

Abstract
The sarcoglycan–sarcospan (SG–SSPN) complex is part of the dystrophin-glycoprotein complex that has been extensively characterized in muscle. To establish the framework for functional studies of sarcoglycans in retina here, we quantified sarcoglycans mRNA levels with real-time reverse transcriptase-polymerase chain reaction (RT-PCR) and performed immunohistochemistry to determine their cellular and subcellular distribution. We showed that the β-, δ-, γ-, ε-sarcoglycans and sarcospan are expressed in mouse retina. They are localized predominantly in the outer and the inner limiting membranes, probably in the Müller cells and also in the ganglion cells axons where the expression of dystrophins have never been reported. We also investigated the status of the sarcoglycans in the retina of mdx3cv mutant mice for all Duchene Muscular Dystrophy (DMD) gene products. The absence of dystrophin did not produce any change in the sarcoglycan–sarcospan components expression and distribution.
Keywords: Animals; Carrier Proteins; metabolism; Cells, Cultured; Dystrophin; metabolism; Gene Expression Regulation; physiology; Glutamate Synthase; metabolism; Immunohistochemistry; methods; Macromolecular Substances; metabolism; Membrane Proteins; metabolism; Mice; Mice, Inbred C57BL; Mice, Inbred mdx; Neoplasm Proteins; metabolism; Neurofilament Proteins; metabolism; Neuroglia; metabolism; RNA, Messenger; biosynthesis; Retina; cytology; metabolism; Retinal Ganglion Cells; metabolism; Reverse Transcriptase Polymerase Chain Reaction; methods; Sarcoglycans; metabolism
Introduction
The dystrophin associated protein complex (DAPC) is a complex conformed by the interaction of multiple proteins that associate intimately with dystrophin at the sarcolemma of skeletal, cardiac and smooth muscle (Campbell, 1995; Straub et al., 1999; Yoshida and Ozawa, 1990). The sarcoglycan–sarcospan (SG–SSPN) and the dystroglycan complexes are essential components of the DAPC, along with syntrophins, dystrobrevins and a diverse group of signaling proteins (Blake et al., 2002). Through its attachment to the extracellular matrix, the DAPC provides a mechanosignaling linkage between the outside compartment of muscle cells and the intracellular cytoskeleton (Ervasti and Campbell, 1993).
The absence of dystrophin leads to the disorganization of the DAPC and especially to the decrease of expression of the sarcoglycans (SGs) in muscle (Chamberlain et al., 1997). The SGs are asparagine-linked glycosylated proteins with single transmembrane domains (Ozawa et al., 1998) that form complex mediating muscle membrane stability (Hack et al., 2000). The integrity of the SG–SSPN complex requires the coordinated translation and assembly of its subunits. Perturbations in this complex lead to limb girdle muscular dystrophies and cardiomyopathies in both humans and mice (Bonnemann et al., 1995; Coral-Vazquez et al., 1999; Lim et al., 1995; Nigro et al., 1996; Noguchi et al., 1995; Roberds et al., 1994). Six sarcoglycan genes and gene products have been identified to date. According to their topology, a-sarcoglycan (a-SG) and e-sarcoglycan (e-SG) are highly similar type I glycosilated transmembrane proteins, whereas g-sarcoglycan (g-SG), d-sarcoglycan (d-SG) and z-sarcoglycan (z-SG) are type II glycosylated transmembrane proteins, highly similar among themselves but weakly similar to b-sarcoglycan, consistent with the idea of multiple gene duplication events (Fanin et al., 1997; Wheeler et al., 2002). Besides, SSPN is a protein with four transmembranal domains, structurally related to the tetra-span family of proteins, a group of proteins thought to be molecular facilitators (Crosbie et al., 1997). SSPN copurifies with the DAPC, and its expression is significantly reduced when the SGs expression is altered, which confirms their association as a complex (Coral-Vazquez et al., 1999).
