

Abstract
β-Lactamases are the primary cause of β-lactam antibiotic resistance in many pathogenic organisms. The β-lactamase catalytic mechanism has been shown to involve a covalent acyl-enzyme. Examination of the structure of the class A β-lactamase from Bacillus licheniformis suggested that replacement of Asn-170 by leucine would disrupt the deacylation reaction by displacing the hydrolytic water molecule. When N170L β-lactamase was reacted with penicillins, a novel product was formed. We postulate that with leucine at position 170 the acyl-enzyme undergoes deacylation by an intramolecular rearrangement (rather than hydrolysis) to form a thiazolidine–oxazolinone as the initial product. The oxazolinone subsequently undergoes rapid breakdown leading to the formation of N-phenylacetylglycine and N-formylpenicillamine. This appears to be the first reported case where a point mutation leads to a change in enzyme mechanism resulting in a substantially altered product, effectively changing the product specificity of β-lactamase into that of d-Ala-d-Ala-carboxypeptidase interacting with benzylpenicillin.
Keywords: mechanism, oxazolinone, intramolecular rearrangement, penicillin
Bacterial resistance to β-lactam antibiotics is primarily due to β-lactamases, which render susceptible drugs inactive (1). X-ray crystallographic structures (2–6), sequence alignments (7), and site-directed mutagenesis studies (ref. 8 and references therein) of class A β-lactamases have resulted in the identification of at least a dozen critical residues for catalysis. The catalytic reaction has been shown to occur via a covalent acyl-enzyme intermediate at Ser-70 (S70) (9). E166 is believed to act as a general base catalyst in deacylation (Scheme I) (3, 8, 10). N170, a highly conserved residue, is situated at the bottom of the substrate binding cavity within hydrogen bonding distance of E166 and the hydrolytic water molecule (4, 6) (see figure 15 in ref. 6). ![]()
Support for the critical role of N170 in β-lactamase catalysis is found in our observation that the N170H, N170D, and N170L mutants of Bacillus licheniformis β-lactamase all showed approximately 1000-fold reductions in kcat relative to wild type (unpublished work). These observations are consistent with large reductions in deacylation (k3), and imply a crucial role for N170 in deacylation [with wild-type β-lactamase the acylation and deacylation rates are similar for first generation penicillins (11)]. Plausible roles for N170 in deacylation, based on structural data, include orientation and/or pKa modulation of E166 and/or the hydrolytic water molecule. Thus, the N170L variant was made with the idea that the leucine would displace the hydrolytic water molecule (6), thus leading to a long-lived acyl-enzyme intermediate.
The enzymatic formation of penicillin degradation products other than penicilloic acid (1) (Scheme I) has not been reported for β-lactamases or their mutants. Examples of novel enzymatic products from point mutations of other enzymes are rare—e.g., a point mutation in T4 lysozyme leading to the opposite anomer of the product (12). We describe here the N170L β-lactamase-mediated cleavage of benzylpenicillin, via the thiazolidine–oxazolinone (2), to N-phenylacetylglycine (5) and N-formylpenicillamine (6). The d-Ala-d-Ala-peptidases, which exhibit considerable similarity in sequence, three-dimensional structure, and catalytic mechanism to the β-lactamases (7, 13, 14), also catalyze a similar fragmentation (15–19).
MATERIALS AND METHODS
Materials.
Benzylpenicillin, cephaloridine, 6-aminopenicillanic acid, phenoxymethylpenicillin, bis-dithionitrobenzoic acid (DTNB) and diacetyl-Lys-d-Ala-d-Ala were purchased from Sigma; 7-(thienyl-2-acetamido)-3-(2-(N,N-dimethyl-aminophenylazo)pyridiniummethyl)-3-cephem-4-carboxylic acid (PADAC) was purchased from Calbiochem; nitrocefin was from Unipath. β-Lactamase production and purification will be described elsewhere. The enzyme preparations used were homogenous by isoelectic focusing-PAGE with Coomassie blue staining.
Kinetics of β-Lactamase Catalysis.
