

Abstract
The sarcoglycan-sarcospan complex (α-, β-, χ-, δ-, ε-, and ζ-SG-SSPN), a component of the dystrophin-associated glycoprotein complex (DAGC), is located at the sarcolemma of muscle fibers where it contributes to maintain cell integrity during contraction-relaxation cycles; χ-and δ-SG are also expressed in the sarcoplasmic reticulum (SR). In this study, we report the identification of a novel isoform of murine δ-SG produced by alternative splicing that we named δ-SG3. This isoform is present at transcript level in several tissues, with its highest expression in skeletal and cardiac muscle. The δ-SG3 protein lacks the last 122 amino acids at the C-terminal, which are replaced by 10 new amino acids (EGFLNMQLAG). Interestingly, double immunofluorescence analysis for δ-SG3 and the dihydropyridine receptor (DHPR) shows a close localization of these two proteins. We propose the subcellular distribution of this novel δ-SG3 isoform at the SR and its involvement in intracellular calcium concentration regulation.
Keywords: Alternative Splicing; Amino Acid Sequence; Animals; Calcium; metabolism; Calcium Channels, L-Type; chemistry; Carrier Proteins; chemistry; Cell Line; Dystrophin; metabolism; Exons; Glycoproteins; chemistry; Introns; Male; Membrane Proteins; chemistry; Mice; Mice, Inbred BALB C; Microscopy, Fluorescence; Molecular Sequence Data; Muscle, Skeletal; metabolism; Neoplasm Proteins; chemistry; Peptides; chemistry; Protein Isoforms; Protein Structure, Tertiary; RNA, Messenger; metabolism; Reverse Transcriptase Polymerase Chain Reaction; Sarcoglycans; chemistry; Sarcoplasmic Reticulum; metabolism; Sequence Homology, Amino Acid; Tissue Distribution
INTRODUCTION
The dystrophin-associated glycoprotein complex (DAGC) is a multimeric array composed of membrane and cytoskeletal proteins that links the extracellular matrix with the cytoskeleton [1]. In skeletal muscle, the DAGC is composed of dystrophin, the syntrophins, the dystroglycans, the sarcoglycans, and sarcospan [2–4]. The importance of the DAGC in normal muscle physiology is clearly demonstrated since the deficiency of almost any of its components constitutes the primary cause of muscular dystrophy [5]. The sarcoglycan-sarcospan complex (SG– SSPN) is a subcomplex of the DAGC composed by the transmembranal proteins α-, β-, χ-, and d-sarcoglycans (SG), as well as by sarcospan (SSPN). Mutations in α-, β-, χ-, and δ-SGs cause autosomal recessive limb girdle muscular dystrophies (LGMD 2D–2F) [6], that have been collectively called sarcoglycanopathies. A fifth member of the sarcoglycan family, ε-SG, is closely related to α-SG, and both are coexpressed in striated muscle as part of different complexes [7–9]. Contrasting with the rest of the sarcoglycans, mutations in the ε-SG gene are not associated with muscular dystrophy but to the myoclonus-distonia syndrome of neurological origin [10]. The most recently described component of the SG-SSPN complex is ζ-SG, which is expressed in skeletal and smooth muscle associated with both a-and ε-SGs [11,12]. To date, ζ-SG has not been associated to any muscular disease.
In striated muscle two different complexes have been recognized on the sarcolemma, both containing β-, χ-, and δ-SGs, and the mutually exclusive components α-and ε-SG [9]. Most of the previous reports focused on the presence of the SG-SSPN complex on the muscle plasma membrane [13]. Nevertheless, a non-sarcolemmal localization of some members of the SG-SSPN complex was first described by Ueda et al. [14], who reported the expression of χ-and δ-SG in the sarcoplasmic reticulum (SR). To date, two human delta sarcoglycan isoforms, δ-SG1 and δ-SG2, have been identified. The δ-SG1 transcript contains 9 exons encompassing 8 kb which are translated in a basic transmembranal 35 kDa protein of 290 amino acids [15,16]; while the δ-SG2 transcript lacks exon 9, terminates at intron 8, and encodes for a protein with a different C-terminal sequence, exchanging the last 57 amino acids of δ-SG1 by 23 different amino acids [17]. In Syrian hamster, three alternative promoters have been found, that produce transcripts with a unique exon 1 which contains 50-untranslated sequences [18]. Herein we describe a new shorter murine δ-SG isoform mainly present in the SR, coexisting with the larger previously reported murine δ-SG isoform. This new isoform, that we named δ-SG3, is originated from alternative splicing of the δ-SG transcript and has 10 new amino acids at its C-terminal that substitute the last 122 amino acids of the reported isoform. The specific localization of this δ-SG isoform in the SR close to the dihydropyridine receptor suggests a possible role in calcium regulation.