Dystrophin is the product of the Duchenne Muscular Dystrophy (DMD) gene, which is located at Xp21 in human (Koenig et al., 1988). Mutations in the DMD gene are responsible of one of the most common inherited myodegenerative diseases, and affect about one in 3500 male newborn. In addition to the severe muscle phenotype, the majority of DMD patients also present certain developmental, behavioral and cognitive deficits indicating that the DGC also play a crucial role during CNS function (Lidov, 1996; Mehler, 2000). Indeed dystrophins, utrophins, dystroglycans and components of the cytoplasmic complex (i.e. syntrophins, dystrobrevins) are found in many tissues, among them the central nervous system (CNS) including brain and retina (Blake and Kroger, 2000; Mehler, 2000; Ueda et al., 2000). In addition to the full-length dystrophin isoforms, internal promoters activation in the DMD gene leads to the expression of four different short products called according to their apparent molecular mass, Dp260, Dp140, Dp116 and Dp71. These products present different tissue-specific pattern of expression. Dp71 is the major DMD gene product in the central nervous system, including retina (Rodius et al., 1997). One of the best characterized deficits in the CNS of DMD patients is the altered dark-adapted electroretinogram (ERG). Eighty percent of DMD patients as well as the mdx3cv mouse strain which suffer a drastic reduction of all the DMD gene products, show a delayed implicit time and a reduced amplitude of the b-wave selectively in the scotopic ERG (Fillers et al., 1995; Sigesmund et al., 1994).
We and others have previously shown that except Dp116, dystrophins and some of the DAPs are expressed in rat, mouse and rabbit retinae (Claudepierre et al., 1999; D’Souza et al., 1995; Koulen et al., 1998; Montanaro et al., 1995; Ueda et al., 1995). In the main glial cell of the retina - the Müller glial cells (MGC) - we have demonstrated the existence of a γ-SG containing complex that includes Dp71, β-dystroglycan and a1-syntrophin (Claudepierre et al., 2000), and suggested that the subcellular distribution of the inwardly rectifying potassium channel Kir4.1 and the water pore aquaporin 4 (AQP4) might depend on the activity of this complex (Dalloz et al., 2003). However, thus far only the expression of δ-and γ-sarcoglycans in retina (Claudepierre et al., 2000) and e-sarcoglycan in brain (Xiao and LeDoux, 2003) has been demonstrated in the CNS. Studying the expression and the cellular localization of the SG–SSPN complex in mouse retina would help us to identify the possible functional correlations between sarcoglycans and the dystrophins–dystroglycan complex, as well as to elucidate the molecular basis of the retinal phenotypes of the DMD. In this study, we employed real-time reverse transcriptase-polymerase chain reaction (RT-PCR), and immunohistochemistry to determine the SGs–SSPN distribution in mouse retina, and by using the mdx3cv mice strain as model, we determined that the sarcoglycans distribution seems to be dystrophins-independent.
Materials and methods
Animals
C57B1/6 control and mdx3cv mutant mice strains were bred in our laboratory. mdx3cv originated from mice generously provided by the late Dr. Verne M. Chapman (Roswell Park Memorial Institute, Buffalo, NY). Mice were identified by PCR analysis as described by Cox et al. (1993). Immunoblot analysis with Dys2 antibody (Novocastra Laboratories) was performed with brain total protein extracts to verify the mdx3cv phenotype. All experiments were in compliance with the European Communities Council Directives (86/609/EEC) for animal care and experimentation.
Real-time RT-PCR
Total RNA from mice retina was extracted using Trizol reagent (Invitrogen-Life Technologies) according to the manufacturer’s instructions. One microgram of total RNA was reverse transcribed using random hexamers and Superscript II (Invitrogen-Life Technologies). Real-time PCR was done using the LightCycler system (Roche Diagnostics, USA) with SYBR Green detection and Tm analysis. LightCycler FastStart DNA Master-PLUS SYBR Green I kit was purchased from Roche Diagnostics (Roche Diagnostics GmbH). Amplification was performed in a total volume of 20 ml containing the Master mix (including Taq DNA polymerase, reaction buffer, MgCl2, dNTP mix and SYBR Green dye), 0.5 mM of each primer (Table 1) and 1/100 of the cDNA previously obtained. Amplification conditions consisted of 40 cycles (95 8C for 0s, 60 8C for 5 s and 72 8C for 20 s).
Table 1.
