sensory deprivation during induction of cortical map plasticity. Nat Neurosci. 2004;7:534–541. doi: 10.1038/nn1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Klingauf J, Tsien RW. Fusion pore modulation as a presynaptic mechanism contributing to expression of long-term potentiation. Philos Trans R Soc Lond B Biol Sci. 2003;358:695–705. doi: 10.1098/rstb.2002.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Morales B, Lee HK, Kirkwood A. Absence of long-term depression in the visual cortex of glutamic Acid decarboxylase-65 knock-out mice. J Neurosci. 2002;22:5271–5276. doi: 10.1523/JNEUROSCI.22-13-05271.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Lester RA. Excitatory amino acid receptors in the vertebrate central nervous system. Pharmacol Rev. 1989;41:143–210. [PubMed] [Google Scholar]
- Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Rose HJ, Metherate R. Auditory thalamocortical synaptic transmission in vitro. J Neurophysiol. 2002;87:361–384. doi: 10.1152/jn.00549.2001. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- Davies SN, Lester RA, Reymann KG, Collingridge GL. Temporally distinct pre- and post-synaptic mechanisms maintain long-term potentiation. Nature. 1989;338:500–503. doi: 10.1038/338500a0. [DOI] [PubMed] [Google Scholar]
- Daw MI, Bannister NV, Isaac JT. Rapid, activity-dependent plasticity in timing precision in neonatal barrel cortex. J Neurosci. 2006;26:4178–4187. doi: 10.1523/JNEUROSCI.0150-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw N, Rao Y, Wang XF, Fischer Q, Yang Y. LTP and LTD vary with layer in rodent visual cortex. Vision Res. 2004;44:3377–3380. doi: 10.1016/j.visres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Daw NW, Gordon B, Fox KD, Flavin HJ, Kirsch JD, Beaver CJ, Ji Q, Reid SN, Czepita D. Injection of MK-801 affects ocular dominance shifts more than visual activity. J Neurophysiol. 1999;81:204–215. doi: 10.1152/jn.1999.81.1.204. [DOI] [PubMed] [Google Scholar]
- Diamond ME, Armstrong-James M, Ebner FF. Experience-dependent plasticity in adult rat barrel cortex. Proc Natl Acad Sci U S A. 1993;90:2082–2086. doi: 10.1073/pnas.90.5.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond ME, Huang W, Ebner FF. Laminar comparison of somatosensory cortical plasticity. Science. 1994;265:1885–1888. doi: 10.1126/science.8091215. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc Natl Acad Sci U S A. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Friedlander MJ. Developmental down-regulation of LTD in cortical layer IV and its independence of modulation by inhibition. Neuron. 1996;16:1097–1106. doi: 10.1016/s0896-6273(00)80136-1. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H. Learning from LTP: a comment on recent attempts to identify cellular and molecular mechanisms of memory. Learn Mem. 1996;3:61–73. doi: 10.1101/lm.3.2-3.61. [DOI] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Feldman DE, Brecht M. Map plasticity in somatosensory cortex. Science. 2005;310:810–815. doi: 10.1126/science.1115807. [DOI] [PubMed] [Google Scholar]
- Feldman DE, Nicoll RA, Malenka RC, Isaac JT. Long-term depression at thalamocortical synapses in developing rat somatosensory cortex. Neuron. 1998;21:347–357. doi: 10.1016/s0896-6273(00)80544-9. [DOI] [PubMed] [Google Scholar]
- Fischer QS, Beaver CJ, Yang Y, Rao Y, Jakobsdottir KB, Storm DR, McKnight GS, Daw NW. Requirement for the RIIbeta isoform of PKA, but not calcium-stimulated adenylyl cyclase, in visual cortical plasticity. J Neurosci. 2004a;24:9049–9058. doi: 10.1523/JNEUROSCI.2409-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer QS, Beaver CJ, Yang Y, Rao Y, Jakobsdottir KB, Storm DR, McKnight GS, Daw NW. Requirement for the RIIbeta isoform of PKA, but not calcium-stimulated adenylyl cyclase, in visual cortical plasticity. J Neurosci. 2004b;24:9049–9058. doi: 10.1523/JNEUROSCI.2409-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. Journal of Neuroscience. 1997;17:2469–2476. doi: 10.1523/JNEUROSCI.17-07-02469.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foeller E, Feldman DE. Synaptic basis for developmental plasticity in somatosensory cortex. Curr Opin Neurobiol. 2004;14:89–95. doi: 10.1016/j.conb.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Foffani G, Tutunculer B, Moxon KA. Role of spike timing in the forelimb somatosensory cortex of the rat. J Neurosci. 2004;24:9266–9271. doi: 10.1523/JNEUROSCI.2523-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K. A critical period for experience-dependent synaptic plasticity in rat barrel cortex. J Neurosci. 1992;12:1826–1838. doi: 10.1523/JNEUROSCI.12-05-01826.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K. Anatomical pathways and molecular mechanisms for plasticity in the barrel cortex. Neuroscience. 2002;111:799–814. doi: 10.1016/s0306-4522(02)00027-1. [DOI] [PubMed] [Google Scholar]