Materials and methods
Animals
Experiments were carried out in male adult Balb C mice. All procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources of the United States as approved in Mexico by the National Academy of Medicine. (http://www.nal.usda.gov/awic/pubs/noawicpubs/careuse.htm)
Antibodies
The peptide corresponding to the specific δ-SG3 C-terminal end, residues 168–177 (EGFLNMQLAG), was synthesized in solution. It was purified by reverse-phase high-performance liquid chromatography on a C18 column and its structure was ascertained by fast atom bombardment mass spectroscopy. The N terminus of the d-SG3 C-terminal sequence was linked to a molecule of aminohexanoic acid (Ahx = NH2-(CH2)5-COOH), functioning as spacer arm, and the amine was substituted by a cysteine. Then, this synthetic peptide was conjugated by the N-terminal cysteine residue to the keyhole limpet hemocyanin (using Kit 77600 inject maleimide KLH, Pierce) using the protocol recommended by the manufacturer, then mixed with Freund’s adjuvant (Sigma) and injected into rabbit. Polyclonal antibodies against the peptide present in the sera were characterized by Western blot using the KLH-conjugated δ-SG3 peptide as described previously [19]. The other antibodies used in this work were: C-terminal δ-SG (C15, polyclonal goat, Santa Cruz Biotechnology), dihydropyridine receptor (polyclonal goat, Santa Cruz Biotechnology). As secondary antibodies, we used Cy3-mouse anti-rabbit (Jackson), Cy3-goat anti-rabbit (Jackson), Cy3-donkey anti-goat (Jackson), and FITC-goat anti-rabbit (Santa Cruz Biotechnology).
Reverse transcription-polymerase chain reaction
Non-quantitative expression analysis of δ-SG1 and δ-SG3 transcripts was carried out by reverse transcription-polymerase chain reaction (RT-PCR) from mice tissues. Total RNA from male adult mice tissues (brain, heart, skeletal muscle, lung, liver, retina, kidney, uterus, small intestine, and testis) as well as from C2C12 cells was extracted with TRIZOL reagent (Invitrogen) according to the manufacturer’s specifications. Reverse transcription reactions were performed with 2 lg of the total RNA using random primers. The obtained cDNA was used for the PCR amplification using a common forward primer and a specific reverse primer for each δ-SG isoform. GAPDH was amplified as positive control: δ-SG1 and δ-SG3:forward 5′-GAACAGTATTCCCACCACAGGAG-3′, δ-SG1 reverse 5′-TCTGCAGATGGCTTCCATATTGC-3′; δ-SG3 reverse 5′-ATGGTGTGTCACCCAGCCAATTG-3′; GAPDH forward 5′-ATCCCATCACCA TCTTCCAG-3′; GAPDH reverse 5′-TGTGGTCATGAGTCCTTCCA-3′. The conditions for PCR were: initial denaturation for 5 min at 94 °C followed by 32 cycles, each cycle consisting of denaturation 30 sec at 94 °C, alignment 30 sec at 60 °C, and extension 1 min at 72 °C; and a final extension during 7 min at 72 °C. PCR products were sequenced with Big Dye Terminator sequencing kit (Applied Biosystems) and ABI 310 genetic analyzer (Applied Biosystems) according to manufacturer’s instructions.
Immunofluorescence
Gastrocnemius and femoral muscles from adult mice were dissected, frozen in liquid nitrogen-isopentane, and cut into 7 lm cryostat sections. Sections were blocked and permeabilized with 5% fetal bovine serum (FBS), 5% bovine serum albumin (BSA), 0.5% gelatin, and 0.5% Triton X-100 diluted in PBS for 1 h. Primary antibodies were diluted in 2.5% FBS, 2.5% BSA, 0.25% gelatin, and 0.5% Tween 20 in PBS, and tissues were incubated for 120 min. After incubation with primary antibody, sections were washed three times with 0.5% Tween 20 in PBS. Secondary antibody coupled to either carboxymethyl indocyanine-3 (Cy3) or fluorescein-isothiocyanate (FITC) was used diluted 1:250 in PBS, 3% NGS, 1% BSA, and 0.5% Tween 20. Then the sections were rinsed three times with PBS, 0.5% Tween 20 and finally they were mounted and cell nuclei were stained using Vectashield-DAPI (Vector Lab). The sections were examined with a fluorescence microscope (Axioplan 2 Imaging Zeiss) under conventional parameters and stacking optical sections to reconstruct 3D images (Apotome Zeiss System).