Primers used for RT-PCR
| Name | Sequence (50 ! 30) | mRNA detected |
|---|---|---|
| AlphaSG-F | GCCGAGTCCCTCTTCCTATT | a-SG |
| AlphaSG-R | CCAGAGACACATTGCACCAG | |
| BetaSG-F | AGCATGGAGTTCCACGAGAG | b-SG |
| BetaSG-R | GCTGGTGATGGAGGTCTTGT | |
| DeltaSG-F | GTCAGAGCAGACCCCTTCAA | d-SG |
| DeltaSG-R | GATCCACGAGGCAGTCTAGC | |
| GamaSG-F | GAGACACCCCTTGTCAGAGC | g-SG |
| GamaSG-R | TGGTCAAACCCACAGTTTCA | |
| EpsilonSGF | TCCATCACAGCTCGATTCAG | e-SG |
| EpsilonSR | TCTGAGTCTGGTGTGGCAAG | |
| SSPN-F | AGAGGACTTGCTGCTCTTGC | SSPN |
| SSPN-R | CCTTTCGGTGTTCACCAAGT | |
| Dp71-F1 | ACCATGAGGGAACACCTCAAAGG | Dp71 |
| Dp71-R1 | TGCAGCTGACAGGCTCAAGCGGAT | |
| Vimentin-F | TTGAGATCGCCACCTACAGGA | Vimentin |
| Vimentin-R | TCCGTCTCTGGTTTCAACCGT | |
| Thy1-F | CAGGACGGAGCTATTGGCACCAT | Thy1 |
| Thy1-R | ACGGCAGTCCAGTCGAAGGTTCT | |
| Actin-F | CTGGGACGACATGGAGAAGATCTG | Actin |
| Actin-R | CGTACTCCTCCTTGGTGATCCAC |
Semi-quantification of the real-time RT-PCR products
To identify PCR products generated in the presence of SYBR Green, a Tm analysis was performed by increasing the temperature from 60 to 90 8C at a linear transition rate of 0.1 8C/s. Fluorescence of the samples was monitored continuously while the temperature was increased. SYBR Green was released upon denaturation, which resulted in a decreasing fluorescence of the signal. The LC software calculated the Tm, i.e. the rate of change in fluorescence (dF/dT) was displayed as a function of temperature. The LC software also calculated the crossing point that indicated the relative expression of each product observed. b-Actin messenger expression was used to normalize the values and the results were calculated as relative expression with respect to actin as 100%.
Antibodies
Antibodies against the SG–SSPN complex used in this study have been previously described (Rivier et al., 1999) and are summarized in the Table 2. The antibodies used against protein cell markers were the anti-glutamine synthetase monoclonal antibody from Chemicon (CA, USA) for Müller glial cells, and the anti-NF68 monoclonal antibody from Sigma (Saint-Quentin Fallavier, France) for ganglion cells.
Table 2.
Sarcoglycans/sarcospan antibodies used for immunohistochemistry assays
| Antibody | Nature | Peptide position | Specificity |
|---|---|---|---|
| Sarco3 | Polyclonal | C-terminal 7aa | α-Sarcoglycan |
| G4 | Polyclonal | Peptide 42–52 | β-Sarcoglycan |
| Nini | Polyclonal | C-terminal 84–290 | δ-Sarcoglycan |
| Peptide 1 | Polyclonal | Peptide 2–13 | γ-Sarcoglycan |
| LG7 | Polyclonal | C-terminal 11aa | ε-Sarcoglycan |
| C5 | Polyclonal | C-terminal 13aa | SSPN |
Tissue preparation and immunohistochemistry
After enucleation and removal of cornea and lens, mice eyes were fixed 3 min in 4% paraformaldehyde. Eyes were cryoprotected before embedding in freezing medium (cryoblock, Labonord) and frozen in liquid nitrogen. They were vertically sliced at 7 mm thickness in a cryostat and placed on gelatin-coated slide glasses. Immunocytochemical labeling was carried out using the indirect immunofluorescence method as previously reported (Dalloz et al., 2003). After mounting in Gel/Mount (Biomeda, Foster City, CA) double labeling were examined and photographed using a confocal laser scanning equipped with an argon-kripton laser (TCS SP1 Leica, Lasertechnik GmbH). Controls were prepared by omitting the primary antibody during the incubation; in these controls, no specific staining could be detected.
Müller cell culture
Primary retinal glial cell cultures were prepared from 2week-old C57B1/6 mice as described by Hicks and Courtois (Hicks and Courtois, 1990).
Ganglion cell culture
Primary cultures of retinal ganglion cells (RGC) were derived from 2-month-old C57B1/6 mice retinae and purified by sequential immunopanning as described for young rat retinae (Barres et al., 1988).
Results and Discussion
In order to investigate the status of the members of the SG–SSPN complex in the retina and to examine the molecular and cellular consequences of the absence of dystrophins for individual SGs expression, here we evaluated the relative sarcoglycan mRNAs and proteins expression in wild-type (wt) C57B1/6 and mdx3cv mice retinae.
Real-time RT-PCR was performed on whole retinal preparation from both strains.