- Fox K, Schlaggar BL, Glazewski S, O'Leary DD. Glutamate receptor blockade at cortical synapses disrupts development of thalamocortical and columnar organization in somatosensory cortex. Proc Natl Acad Sci U S A. 1996;93:5584–5589. doi: 10.1073/pnas.93.11.5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker D, Miles R. EPSP amplification and the precision of spike timing in hippocampal neurons. Neuron. 2000;28:559–569. doi: 10.1016/s0896-6273(00)00133-1. [DOI] [PubMed] [Google Scholar]
- Fricker D, Miles R. Interneurons, spike timing, and perception. Neuron. 2001;32:771–774. doi: 10.1016/s0896-6273(01)00528-1. [DOI] [PubMed] [Google Scholar]
- Genoud C, Knott GW, Sakata K, Lu B, Welker E. Altered synapse formation in the adult somatosensory cortex of brain-derived neurotrophic factor heterozygote mice. J Neurosci. 2004;24:2394–2400. doi: 10.1523/JNEUROSCI.4040-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazanfar AA, Stambaugh CR, Nicolelis MA. Encoding of tactile stimulus location by somatosensory thalamocortical ensembles. J Neurosci. 2000;20:3761–3775. doi: 10.1523/JNEUROSCI.20-10-03761.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Differential Regulation of Neocortical Synapses by Neuromodulators and Activity. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Efficacy of thalamocortical and intracortical synaptic connections: quanta, innervation, and reliability. Neuron. 1999;23:385–397. doi: 10.1016/s0896-6273(00)80788-6. [DOI] [PubMed] [Google Scholar]
- Glazewski S, Chen CM, Silva A, Fox K. Requirement for alpha-CaMKII in experience-dependent plasticity of the barrel cortex. Science. 1996;272:421–423. doi: 10.1126/science.272.5260.421. [DOI] [PubMed] [Google Scholar]
- Glazewski S, Giese KP, Silva A, Fox K. The role of alpha-CaMKII autophosphorylation in neocortical experience-dependent plasticity. Nat Neurosci. 2000;3:911–918. doi: 10.1038/78820. [DOI] [PubMed] [Google Scholar]
- Glazewski S, McKenna M, Jacquin M, Fox K. Experience-dependent depression of vibrissae responses in adolescent rat barrel cortex. Eur J Neurosci. 1998;10:2107–2116. doi: 10.1046/j.1460-9568.1998.00222.x. [DOI] [PubMed] [Google Scholar]
- Goda Y, Stevens CF. Two components of transmitter release at a central synapse. Proc Natl Acad Sci U S A. 1994;91:12942–12946. doi: 10.1073/pnas.91.26.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda Y, Stevens CF. Long-term depression properties in a simple system. Neuron. 1996;16:103–111. doi: 10.1016/s0896-6273(00)80027-6. [DOI] [PubMed] [Google Scholar]
- Goodman CS, Shatz CJ. Developmental mechanisms that generate precise patterns of neuronal connectivity. Cell. 1993;72:77–98. doi: 10.1016/s0092-8674(05)80030-3. [DOI] [PubMed] [Google Scholar]
- Hardingham N, Glazewski S, Pakhotin P, Mizuno K, Chapman PF, Giese KP, Fox K. Neocortical long-term potentiation and experience-dependent synaptic plasticity require alpha-calcium/calmodulin-dependent protein kinase II autophosphorylation. J Neurosci. 2003;23:4428–4436. doi: 10.1523/JNEUROSCI.23-11-04428.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK. Critical period regulation. Annu Rev Neurosci. 2004;27:549–579. doi: 10.1146/annurev.neuro.27.070203.144327. [DOI] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science. 1998a;282:1504–1508. doi: 10.1126/science.282.5393.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK, Gordon JA, Brandon EP, McKnight GS, Idzerda RL, Stryker MP. Comparison of plasticity in vivo and in vitro in the developing visual cortex of normal and protein kinase A RIbeta-deficient mice. J Neurosci. 1998b;18:2108–2117. doi: 10.1523/JNEUROSCI.18-06-02108.