Reactions were monitored with a Hewlett–Packard 8452A UV/Vis spectrophotometer at 240 and 257 nm. Typical experimental conditions were: 50 mM potassium phosphate (pH 7.0), 22°C, [benzylpenicillin]initial = 5.4 × 10−4 M, [wild-type β-lactamase] = 3 × 10−10 M, [N170L β-lactamase] = 1.5 × 10−6 M, 22°C.
HPLC Analyses.
The following HPLC system was used: column, C18 (10 μm spheres); mobile phase, 10% acetonitrile/90% 50 mM potassium phosphate (apparent pH, pH*, 7.3); flow rate, 0.8 ml/min; detector, absorbance at 257 nm. For analysis of the first phase, 200-μl samples containing 5.4 × 10−4 M benzylpenicillin, 1.5 × 10−6 M N170L, 2.7 × 10−6 M uracil (internal standard) in 50 mM potassium phosphate (pH 7.3) were incubated at 22°C. At various times, 20-μl aliquots were injected directly onto the HPLC. For the second phase, a 1-ml sample containing 5.4 × 10−4 M benzylpenicillin, 1.5 × 10−6 M N170L, 2.7 × 10−6 M uracil (internal standard) in 50 mM potassium phosphate (pH 7.3) was incubated at 22°C until the absorbance at 257 nm reached a maximum (taken as time zero): 20-μl aliquots were then injected directly into the HPLC at various times. Rate constants were calculated by plotting peak area (normalized to the internal standard peak area) vs. incubation time and curve-fitting. Retention times for uracil, d-5,5-dimethyl-Δ2-thiazoline-4-carboxylic acid, 5R,6R-penicilloic acid, N-formylpenicillamine, and N-phenylacetylglycine were 3.46, 4.27, 4.98, 5.40, and 6.92 min, respectively.
NMR.
1H-NMR spectra were recorded on a Varian Unity Plus 500 MHz spectrometer (25°C): 48 acquisitions were averaged for each spectrum. Solutions of 1 ml of 3 × 10−5 M N170L β-lactamase in 50 mM potassium phosphate (pH 7.0) and 350 μl of 100 mM potassium phosphate (pH 7.4) were evaporated to dryness under vacuum. Each sample was reconstituted with 350 μl of 2H2O (D2O). The two reconstituted samples and 525 mg of potassium benzylpenicillin were combined in an NMR tube (final pD 7.7), immediately placed in the NMR sample probe and scanned at various times.
HPLC–Electrospray Ionization Mass Spectrometry (LC/MS).
The following LC/MS system was used: column, C18 (10 μm spheres); mobile phase, 7% acetonitrile/93% 20 mM NH4HCO3 (pH* 7.1); flow rate, 0.8 ml/min; MS detector, Perkin–Elmer SCIEX API 300 triple quadrupole mass spectrometer equipped with electrospray interface. A sample containing 5.4 × 10−4 M benzylpenicillin (initial) and 1.5 × 10−6 M N170L in 50 mM sodium pyrophosphate (pH 8.0) was incubated at 22°C for 15 min (to allow benzylpenicillin depletion). A 20-μl sample was injected into the LC/MS. Retention time of N-phenylacetylglycine was 8.13 min.
Determination of Free Thiol.
Ellman’s method was used to determine free thiol produced during the reaction of N170L with benzylpenicillin. A 1-ml sample containing 5.4 × 10−4 M benzylpenicillin, 2.1 × 10−3 M DTNB, and 1.5 × 10−6 M N170L in 50 mM potassium phosphate (pH 7.3) was incubated at 22°C for 15 min (to allow benzylpenicillin depletion), absorbance at 412 nm was then monitored.
d-Ala-d-Ala-Carboxypeptidase Activity Determination.
d-Ala-d-Ala-carboxypeptidase activity was measured as described by Georgopapadakou and coworkers (20).