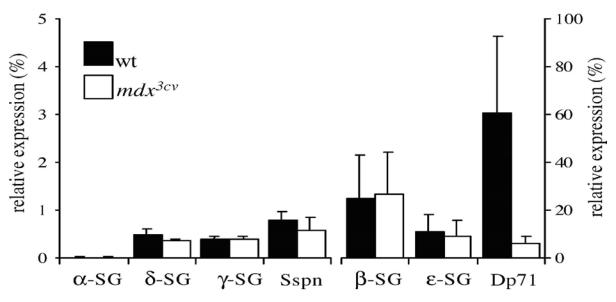
Positive controls were performed by classical RT-PCR with mice muscle extracts. All the amplification products had the expected size (data not shown). Fig. 1 revealed that the mRNAs of β-, δ-, γ-, ε-SGs and SSPN were expressed in wt mice retinae at various levels. The α-and ε-SGs transcripts were the most abundantly expressed. Comparative analysis of the transcript levels between the wt and the mdx3cv strains showed that there was not a significant difference in levels of expression of neither the SGs nor the SSPN mRNAs. The analysis of protein expression by Western blot show similar results to those found for transcripts (data not shown). It should be noted that no a-SG transcript or protein was detected in mice retinae from wt and mdx3cv strains. On the other hand, as previously reported, we noted a great reduction of the Dp71 mRNA in mdx3cv with respect to the wt (Fig. 1), whereas smaller perturbations were observed for the other DMD gene products expressed in retina (data not shown).
Fig. 1.
To assess the retinal distribution of SGs in presence or absence of dystrophins, we performed immunostaining assays with polyclonal antisera specific for single SGs and SSPN in serial retinal sections of wt and mdx3cv mice strains. Fig. 2 shows that the pan-specific dystrophins antibody H4, revealed in retinae of wt mice a punctuate signal in the outer plexiform layer (OPL, arrows), a staining around the blood vessels and at the inner limiting membrane (ILM, arrow heads). This labeling disappears in the mdx3cv mice strain, which confirmed that in spite of the fact that the dystrophins mRNAs are detected at different levels in the mdx3cv strain, this mutation resulted in a dramatic reduction of protein expression.
Fig. 2.

Immunohistochemical analysis of the dystrophins and sarcoglycan–sarcospan complex components in wild-type (C57B1/6) and mdx3cv mice retinae. Fluorescence photomicrographs of vertical sections through mouse retinae immunostained with antibodies directed against dystrophins, e-SG, b-SG, d-SG, g-SG and SSPN, respectively. OLM, outer limiting membrane; ONL, outer nuclear layer; OPL, outer plexiform layer; ENL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer; ILM, inner limiting membrane. Scale bar: 20 mm. Arrows indicate punctuate dystrophins staining in the OPL while arrowheads indicate staining around blood vessels and at the ILM. Small arrows indicate SGs–SSPN staining at radial processes terminating at the OLM.
It has been shown that from the SG-family members, which are largely restricted to muscle, the e-SG is highly expressed in many tissues, including brain (McNally et al., 1998). In mice retinae, the anti e-SG antibody, LG7, clearly revealed that e-SG is highly expressed at the vitreal surface in the ganglion cell layer (GCL) and ILM. This localization could correspond to a distribution at the soma of ganglion cells, the nerve fiber layer (NFL) and/or at the MGC end feet in both wt and mdx3cv mice strains. It was also observed a diffuse labeling at the inner nuclear layer (INL), at the OPL and at radial processes terminating at the outer limiting membrane (OLM, small arrows). As shown in Figs. 2 and 3, the overall distribution pattern of b-, d-, g-SGs and SSPN immunoreactivity did not differ from the one observed for e-SG, with the particularity that e-SG is the most highly expressed at both edges of the retina. In addition, as expected from the results obtained by real-time RT-PCR (Fig. 1), no positive staining was detected for the a-SG neither in wt nor in mdx3cv mice (data not shown).
Fig. 3. Immunohistochemical analysis of the cellular localization of dystrophins, sarcoglycans and sarcospan in wild-type (C57B1/6) mouse retina.