Cell culture
C2C12 cells were cultured in growing conditions in DMEM supplemented with 10% fetal bovine serum in a humidified atmosphere at 37 °C and 5% CO2. Once cell confluence was reached, differentiation was induced by substituting growing medium by differentiation DMEM supplemented with 1% horse serum.
Results
Structure of the novel isoform of delta-sarcoglycan
By screening of a mouse skeletal muscle cDNA library, we isolated cDNA clones of the δ-SG gene. The DNA sequence analysis of one of this clones showed a cDNA sequence with a different 30-end than the reported one for δ-SG mRNA. To determine the origin of this new sequence, we aligned it with the complete mouse δ-SG gene sequence. The analysis showed us a new δ-SG transcript identical to the reported transcript (δ-SG1) (GenBank Accession No. NM011891) from exon 1 to exon 5, but with a portion of intron 5 added to the 30-end sequence of exon 5(Figs. 1A and C). The analysis of this new δ-SG cDNA sequence by a computer program (DNA MAN Lynnon BioSoft) predicted the translation of a shorter protein, in which the 122 amino acids at the carboxy end of the δ-SG1 isoform were replaced by 10 new amino acids (EGFLNMQLAG) and a stop codon at position 532 of the coding sequence, codified by a portion of intron 5 (Fig. 1B). We named this new isoform δ-SG3. The δ-SD3 isoform inferred is composed of 177 amino acids, compared to the 289 amino acids of the full-length δ-SG1. It conserves the first two motifs of the three asparagine-linked N-glycosylation sites and lacks a portion of the C-terminal, that contains four cysteine residues that are a common feature of Cy3-mouse anti-rabbit (Jackson) of the sarcoglycans.
Fig. 1.
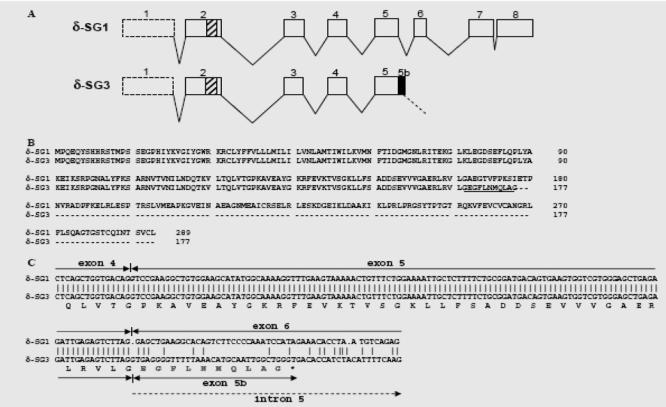
Gene structure of mouse SGCD gene and comparison of δ-SG1 and δ-SG3 transcript and protein sequences. (A) Structure of the mouse SGCD gene. The different exons (boxes) of δ-SG1 and δ-SG3 are represented. The black box indicates alternative splicing for exon 5 (exon 5b) in δ-SG3 isoform. Striped box in exon 2 indicates the region encoding the transmembrane domain. Untranslated exon 1 is indicated with dotted box. (B) Alignment of amino acid sequences of δ-SG1 and δ-SG3. The open-reading frame of d-SG1 encodes a polypeptide of 258 amino acids while δ-SG3 encodes a polypeptide of 177 amino acids. The new 10 amino acids at the carboxy end terminal of δ-SG3 are underlined. (C) Alignment of partial transcript sequence of δ-SG1 and δ-SG3 isoforms and predicted peptide sequence. Both isoforms share the same DNA sequence from exon 4 to exon 5, but in δ-SG3 this exon part of intron 5 is added by alternative splicing, exon 5b. The new 10 amino acids codified by this addition are shaded.