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol. 1970;206:419–436. doi: 10.1113/jphysiol.1970.sp009022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inan M, Lu HC, Albright MJ, She WC, Crair MC. Barrel map development relies on protein kinase A regulatory subunit II beta-mediated cAMP signaling. J Neurosci. 2006;26:4338–4349. doi: 10.1523/JNEUROSCI.3745-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT. Postsynaptic silent synapses: evidence and mechanisms. Neuropharmacology. 2003;45:450–460. doi: 10.1016/s0028-3908(03)00229-6. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Hjelmstad GO, Nicoll RA, Malenka RC. Long-term potentiation at single fiber inputs to hippocampal CA1 pyramidal cells. Proc Natl Acad Sci U S A. 1996;93:8710–8715. doi: 10.1073/pnas.93.16.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Itami C, Kimura F, Kohno T, Matsuoka M, Ichikawa M, Tsumoto T, Nakamura S. Brain-derived neurotrophic factor-dependent unmasking of "silent" synapses in the developing mouse barrel cortex. Proc Natl Acad Sci U S A. 2003;100:13069–13074. doi: 10.1073/pnas.2131948100. Epub 12003 Oct 13013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itami C, Mizuno K, Kohno T, Nakamura S. Brain-derived neurotrophic factor requirement for activity-dependent maturation of glutamatergic synapse in developing mouse somatosensory cortex. Brain Res. 2000;857:141–150. doi: 10.1016/s0006-8993(99)02352-5. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, Itohara S. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature. 2000;406:726–731. doi: 10.1038/35021059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Bischofberger J, Fricker D, Miles R. Interneuron Diversity series: Fast in, fast out--temporal and spatial signal processing in hippocampal interneurons. Trends Neurosci. 2004;27:30–40. doi: 10.1016/j.tins.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Kamboj SK, Swanson GT, Cull-Candy SG. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. J Physiol. 1995;486:297–303. doi: 10.1113/jphysiol.1995.sp020812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kidd FL, Coumis U, Collingridge GL, Crabtree JW, Isaac JT. A presynaptic kainate receptor is involved in regulating the dynamic properties of thalamocortical synapses during development. Neuron. 2002;34:635–646. doi: 10.1016/s0896-6273(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Kidd FL, Isaac JT. Developmental and activity-dependent regulation of kainate receptors at thalamocortical synapses. Nature. 1999;400:569–573. doi: 10.1038/23040. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Bear MF. Hebbian synapses in visual cortex. J Neurosci. 1994;14:1634–1645. doi: 10.1523/JNEUROSCI.14-03-01634.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Lee HK, Bear MF. Co-regulation of long-term potentiation and experience-dependent synaptic plasticity in visual cortex by age and experience. Nature. 1995;375:328–331. doi: 10.1038/375328a0. [DOI] [PubMed] [Google Scholar]
- Kullmann DM. Amplitude fluctuations of dual-component EPSCs in hippocampal pyramidal cells: implications for long-term potentiation. Neuron. 1994;12:1111–1120. doi: 10.1016/0896-6273(94)90318-2. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Asztely F. Extrasynaptic glutamate spillover in the hippocampus: evidence and implications. Trends Neurosci. 1998;21:8–14. doi: 10.1016/s0166-2236(97)01150-8. [DOI] [PubMed] [Google Scholar]
- Lerma J. Roles and rules of kainate receptors in synaptic transmission. Nat Rev Neurosci. 2003;4:481–495. doi: 10.1038/nrn1118. [DOI] [PubMed] [Google Scholar]
- LeVay S, Wiesel TN, Hubel DH. The development of ocular dominance columns in normal and visually deprived monkeys. J Comp Neurol. 1980;191:1–51. doi: 10.1002/cne.901910102. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Liu S, Rao Y, Daw N. Roles of protein kinase A and protein kinase G in synaptic plasticity in the visual cortex. Cereb Cortex. 2003a;13:864–869. doi: 10.1093/cercor/13.8.864. [DOI] [PubMed] [Google Scholar]
- Liu S, Rao Y, Daw N. Roles of protein kinase A and protein kinase G in synaptic plasticity in the visual cortex. Cereb Cortex. 2003b;13:864–869. doi: 10.1093/cercor/13.8.864. [DOI] [PubMed] [Google Scholar]