RESULTS
The altered product specificity of the N170L mutant was discovered during steady-state kinetic analysis of the N170L-catalyzed hydrolysis of benzylpenicillin at pH 7 (30°C). Rather than the normal Michaelis–Menten progress curve found with the wild-type and N170H and N170D mutants, the biphasic progress curve shown in Fig. 1A was obtained; similar progress curves were also found for the action of N170L on other penicillins such as ampicillin and phenoxymethylpenicillin, but, interestingly, not with 6-aminopenicillanic acid or cephalosporins (e.g., nitrocefin, PADAC, cephaloridine). Absorbance spectra of the N170L/benzylpenicillin reaction mixture at various times revealed formation and subsequent decay of a new peak at 257 nm (Fig. 1C). Monitoring the reaction at 257 nm clearly shows the buildup of a transient species (Fig. 1B). The kinetics of the first phase of the reaction (increase in A257) are zero-order in substrate, those of the second phase are first-order in substrate. The amplitude for the first phase was proportional to [So], and the rate constant was proportional to [N170L], as expected for substrate saturation. In the second phase (decrease in A257), the amplitude was again proportional to [So], whereas the first-order rate constant was independent of the enzyme concentration. The observations suggest enzymatic formation of an atypical product during the first phase of the reaction, followed by its nonenzymatic degradation in the second. Since the N170L enzyme was homogenous by isoelectric focusing and showed the same effects from different preparations, we conclude that there is only one form of the enzyme present. This is also consistent with the apparently normal kinetics observed with cephalosporins. That zero-order (i.e., saturation) kinetics were observed throughout the first phase of the reaction indicates that Km ≪ 10−5 M; for the wild-type enzyme Km = 1.2 × 10−4 M. The decreased Km for the mutant is consistent with rate-limiting deacylation.
Figure 1.

Kinetics of wild-type (solid line) and N170L (dotted line) B. licheniformis β-lactamase-catalyzed degradation of benzylpenicillin (A) at 240 nm, (B) at 257 nm. (C) Spectra of benzylpenicillin (solid line), benzylpenicillin after hydrolysis with wild-type β-lactamase (dotted line), and benzylpenicillin after degradation with N170L β-lactamase (broken line): absorbance due to the enzyme has been subtracted.
Reverse-phase HPLC analysis of the enzymatic (first) phase of the N170L/benzylpenicillin reaction revealed simultaneous zero-order production of 5R,6R-benzylpenicilloic acid (1, the normal, wild-type enzymatic product) and two unknown products (P1′ and P2′); formation of all three products ceased upon substrate depletion (the end of the first phase). During the nonenzymatic second phase, simultaneous first-order disappearance of one of the atypical products (P1′) (k = 1.0 ± 0.6 × 10−4·s−1) and first-order appearance of another species (P1") (k = 0.3 ± 1.0 × 10−4·s−1) was observed. Incubation of 5R,6R-benzylpenicilloic acid under the same conditions with or without N170L did not result in P1′ or P2′ formation. Thus, the possibility of an N170L-catalyzed conversion of the normal penicilloic acid product into the abnormal products is eliminated. LC/MS analysis of the reaction mixture gave m/e = 193 for P2′; m/e values for P1′ and P1" were not obtained due to difficulties in resolving the P1′, P1", and 5R,6R-benzylpenicilloic acid peaks under HPLC mobile-phase conditions amenable to electrospray-MS detection.
1H-NMR was also used to examine the N170L-catalyzed degradation of benzylpenicillin. Comparison of methyl peak integration values from a spectrum taken just after substrate depletion (Fig. 2A) shows the original benzylpenicillin partitioned into 79% 5R,6R-benzylpenicilloic and 21% P1′/P2′. Later spectra (Fig. 2B) show the disappearance of P1′ and the buildup of 5S,6R-benzylpenicilloic acid and P1". The 5S,6R-benzylpenicilloic acid is a known nonenzymatic epimerization product of 5R,6R-benzylpenicilloic acid. We confirmed, by 1H-NMR, 5S,6R-benzylpenicilloic acid formation under identical buffer conditions from 5R,6R-benzylpenicilloic acid, freshly generated from benzylpenicillin by hydrolysis with wild-type β-lactamase. Additionally, the sum of the P1′ and P1" methyl-peak integration values remained approximately constant after substrate depletion, whereas that of the 5S,6R-benzylpenicilloic acid increased as the 5R,6R-benzylpenicilloic acid decreased. Thus, of the observed compounds in the reaction mixture, only P1" originated from breakdown of P1′.