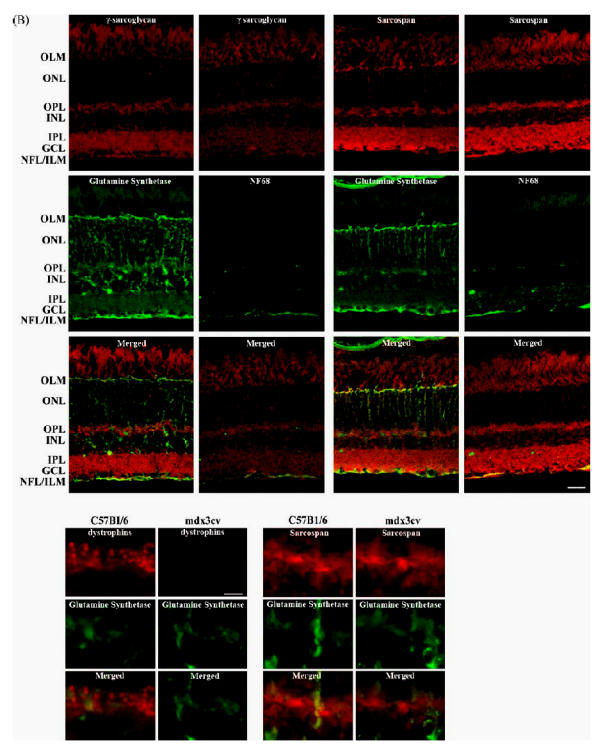
(A) Fluorescence photomicrographs of vertical sections through mouse retina double immunolabeled for dystrophins, ε-SG, β-SG, δ-SG, γ-SG and SSPN in red and a cell marker of Müller glial cells, glutamine synthetase; or of ganglion cells axons, neuro-filament 68 (NF68) in green. OLM, outer limiting membrane; OPL outer plexiform layer; ENL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer; NFL, nerve fiber layer; ILM, inner limiting membrane. Scale bar: 20 mm.
Arrows indicate the considerable overlap between dystrophins or ε-SG and the glutamine synthetase staining at the ILM while arrowheads indicate the overlap between ε-SG and the glutamine synthetase staining observed at the MGC fibers and at the OLM. Stars indicate the overlap of ε-SG and NF68 staining at the NFL.
(B) Dystrophins and SSPN immunolocalizations in the OPL are shown at higher magnification. The bars and puncta staining of dystrophins observed in wt mice retinae clearly disappear in mdx3cv mice retinae. On the contrary, no differences could be established between the SSPN staining from wt and mdx3cv mice retinae. Scale bar: 6 mm.
It has been established that in the mouse retina, the Dp427, Dp260 and Dp140 are concentrated within the OPL (Howard et al., 1998), in two morphological shapes, bars and puncta most likely corresponding to the labeling of photoreceptors terminals and/or dendritic processes of bipolar neurons. In this study, we were unable to neither detect this kind of specific immunoreactivity with antibodies against SGs and SSPN, in the wt nor in the mdx3cv mice (compare immunostaining in Figs. 2 and 3B for dystrophins and SSPN). Although under the basis of this result, we cannot completely exclude the participation of SGs and SSPN at the formation of functional complex with dystrophins at the OPL, this observation is not in favor of the constitution of such a complex.
Thus, our results suggest that opposite to what is known for skeletal muscle (Chamberlain et al., 1997), the expression and localization of SGs and SSPN in retina are if not totally, at least partially independent of the dystrophins-expression. In order to explore in more detail this hypothesis, we performed double labeling experiments in which the immunostaining of dystrophins, SGs and SSPN was combined with the labeling of glutamine synthetase or the neurofilament subunit of 68 kDa (NF68), specific markers of MGC and axons of ganglion cells, respectively. In wt retina, double immunolabeling with the pan-specific dystrophins antibody H4 and the anti-glutamine synthetase shows strongly stained glutamine synthetase positive MGC spanning the whole thickness of the retina from the OLM to the ILM. A considerable overlap was observed at the ILM (Fig. 3, arrows) confirming our previous observation of the localization of the Dp71 at the end feet of the MGC (Dalloz et al., 2003). It should be noted that no staining of dystrophins was evident at the OLM or in the MGC fibers stained by the glutamine synthetase antibody in the outer retina. We therefore investigated if the dystrophins may be colocalized with the NF68 at ganglion cell fibers. In wt retina, double immunolabeling with the pan-specific dystrophins antibody H4 and the anti-NF68 shows strongly stained NF68 positive nerve fibers without overlapping with the dystrophins staining. To ascertain whether or not the dystrophins were expressed by the MGC and the ganglion cells, we performed a comparative study by RT-PCR of their mRNA expression in whole retina, cultured MGC and cultured ganglion cells. We failed to detect the expression of Dp427, Dp260 and Dp140 in both cultured cell types, whereas the transcript of Dp71 was not expressed in the cultured ganglion cells, but was detected in the cultured MGC (data not shown).