- Lu HC, Butts DA, Kaeser PS, She WC, Janz R, Crair MC. Role of efficient neurotransmitter release in barrel map development. J Neurosci. 2006;26:2692–2703. doi: 10.1523/JNEUROSCI.3956-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HC, Gonzalez E, Crair MC. Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron. 2001;32:619–634. doi: 10.1016/s0896-6273(01)00501-3. [DOI] [PubMed] [Google Scholar]
- Lu HC, She WC, Plas DT, Neumann PE, Janz R, Crair MC. Adenylyl cyclase I regulates AMPA receptor trafficking during mouse cortical 'barrel' map development. Nat Neurosci. 2003;6:939–947. doi: 10.1038/nn1106. [DOI] [PubMed] [Google Scholar]
- MacLean JN, Fenstermaker V, Watson BO, Yuste R. A visual thalamocortical slice. Nat Methods. 2006;3:129–134. doi: 10.1038/nmeth849. [DOI] [PubMed] [Google Scholar]
- Maffei L, Berardi N, Domenici L, Parisi V, Pizzorusso T. Nerve growth factor (NGF) prevents the shift in ocular dominance distribution of visual cortical neurons in monocularly deprived rats. J Neurosci. 1992;12:4651–4662. doi: 10.1523/JNEUROSCI.12-12-04651.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation--a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Montgomery JM, Pavlidis P, Madison DV. Pair recordings reveal all-silent synaptic connections and the postsynaptic expression of long-term potentiation. Neuron. 2001;29:691–701. doi: 10.1016/s0896-6273(01)00244-6. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- Panzeri S, Petersen RS, Schultz SR, Lebedev M, Diamond ME. The role of spike timing in the coding of stimulus location in rat somatosensory cortex. Neuron. 2001;29:769–777. doi: 10.1016/s0896-6273(01)00251-3. [DOI] [PubMed] [Google Scholar]
- Persico AM, Mengual E, Moessner R, Hall FS, Revay RS, Sora I, Arellano J, DeFelipe J, Gimenez-Amaya JM, Conciatori M, Marino R, Baldi A, Cabib S, Pascucci T, Uhl GR, Murphy DL, Lesch KP, Keller F. Barrel pattern formation requires serotonin uptake by thalamocortical afferents, and not vesicular monoamine release. J Neurosci. 2001;21:6862–6873. doi: 10.1523/JNEUROSCI.21-17-06862.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen CC, Sakmann B. The excitatory neuronal network of rat layer 4 barrel cortex. J Neurosci. 2000;20:7579–7586. doi: 10.1523/JNEUROSCI.20-20-07579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Sekhar AK, Shouval HZ, Bear MF. Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron. 2001;29:157–169. doi: 10.1016/s0896-6273(01)00187-8. [DOI] [PubMed] [Google Scholar]
- Prakash N, Cohen-Cory S, Frostig RD. RAPID and opposite effects of BDNF and NGF on the functional organization of the adult cortex in vivo. Nature. 1996;381:702–706. doi: 10.1038/381702a0. [DOI] [PubMed] [Google Scholar]
- Prakash N, Cohen-Cory S, Penschuck S, Frostig RD. Basal forebrain cholinergic system is involved in rapid nerve growth factor (NGF)-induced plasticity in the barrel cortex of adult rats. J Neurophysiol. 2004;91:424–437. doi: 10.1152/jn.00489.2003. Epub 2003 Sep 2024. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, Bear MF. Bidirectional, experience-dependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci U S A. 1999a;96:12876–12880. doi: 10.1073/pnas.96.22.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci. 1999b;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Renger JJ, Hartman KN, Tsuchimoto Y, Yokoi M, Nakanishi S, Hensch TK. Experience-dependent plasticity without long-term depression by type 2 metabotropic glutamate receptors in developing visual cortex. Proc Natl Acad Sci U S A. 2002;99:1041–1046. doi: 10.1073/pnas.022618799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EB, Meredith MA, Ramoa AS. Suppression of NMDA receptor function using antisense DNA block ocular dominance plasticity while preserving visual responses. J Neurophysiol. 1998;80:1021–1032. doi: 10.1152/jn.1998.80.3.1021. [DOI] [PubMed] [Google Scholar]
- Roberts EB, Ramoa AS. Enhanced NR2A subunit expression and decreased NMDA receptor decay time at the onset of ocular dominance plasticity in the ferret. Journal Of Neurophysiology. 1999;81:2587–2591. doi: 10.1152/jn.1999.81.5.2587. [DOI] [PubMed] [Google Scholar]
- Rocamora N, Welker E, Pascual M, Soriano E. Upregulation of BDNF mRNA expression in the barrel cortex of adult mice after sensory stimulation. J Neurosci. 1996;16:4411–4419. doi: 10.1523/JNEUROSCI.16-14-04411.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, Tingley WG, Huganir RL. Glutamate receptor phosphorylation and synaptic plasticity. Curr Opin Neurobiol. 1994;4:383–388. doi: 10.1016/0959-4388(94)90100-7. [DOI] [PubMed] [Google Scholar]