Figure 2.

Thiazolidine methyl region of the 1H-NMR spectra of the N170L B. licheniformis β-lactamase catalyzed degradation of benzylpenicillin. (A) Just after completion of the first kinetic phase, and (B) 2.3 hr after completion of the first phase. Peak assignments: A, d-5,5-dimethyl-Δ2-thiazoline-4-carboxylic acid, Me; B, 5S,6R-benzylpenicilloic acid, Me; C, 5R,6R-benzylpenicilloic acid, Me; D, N-formylpenicillamine, Me; E, not identified, possibly benzylpenillic acid; F, d-5,5-dimethyl-Δ2-thiazoline-4-carboxylic acid and N-formyl-penicillamine, Me; G, 5R,6R-benzylpenicilloic acid, Me; H, 5S,6R-benzylpenicilloic acid, Me. Resonances from A and B not shown: δ 3.44 ppm, s, 5S,6R-benzylpenicilloic acid, H-9; δ 3.46 ppm, s, 5R,6R-benzylpenicilloic acid, H-9; δ 3.71 ppm, s, N-phenylacetylglycine, H-9; δ 3.74 ppm, dd, 12 Hz, 42 Hz, 5R,6R-benzylpenicilloic acid, H-9; δ 3.83 ppm, dd, 5 Hz, 26 Hz, 5S,6R-benzylpenicilloic acid, H-9; δ 4.27 ppm, d, 6 Hz, 5R,6R-benzylpenicilloic acid, H-6; δ 4.35 ppm, s, N-formylpenicillamine, H-3; δ 4.53 ppm, d, 2 Hz, d-5,5-dimethyl-Δ2-thiazoline-4-carboxylic acid, H-3; δ 5.09 ppm, d, 6 Hz, 5R,6R-benzylpenicilloic acid, H-5; δ 5.10 ppm, s, 5S,6R-benzylpenicilloic acid, H-5; δ 7.35–7.48 ppm, aromatics; δ 8.18 ppm, s, N-formylpenicillamine, H-5; δ 8.23 ppm, d, 2 Hz, d-5,5-dimethyl-Δ2-thiazoline-4-carboxylic acid, H-3.
To determine if a free thiol group was being generated during either phase, DTNB (21) was added to the N170L/benzylpenicillin reaction mixture: a lag corresponding to the first phase was observed, followed by a first-order increase in A412 vs. time which, when extrapolated to infinite time, gave a (molar) amplitude equal to 22% of the original benzylpenicillin concentration. This compares favorably with the 21% conversion of benzylpenicillin to P1′/P2′ seen by 1H-NMR. The first-order rate constant, 1.5 ± 0.01 × 10−4 s−1, was in excellent agreement with the value of 1.6 ± 0.01 × 10−4 s−1 observed for the A255 vs. time decay for the same reaction without DTNB. These results indicate that P1", but not P1′, contains a free SH group.
DISCUSSION
Establishment and Identification of a Unique Enzymatic Product.
The structures of P1′, P2′, and P1" were identified as d-5,5-dimethyl-Δ2-thiazoline-4-carboxylic acid (4), N-phenylacetylglycine (5), and N-formylpenicillamine (6), respectively, based primarily on the MS and 1H-NMR data. The m/e value of 193 observed for P2′ suggested its identity might be N-phenylacetylglycine, later confirmed by 1H-NMR of an authentic sample. Published chemical shift values for d-5,5-dimethyl-Δ2-thiazoline-4-carboxylic acid and N-formylpenicillamine (22) are in excellent agreement with our peak assignments for P1′ and P1", respectively. The appearance and disappearance of the absorbance at 257 nm, concomitant with the appearance and disappearance of P1′, is consistent with the known absorbance spectra of d-5,5-dimethyl-Δ2-thiazoline-4-carboxylic acid and N-formylpenicillamine (22). Additionally, the simultaneous formation of P1" and a free thiol (as measured in the DTNB experiment) are in accord with the identification of P1" as N-formylpenicillamine.