We further compared in wt retinae the double immunolabeling of each one of the SGs and SSPN with either the labeling of the glutamine synthetase or the NF68. Similarly to what was found with dystrophins, a considerable overlapping was observed at the ILM between the ε-SG and the glutamine synthetase (Fig. 3, arrows). However, in this case, ε-SG expression was also noted within the external retina, where an overlap of the staining was also observed at the MGC fibers and the OLM (Fig. 3, arrowheads). A similar localization pattern was observed in thecaseof β-, δ-, γ-SGs and SSPN (Fig. 3). These results show that whereas Dp71 and all the SGs and SSPN are localized together at the end feet of the MGC, only the SGs and SSPN are present in the outer retina. It is tempting to speculate that as reported in skeletal and cardiac muscles situation (Straub et al., 1999; Wheeler et al., 2002), it would be possible that two distinct sarcoglycans complexes co-exist in MGC, one composed of Dp71 and the well-characterized tetrameric unit of ε-, β-, γ-and δ-SGs, and a second one that would not be associated with Dp71, composed of SGs, and yet unidentified proteins.
We thereafter investigated whether the ε-SG immunostaining overlaps or not with NF68. On the contrary to what was observed in the case of dystrophins, both immunoreactivities were detected at the NFL, thus suggesting that ε-SG is expressed by the ganglion cells (Fig. 3, stars). As expected, the comparison of the double immunolabeling of β- δ-, γ-SGs or SSPN and NF68 revealed a similar localization to that of ε-SG for all these proteins (Fig. 3). However, it should be remarked that the high immunofluorescence observed for ε-SG at the nerve fibers could mask the ganglion cells soma immunolabeling that is more evident for the other SGs and the SSPN. Thus, in retina, the dystrophins and the SGs–SSPN complex not only differ in their intracellular localization but they are also differentially expressed between glial and neuron cells. Since the dystrophins are not expressed by the ganglion cells, the SG–SSPN complex should be associated to yet unidentified partners.
It is known that at the sarcolemma, the SGs associate with the dystroglycan complex (Ibraghimov-Beskrovnaya et al.,, 1992). Because dystrophin and dystroglycan complex SSPN in retina may help to explain why the absence of interact with actin and laminin, respectively, the absence dystrophins in retina produces a relatively mild phenotype, of dystrophin from this molecular linkage has direct compared with the devastating muscle pathology. On the molecular consequences for the assembly of the DGC. basis of these observations, it could be possible to propose Thereafter, these molecular differences are associated with that a similar phenomenon could occur in other structures of progressive muscle degeneration that leads to skeletal and the CNS. Moreover, the absence of perturbations in the cardiac muscle weakness. The lack of an apparent expression and localization of the SG–SSPN complex in molecular association between dystrophins and SGs and retina observed here suggests that SGs and SSPN are not implicated in the ERG phenotype observed in mdx3cv mice strain.
Although the precise function of the SG–SSPN complex is not known, it is thought to have mechanical and non-mechanical roles that mediate interactions between the extracellular matrix, the membrane and the cytoskeleton (Hack et al., 1999). Interestingly, in retina, the expression of all the subunits is mostly restricted to the ganglion cell layer, including ganglion cells soma, optic nerve fibers and MGC end feet, but also the apical end of MGC forming the OLM. This localization could reflect a role of the SG–SSPN complex in the membrane of ganglion cells to form the optic nerve fiber and in the MGC to stabilize the limiting membranes of the retina with the retinal endothelium on one hand and the vitreous body on the other hand in order to stabilize the global structure of the retina, functional roles that still to be determined.
Conclusion
This study reveals the pattern of expression and localization of sarcoglycans and sarcospan mRNAs and proteins in the adult mouse retina. We showed here that b-, d-, g-, e-SGs and SSPN are expressed in mouse retina. Moreover, we determined that the sarcoglycans are localized predominantly at the outer and the inner edges of the retina, probably in the Müller glial cells and also in the ganglion cells soma and axons where the expression of dystrophins has never been reported. The use of the mdx3cv strain allowed us to conclude that the SG–SSPN complex expression and localization could be at least partially dystrophins independent. Clarification of the role of sarcoglycans and sarcospan in nervous system and particularly in retina awaits further investigation.