Expression profile of the d-SG1 and d-SG3 isoforms in different mouse tissues and cell lines
RT-PCRs were performed with total RNA from different mouse tissues to determine the mRNA expression of δ-SG3 (Fig. 2). As shown in Fig. 2A, both isoforms were detected in all tissues examined. The mRNA expression of both isoforms was most abundant in skeletal and cardiac muscle, where δ-SG1 and δ-SG3 show a similar expression level. In most other tissues, the δ-SG1 isoform is predominant, and in liver, small intestine, and brain, δ-SG3 is barely detectable. A middle expression was detected for both isoforms at lung and retina. To evaluate the differentiation-dependent expression of δ-SG3, we performed RTPCR using total RNA from myoblasts and myotubes of the C2C12 cell line (data not shown). The expression of δ-SG3 was only evident in myotubes, suggesting a development-dependent regulation.
Fig. 2.
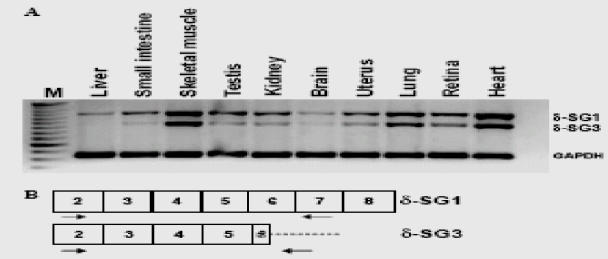
Transcript expression of δ-SG isoforms 1 and 3 in different mouse tissues. (A) RT-PCR results separately using δ-SG1- and δ-SG3 specific primer pair. Note that all tissues show more expression of δ-SG1 than δ-SG3, except for skeletal muscle and heart where both isoforms showed similar expression level. RT-PCR for δ-SG1 and δ-SG3 produced products of 654 and 530 bp, respectively. GAPDH was amplified as positive control and produced a PCR product of 275 bp. Lane M, 50 bp ladder; (−), negative control. (B) Schematic representation of the exon (boxes) primer localization used for the specific RT-PCR of δ-SG1-and δ-SG3 transcripts. Forward primer was the same for both isoforms, reverse primer for δ-SG1 was located in exon 7, and reverse primer for δ-SG3 was located in a portion of exon 5b.
Localization of the d-SG3 isoform in mouse skeletal muscle
To investigate the localization of the δ-SG3 isoform in mouse skeletal muscle, a specific rabbit polyclonal antibody was generated. The immunolabeling of mouse gastrocnemius cryostat longitudinal sections with this antiserum showed a striated pattern, resembling the sarcoplasmic reticulum structure (Figs. 3A, C and E). Furthermore, in skeletal muscle cross-sections a patched staining pattern was observed on the sarcolemma (Fig. 3F). Specific labeling was eliminated when the δ-SG3 isoform antiserum was preabsorbed with the 10-fold excess immunogenic peptide (Figs. 3B and D).
Fig. 3.
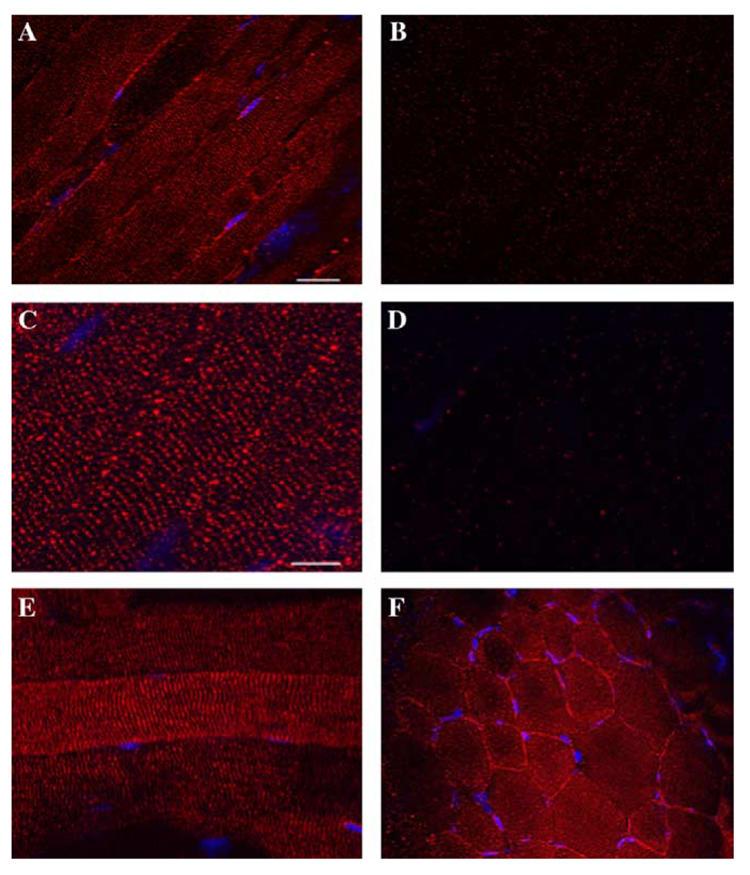
Immunolocalization of δ-SG3 protein in mouse skeletal and cardiac muscle. Longitudinal (A–C, and E) and cross-sections (F) from mouse skeletal muscle; and were immunolabeled with rabbit serum anti-δ-SG3. The longitudinal sections staining (A–C) shows a striated pattern. Note there is also a faint staining at the sarcolemma (A,C). The sarcolemma staining shows a punctate pattern in cross-sections (C). Immunolabeling of longitudinal sections with the antiserum preabsorbed with the synthetic peptide used to immunize the rabbit did not show staining (B,C).