- Rozas C, Frank H, Heynen AJ, Morales B, Bear MF, Kirkwood A. Developmental inhibitory gate controls the relay of activity to the superficial layers of the visual cortex. J Neurosci. 2001;21:6791–6801. doi: 10.1523/JNEUROSCI.21-17-06791.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruano D, Lambolez B, Rossier J, Paternain AV, Lerma J. Kainate receptor subunits expressed in single cultured hippocampal neurons: molecular and functional variants by RNA editing. Neuron. 1995;14:1009–1017. doi: 10.1016/0896-6273(95)90339-9. [DOI] [PubMed] [Google Scholar]
- Schlaggar BL, Fox K, O'Leary DD. Postsynaptic control of plasticity in developing somatosensory cortex. Nature. 1993;364:623–626. doi: 10.1038/364623a0. [DOI] [PubMed] [Google Scholar]
- Schlaggar BL, O'Leary DD. Potential of visual cortex to develop an array of functional units unique to somatosensory cortex. Science. 1991;252:1556–1560. doi: 10.1126/science.2047863. [DOI] [PubMed] [Google Scholar]
- Scott HL, Braud S, Bannister NJ, Isaac JT. Synaptic strength at the thalamocortical input to layer IV neonatal barrel cortex is regulated by protein kinase C. Neuropharmacology. 2006 doi: 10.1016/j.neuropharm.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Soderling TR, Derkach VA. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000;23:75–80. doi: 10.1016/s0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- Vicini S, Jian Feng W, Jin Hong L, Wei Jian Z, Yue Hua W, Jian Hong L, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N- methyl-D-aspartate receptors. Journal of Neurophysiology. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- Vignes M, Collingridge GL. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- Wallace H, Fox K. The effect of vibrissa deprivation pattern on the form of plasticity induced in rat barrel cortex. Somatosens Mot Res. 1999;16:122–138. doi: 10.1080/08990229970564. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Inoue Y, Sakimura K, Mishina M. Developmental changes in distribution of NMDA receptor channel subunit mRNAs. Neuroreport. 1992;3:1138–1140. doi: 10.1097/00001756-199212000-00027. [DOI] [PubMed] [Google Scholar]
- Watson RF, Abdel-Majid RM, Barnett MW, Willis BS, Katsnelson A, Gillingwater TH, McKnight GS, Kind PC, Neumann PE. Involvement of protein kinase A in patterning of the mouse somatosensory cortex. J Neurosci. 2006;26:5393–5401. doi: 10.1523/JNEUROSCI.0750-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker C. Microelectrode delineation of fine grain somatotopic organization of (SmI) cerebral neocortex in albino rat. Brain Res. 1971;26:259–275. [PubMed] [Google Scholar]
- Welker E, Armstrong-James M, Bronchti G, Ourednik W, Gheorghita-Baechler F, Dubois R, Guernsey DL, Van der Loos H, Neumann PE. Altered sensory processing in the somatosensory cortex of the mouse mutant barrelless. Science. 1996;271:1864–1867. doi: 10.1126/science.271.5257.1864. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Prybylowski K, Standley S, Sans N, Petralia RS. Trafficking of NMDA receptors. Annu Rev Pharmacol Toxicol. 2003;43:335–358. doi: 10.1146/annurev.pharmtox.43.100901.135803. [DOI] [PubMed] [Google Scholar]
- Wilding TJ, Huettner JE. Differential antagonism of alpha-amino-3-hydroxy-5-methyl-4- isoxazolepropionic acid-preferring and kainate-preferring receptors by 2,3-benzodiazepines. Mol Pharmacol. 1995;47:582–587. [PubMed] [Google Scholar]
- Woolsey TA, Van der Loos H. The structural organization of layer IV in the somatosensory region (SI) of mouse cerebral cortex. The description of a cortical field composed of discrete cytoarchitectonic units. Brain Res. 1970;17:205–242. doi: 10.1016/0006-8993(70)90079-x. [DOI] [PubMed] [Google Scholar]
- Yanagisawa T, Tsumoto T, Kimura F. Transiently higher release probability during critical period at thalamocortical synapses in the mouse barrel cortex: relevance to differential short-term plasticity of AMPA and NMDA EPSCs and possible involvement of silent synapses. Eur J Neurosci. 2004;20:3006–3018. doi: 10.1111/j.1460-9568.2004.03756.x. [DOI] [PubMed] [Google Scholar]