Acknowledgments
This work was supported by Institut National de la Santé Et de la Recherche Médicale (INSERM) and by grants from the Association Française contre les Myopathies (AFM) to A.R. and D.M.; P.P. received financial support from Fédération des Aveugles et handicapés visuels de France (FAF). P.P. and F.J.E from the AFM. A.B. received financial support from Retina France.
References
- Barres BA, Silverstein BE, Corey DP, Chun LL. Neuron. 1988;1:791–803. doi: 10.1016/0896-6273(88)90127-4. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Kroger S. Trends Neurosci. 2000;23:92–99. doi: 10.1016/s0166-2236(99)01510-6. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey SE, Davies KE. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- Bonnemann CG, Modi R, Noguchi S, Mizuno Y, Yoshida M, Gussoni E, McNally EM, Duggan DJ, Angelini C, Hoffman EP. Nat Genet. 1995;11:266–273. doi: 10.1038/ng1195-266. [DOI] [PubMed] [Google Scholar]
- Campbell KP. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Chamberlain JS, Corrado K, Rafael JA, Cox GA, Hauser M, Lumeng C. Soc Gen Physiol Ser. 1997;52:19–29. [PubMed] [Google Scholar]
- Claudepierre T, Dalloz C, Mornet D, Matsumura K, Sahel J, Rendon A. J Cell Sci. 2000;113(Pt 19):3409–3417. doi: 10.1242/jcs.113.19.3409. [DOI] [PubMed] [Google Scholar]
- Claudepierre T, Rodius F, Frasson M, Fontaine V, Picaud S, Dreyfus H, Mornet D, Rendon A. Invest Ophthalmol Vis Sci. 1999;40:1520–1529. [PubMed] [Google Scholar]
- Coral-Vazquez R, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, Straub V, Barresi R, Bansal D, Hrstka RF, Williamson R, Campbell KP. Cell. 1999;98:465–474. doi: 10.1016/s0092-8674(00)81975-3. [DOI] [PubMed] [Google Scholar]
- Cox GA, Phelps SF, Chapman VM, Chamberlian JS. Nat Genet. 1993;4:87–93. doi: 10.1038/ng0593-87. [DOI] [PubMed] [Google Scholar]
- Crosbie RH, Heighway J, Venzke DP, Lee JC, Campbell KP. J Biol Chem. 1997;272:31221–31224. doi: 10.1074/jbc.272.50.31221. [DOI] [PubMed] [Google Scholar]
- D’Souza VN, Nguyen TM, Morris GE, Karges W, Fillers DA, Ray PN. Hum Mol Genet. 1995;4:837–842. doi: 10.1093/hmg/4.5.837. [DOI] [PubMed] [Google Scholar]
- Dalloz C, Sarig R, Fort P, Yaffe D, Bordais A, Pannicke T, Grosche J, Mornet D, Reichenbach A, Sahel J, Nudel U, Rendon A. Hum Mol Genet. 2003;12:1543–1554. doi: 10.1093/hmg/ddg170. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. Mol Cell Biol Hum Dis Ser. 1993;3:139–166. doi: 10.1007/978-94-011-1528-5_6. [DOI] [PubMed] [Google Scholar]
- Fanin M, Duggan DJ, Mostacciuolo ML, Martinello F, Freda MP, Soraru G, Trevisan CP, Hoffman EP, Angelini C. J Med Genet. 1997;34:973–977. doi: 10.1136/jmg.34.12.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack AA, Cordier L, Shoturma DI, Lam MY, Sweeney HL, McNally EM. Proc Natl Acad Sci USA. 1999;96:10723–10728. doi: 10.1073/pnas.96.19.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack AA, Groh ME, McNally EM. Microsc Res Tech. 2000;48:167–180. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<167::AID-JEMT5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Hicks D, Courtois Y. Exp Eye Res. 1990;51:119–129. doi: 10.1016/0014-4835(90)90063-z. [DOI] [PubMed] [Google Scholar]