A previous report showed the presence of δ-SG in SR [14] similar to that observed by us in the case of d-SG3. However, those authors used an anti-δ-SG antibody that recognizes the N-terminal of both isoforms. This fact makes it impossible to distinguish between δ-SG1 and δ-SG3. To assess this situation, we performed double-labeling with δ-SG3 antiserum and an antibody that recognizes the C-terminal of δ-SG1. While staining of δ-SG3 showed a scarce and irregular distribution along the sarcolemma (Figs. 4A and D), the δ-SG1 signal was more intense and with a regular distribution (Figs. 4B and E). In longitudinal sections, it was observed that the δ-SG isoforms are distributed in a similar striated pattern (Figs. 4D–F), suggesting that both δ-SG isoforms are localized in SR. Merge image revealed a close positioning, but no colocalization of δ-SG1 and δ-SG3 in pairs (Fig. 4F). This pattern was confirmed through the acquisition of 3D images (AxioVision, Carl Zeiss) (Figs. 4G and H). Interestingly, the δ-SG3 isoform seems to be more abundant in SR than δ-SG1 (Fig. 4H).
Fig. 4.
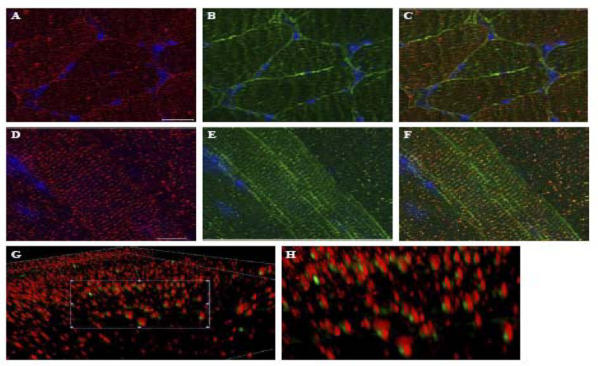
Double immunofluorescence analysis for δ-SG3 and δ-SG1. Transversal (A–C) and longitudinal (D–G) cryosections of mouse skeletal muscle were double labeled with antibodies against δ-SG3 and δ-SG1. Panels show images obtained for δ-SG3 (A,D), δ-SG3 (B,E), and merge d-SG3/d-SG1 (C,F). 3D images in two magnifications of the merge longitudinal section staining (G,H) show the nearness of both isoforms, and that δ-SG1 is always accompanied
To explore the striated pattern of δ-SG3 in skeletal muscle more in detail, double-labeling experiments were carried out with antibodies against δ-SG3 and the DHPR (a transverse tubule marker). As observed in Figs. 5A–D, δ-SG3 and DHPR presented a similar distribution. A merged image shows that they do not colocalize (Fig. 5C). Nevertheless, 3D images of the same region, acquired with a higher magnification (AxioVision, Carl Zeiss), showed that δ-SG3 is located in SR, next to the DHPR (Fig. 5D).
Fig. 5.
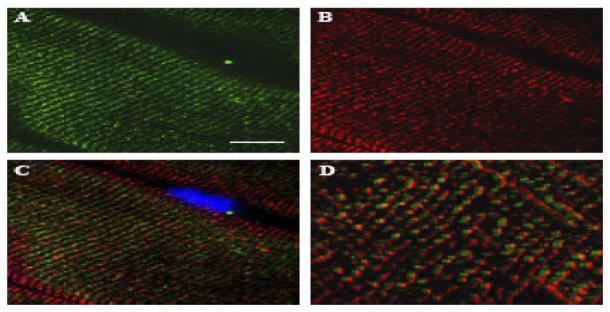
Double immunofluorescence analysis for δ-SG3 and DHPR. Longitudinal cryosections of mouse skeletal muscle were double labeled with antibodies against δ-SG3 and DHPR. Panels show images obtained for DHPR (A), δ-SG3 (B), and merge DHPR/δ-SG3 (C). A higher magnification of the merge image (D) shows that δ-SG3 is always accompanied by DHPR.
Discussion
In the present study, we identified a novel isoform of δ-SG (δ-SG3) expressed in mouse skeletal and cardiac muscle. The δ-SG3 isoform is produced by an alternative splicing that unprocessed part of intron 5 in the 3′-end sequence of exon 5. The predicted peptide sequence of the new isoform conserves the N-terminal, the transmembranal domain, misses the last 122 amino acids of the C-terminal, and has instead 10 new amino acids. The loss of the amino acids at the C-terminal causes the elimination of one N-glycosylation site and the four cysteine residues. Holt and Campbell [20] have shown that glycosylation and a proper folding of the sarcoglycans is important for the assembly of the sarcoglycan complex. Therefore, the absence of one Nglycosylation site in the δ-SG3 isoform could modify its folding properties and its capabilities to interact with other members of the SG-SSPN complex. On the other hand, it has been suggested that the cysteine residues at the carboxy end of β-, χ-, and δ-SG may function as receptors for some by δ-SG3 unknown ligands [13,21]. Consequently, the elimination of these residues in the δ-SG3 isoform could modify its capacity of interaction with other proteins.
Previously, Ueda et al. [14] demonstrated that the expression of δ-and χ-SGs in the SR of skeletal muscle is independent of dystrophin and the rest of the sarcoglycans. In concert with our results, this suggests that the lack of the δ-SG1-C-terminal in δ-SG3 could confer to the protein the capacity to form an alternative complex, at least in skeletal muscle. We also suggest that in this tissue the association of the δ-SG isoforms with β-SG is not required for the maturation of the complex with χ-SG. This is opposite to what Shi et al. [22] reported. They demonstrated that the assembly pathway of the sarcoglycans depends on β-and δ-SG. Thus, it will be interesting to explore the biochemical interactions of δ-SG3 with the other components of the DAGC and to compare the complexes formed with those formed by δ-SG1.
Immunofluorescence images of longitudinal mouse skeletal muscle cryosections revealed that δ-SG3 is mostly expressed in a striated pattern excluded from the sarcolemma, in clear contrast with δ-SG1 that is detected along the muscle membrane. A similar localization of δ-SG has been observed by Ueda et al. [14] using a pan-specific antibody. By confocal and electronic microscopy they demonstrated that δ-SG is located at the central portion of the I bands, in the terminal cisternae of the SR. Our double immunofluorescence assays with an antiserum against δ-SG3 and a polyclonal antibody against the carboxy terminus of δ-SG1 suggest that both, δ-SG3 and d-SG1, are located at the SR. Besides, double immunofluorescence with anti-δ-SG3 and anti-DHPR antibodies showed that δ-SG3 localizes very close to the DHPR. As is well known, contraction is initiated by the depolarization of the T-tubular membrane, where the DHPR act as voltage sensors. This in turn causes Ca2+ to be released by the ryanodyne receptor (RyR) Ca2+ channels of the SR [23–25]. Recently, Iwata et al. [26] demonstrated that the damage to skeletal muscle in BIO14.6 hamsters that lack δ-SG is due mainly to the deregulation of the intracellular Ca2+ homeostasis that can be significantly prevented by Ca2+ handling drugs. Besides, Ueyama et al. [27] reported that the BIO14.6 hamster has an abnormal RyR expression in the cardiac muscle, suggesting abnormal calcium regulation caused by δ-sarcoglycan deficiency. Taking in account all this results, we consider a possible role of δ-SG3 in muscle contraction and/or in Ca2+ regulation mechanisms. Our hypothesis points towards a structural role related to the correct localization and maintenance of the proteins that participate in the regulation of the calcium concentration during the contraction-relaxation cycles at the terminal cisternae of the SR and very likely through an interaction with the Ca2+ channels. Further studies will be necessary to clarify this issue, localizing δ-SG3 more accurately, specifically at the triad junction, where it seems to form a complex with a different composition than in sarcolemma.