- Howard PL, Dally GY, Wong MH, Ho A, Weleber RG, Fillers DA, Ray PN. Hum Mol Genet. 1998;7:1385–1391. doi: 10.1093/hmg/7.9.1385. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Koenig M, Monaco AP, Kunkel LM. Cell. 1988;53:219–226. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- Koulen P, Blank M, Kroger S. J Neurosci Res. 1998;51:735–747. doi: 10.1002/(SICI)1097-4547(19980315)51:6<735::AID-JNR7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Lidov HG. Brain Pathol. 1996;6:63–77. doi: 10.1111/j.1750-3639.1996.tb00783.x. [DOI] [PubMed] [Google Scholar]
- Lim YC, Doblar DD, Frenette L, Fan PH, Poplawski S, Nanda NC. J Clin Anesth. 1995;7:245–249. doi: 10.1016/0952-8180(94)00049-a. [DOI] [PubMed] [Google Scholar]
- McNally EM, Ly CT, Kunkel LM. FEBS Lett. 1998;422:27–32. doi: 10.1016/s0014-5793(97)01593-7. [DOI] [PubMed] [Google Scholar]
- Mehler MF. Brain Res Brain Res Rev. 2000;32:277–307. doi: 10.1016/s0165-0173(99)00090-9. [DOI] [PubMed] [Google Scholar]
- Montanaro F, Carbonetto S, Campbell KP, Lindenbaum M. J Neurosci Res. 1995;42:528–538. doi: 10.1002/jnr.490420411. [DOI] [PubMed] [Google Scholar]
- Nigro V, Piluso G, Belsito A, Politano L, Puca AA, Papparella S, Rossi E, Viglietto G, Esposito MG, Abbondanza C, Medici N, Molinari AM, Nigro G, Puca GA. Hum Mol Genet. 1996;5:1179–1186. doi: 10.1093/hmg/5.8.1179. [DOI] [PubMed] [Google Scholar]
- Noguchi S, McNally EM, Ben Othmane K, Hagiwara Y, Mizuno Y, Yoshida M, Yamamoto H, Bonnemann CG, Gussoni E, Denton PH, et al. Science. 1995;270:819–822. doi: 10.1126/science.270.5237.819. [DOI] [PubMed] [Google Scholar]
- Ozawa E, Noguchi S, Mizuno Y, Hagiwara Y, Yoshida M. Muscle Nerve. 1998;21:421–438. doi: 10.1002/(sici)1097-4598(199804)21:4<421::aid-mus1>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Pillers DM, Weleber RG, Woodward WR, Green DG, Chapman VM, Ray PN. Invest Ophthalmol Vis Sci. 1995;36:462–466. [PubMed] [Google Scholar]
- Rivier F, Robert A, Royuela M, Hugon G, Bonet-Kerrache A, Mornet D. J Muscle Res Cell Motil. 1999;20:305–314. doi: 10.1023/a:1005426920070. [DOI] [PubMed] [Google Scholar]
- Roberds SL, Leturcq F, Allamand V, Piccolo F, Jeanpierre M, Anderson RD, Lim LE, Lee JC, Tome FM, Romero NB, et al. Cell. 1994;78:625–633. doi: 10.1016/0092-8674(94)90527-4. [DOI] [PubMed] [Google Scholar]
- Rodius F, Claudepierre T, Rosas-Vargas H, Cisneros B, Montanez C, Dreyfus H, Mornet D, Rendon A. Neuroreport. 1997;8:2383–2387. doi: 10.1097/00001756-199707070-00056. [DOI] [PubMed] [Google Scholar]
- Sigesmund DA, Weleber RG, Pillers DA, Westall CA, Panton CM, Powell BR, Heon E, Murphey WH, Musarella MA, Ray PN. Ophthalmology. 1994;101:856–865. doi: 10.1016/s0161-6420(13)31249-4. [DOI] [PubMed] [Google Scholar]
- Straub V, Ettinger AJ, Durbeej M, Venzke DP, Cutshall S, Sanes Wheeler MT, Allikian MJ, Heydemann A, McNally EM. Cold Spring Harb Symp Quant Biol. 2002;67:389–397. doi: 10.1101/sqb.2002.67.389. [DOI] [PubMed] [Google Scholar]
- Campbell JRKP. J Biol Chem. 1999;274:27989–27996. doi: 10.1074/jbc.274.39.27989. [DOI] [PubMed] [Google Scholar]
- Ueda H, Baba T, Ohno S. Histol Histopathol. 2000;15:753–760. doi: 10.14670/HH-15.753. [DOI] [PubMed] [Google Scholar]
- Xiao J, LeDoux MS. Brain Res Mol Brain Res. 2003;119:132–143. doi: 10.1016/j.molbrainres.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Ueda H, Tsukahara S, Kobayashi T, Ohno S. Ophthalmic Res. 1995;27:219–226. doi: 10.1159/000267709. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Ozawa E. J Biochem (Tokyo) 1990;108:748–752. doi: 10.1093/oxfordjournals.jbchem.a123276. [DOI] [PubMed] [Google Scholar]