Acknowledgments
F. J. Estrada was supported by the Consejo Nacional de Ciencia y Tecnología (CONACYT) Register Number 165401.
Abbreviations
- SG
sarcoglycan
- DAGC
dystrophin-associated glycoprotein complex
- SSPN
sarcospan
- SR
sarcoplasmic reticulum
- DHPR
dihydropyridine receptor
- LGMD
limb girdle muscular dystrophy
- RyR
ryanodyne receptor
References
- 1.Angelini C. Advances and perspectives in muscular dystrophies. Basic Appl Myol. 2002;12:17–25. [Google Scholar]
- 2.Coral-Vazquez RM, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, Straub V, Barresi R, Bansal D, Hrstka RF, Williamson R, Campbell KP. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell. 1999;98:465–474. doi: 10.1016/s0092-8674(00)81975-3. [DOI] [PubMed] [Google Scholar]
- 3.Crosbie RH, Heighway J, Venzke DP, Lee JC, Campbell KP. Sarcospan, the 25-kDa transmembrane component of the dystrophin-glycoprotein complex. J Biol Chem. 1997;272:31221–31224. doi: 10.1074/jbc.272.50.31221. [DOI] [PubMed] [Google Scholar]
- 4.Straub V, Campbell KP. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr Opin Neurol. 1997;2:168–175. doi: 10.1097/00019052-199704000-00016. [DOI] [PubMed] [Google Scholar]
- 5.Sciandra F, Bozzi M, Bianchi M, Pavoni E, Giardina B, Brancaccio A. Dystroglycan and muscular dystrophies related to the dystrophin-glycoprotein complex. Ann Ist Super Sanita. 2003;39:173–181. [PubMed] [Google Scholar]
- 6.Hack AA, Groh ME, McNally EM. Sarcoglycans in muscular dystrophy. Microsc Res Tech. 2000;48:167–180. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<167::AID-JEMT5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 7.Ettinger AJ, Feng G, Sanes JR. Epsilon-Sarcoglycan, a broadly expressed homologue of the gene mutated in limb-girdle muscular dystrophy. J Biol Chem. 1997;272:32534–32538. doi: 10.1074/jbc.272.51.32534. [DOI] [PubMed] [Google Scholar]
- 8.McNally EM, Ly CT, Kunkel LM. Human epsilon-sarcoglycan is highly related to alpha-sarcoglycan (adhalin), the limb girdle muscular dystrophy 2D gene. FEBS Lett. 1998;422:27–32. doi: 10.1016/s0014-5793(97)01593-7. [DOI] [PubMed] [Google Scholar]
- 9.Straub V, Ettinger AJ, Durbeej M, Venzke DP, Cutshall S, Sanes JR, Campbell KP. e-Sarcoglycan replaces a-sarcoglycan in smooth muscle to form a unique dystrophin-glycoprotein complex. J Biol Chem. 1999;274:27989–27996. doi: 10.1074/jbc.274.39.27989. [DOI] [PubMed] [Google Scholar]
- 10.Zimprich A, Grabowski M, Asmus F, Naumann M, Berg D, Bertram M, Scheidtmann K, Kern P, Winkelmann J, Muller-Myhsok B, et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet. 2001;29:66–69. doi: 10.1038/ng709. [DOI] [PubMed] [Google Scholar]
- 11.Wheeler MT, Zarnegar S, McNally EM. f-Sarcoglycan, a novel component of the sarcoglycan complex, is reduced in muscular dystrophy. Hum Mol Genet. 2002;11:2147–2154. doi: 10.1093/hmg/11.18.2147. [DOI] [PubMed] [Google Scholar]
- 12.Anastasi G, Cutroneo G, Sidoti A, Santoro G, D’Angelo R, Rizzo G, Rinaldi C, Giacobbe O, Bramanti P, Navarra G, Amato A, Favaloro A. Sarcoglycan subcomplex in normal human smooth muscle: an immunohistochemical and molecular study. Int J Mol Med. 2005;16:367–374. [PubMed] [Google Scholar]
- 13.Chan YM, Bonnemann CG, Lidov HGW, Kunkel LM. Molecular organization of sarcoglycan complex in mouse myotubes in culture. J Cell Biol. 1998;143:2033–2044. doi: 10.1083/jcb.143.7.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ueda H, Ueda K, Baba T, Ohno S. d-and c-Sarcoglycan localization in the sarcoplasmic reticulum of skeletal muscle. J Histochem Cytochem. 2001;49:529–537. doi: 10.1177/002215540104900413. [DOI] [PubMed] [Google Scholar]
- 15.Nigro V, Piluso G, Belsito A, Politano L, Puca AA, Papparella S, Rossi E, Viglietto G, Esposito MG, Abbondanza C, Medici N, Molinari AM, Nigro G, Puca GA. Identification of a novel sarcoglycan gene at 5q33 encoding a sarcolemmal 35 kDa glycoprotein. Hum Mol Genet. 1996;5:1179–1186. doi: 10.1093/hmg/5.8.1179. [DOI] [PubMed] [Google Scholar]
- 16.Jung D, Duclos F, Apostol B, Straub V, Lee JC, Allamand V, Venzke DP, Sunada Y, Moomaw CR, Leveille CJ, Slaughter CA, Crawford TO, McPherson JD, Campbell KP. Characterization of d-sarcoglycan, a novel component of the oligomeric sarcoglycan complex involved in limb-girdle muscular dystrophy. J Biol Chem. 1996;271:32321–32329. doi: 10.1074/jbc.271.50.32321. [DOI] [PubMed] [Google Scholar]
- 17.Leiden Muscular Dystrophy pages. Available from: < http://www.dmd.nl/>.
- 18.Sakamoto A, Abe M, Masaka T. Delineation of genomic deletion in cardiomyopathic hamster. FEBS Lett. 1999;447:124–128. doi: 10.1016/s0014-5793(99)00267-7. [DOI] [PubMed] [Google Scholar]
- 19.Rivier F, Robert A, Royuela M, Hugon G, Bonet-Kerrache A, Mornet D. Utrophin and dystrophin-associated glycoproteins in normal and dystrophin deficient cardiac muscle. J Muscle Res Cell Motil. 1999;20:305–314. doi: 10.1023/a:1005426920070. [DOI] [PubMed] [Google Scholar]
- 20.Holt KH, Campbell KP. Assembly of the sarcoglycan complex. Insights for muscular dystrophy. J Biol Chem. 1998;273:34667–34670. doi: 10.1074/jbc.273.52.34667. [DOI] [PubMed] [Google Scholar]
- 21.McNally EM, Duggan D, Gorospe JR, Bonnemann CG, Fanin M, Pegorato E, Lidov HG, Noguchi S, Ozawa E, Finkel RS, Cruse RP, Angelini C, Kunkel LM, Hoffman EP. Mutations that disrupt the carboxyl-terminus of gamma-sarcoglycan cause muscular dystrophy. Hum Mol Genet. 1996;5:1841–1847. doi: 10.1093/hmg/5.11.1841. [DOI] [PubMed] [Google Scholar]
- 22.Shi W, Chen Z, Schottenfeld J, Stahl RC, Kunkel LM, Chan YM. Specific assembly pathway of sarcoglycans is dependent on beta-and delta-sarcoglycan. Muscle Nerve. 2004;29:409–19. doi: 10.1002/mus.10566. [DOI] [PubMed] [Google Scholar]
- 23.Manttari S, Jarvilehto M. Comparative analysis of mouse skeletal muscle fibre type composition and contractile responses to calcium channel blocker. BMC Physiol. 2005;5:4. doi: 10.1186/1472-6793-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beam KG, Tanabe T, Numa S. Structure, function and regulation of skeletal muscle dihydropyridine receptor. Ann NY Acad Sci. 1989;560:127–137. doi: 10.1111/j.1749-6632.1989.tb24090.x. [DOI] [PubMed] [Google Scholar]
- 25.Manttari S. Oulu University Press; 2005. Dihidropyridine receptors in skeletal muscle with comparative reference to muscle development and exercise in mouse and salmon. PDF < http://herkules.oulu.fi/isbn9514277201/>. [Google Scholar]
- 26.Iwata Y, Katanosaka Y, Shijun Z, Kobayashi Y, Hanada H, Shigekawa M, Wakabayashi S. Protective effect of Ca2+ handling drugs against abnormal Ca2+ homeostasis and cell damage in myopathic skeletal muscle cells. Biochem Pharmacol. 2005;70:740–751. doi: 10.1016/j.bcp.2005.05.034. [DOI] [PubMed] [Google Scholar]
- 27.Ueyama T, Ohkusa T, Hisamatsu Y, Nakamura Y, Yamamoto T, Yano M, Maatsuzaki M. Alterations in cardiac SR Ca(2+)-release channels during development of heart-failure in cardiomyopathic hamsters. Am J Physiol. 1998;274:1–7. doi: 10.1152/ajpheart.1998.274.1.H1. [DOI] [PubMed] [Google Scholar]