Experiments in which P1′ was generated at pH 7 and then allowed to degrade at more acidic pHs (22°C) demonstrated a linear dependence of the first-order rate constant on hydronium ion concentration, log kobs = −0.82pH + 2.2. Such acid-catalyzed hydrolysis has been previously observed for d-5,5-dimethyl-Δ2-thiazoline-4-carboxylic acid (22).
Proposed Catalytic Mechanism Leading to Formation of d-5,5-dimethyl-Δ2-thiazoline-4-carboxylic Acid and N-Phenylacetylglycine.
Mutation of N170 to hydrophobic leucine eliminates H-bonds between residue 170 and E166 and the hydrolytic water molecule. Loss of these hydrogen bonds could cause repositioning of E166 and/or the hydrolytic water molecule (even complete displacement of the latter), thus compromising the normal deacylation pathway. The observed decrease in deacylation rate for the N170L mutant is consistent with such effects, and could make alternate deacylation pathways kinetically accessible.
We propose the following mechanism (Scheme II) for N170L-mediated formation of N-phenylacetylglycine (5) and N-formylpenicillamine (6) [via d-5,5-dimethyl-Δ2-thiazoline-4-carboxylic acid (4)] from benzylpenicillin. Formation of a relatively long-lived covalent acyl-enzyme intermediate proceeds as with the wild type (although we cannot rule out an effect of mutation at N170 on the rate of acylation). In contrast to the normal hydrolytic pathway, deacylation occurs by an intramolecular rearrangement to yield the thiazolidine–oxazolinone 2. This molecule, 2, is very unstable under aqueous conditions (23), and decomposes by two competing pathways: (i) hydrolysis to afford 5R,6R-benzylpenicilloic acid (1), and (ii) fragmentation into 3 and 4 in the manner proposed for d-Ala-d-Ala-peptidase-catalyzed degradation of benzylpenicillin (24). Hydrolysis of 2 will be favored in aqueous solution, whereas fragmentation will be favored in the noncovalent complex with the enzyme. The ratio of hydrolysis to fragmentation is thus determined by the rate of dissociation of 2 from the enzyme: longer-lived complexes will favor fragmentation. This is supported by the observation that when nafcillin was used as substrate, 75% of the substrate was converted to cleavage products, reflecting slower release of the oxazolinone–thiazolidine from the enzyme due to the larger aromatic side-chain substituent. Oxazolinone 3 is also very labile and rapidly hydrolyzes to the observed product, N-phenylacetylglycine (5). The existence of thiazoline 4 as the precursor of the observed product, N-formylpenicillamine (6), in the d-Ala-d-Ala-peptidase-catalyzed fragmentation of benzylpenicillin has been questioned (22). We, however, clearly observed formation of 4 by 1H-NMR from the action of the N170L mutant on benzylpenicillin. The possibility that the acyl-enzyme deacylates via both formation of the oxazolinone and hydrolysis cannot be ruled out; however, the known hydrolytic instability of 2 is consistent with the mechanism proposed in Scheme II. 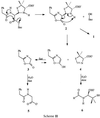
The mechanism in Scheme III, noted as an alternative for d-Ala-d-Ala-peptidase-catalyzed fragmentation (24), can be eliminated on the following basis. When the N170L-catalyzed fragmentation of benzylpenicillin was monitored by 1H-NMR, no signal for the proton derived from H6 of the parent benzylpenicillin was observed for N-phenylacetylglycine, indicating rapid solvent exchange. No exchange is predicted on the basis of Scheme III (but is for Scheme II). Additionally, when 6-aminopenicillanic acid was reacted with N170L β-lactamase the only degradation product detected by 1H-NMR was 5R,6R-penicilloic acid, demonstrating the requirement of 6-amino substitution for fragmentation to occur. 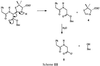
Thus, we believe that replacement of Asn-170 by leucine leads to displacement of the hydrolytic water such that the acyl-enzyme undergoes deacylation by intramolecular oxazolinone formation with 2 as the sole product. With the cephalosporins, the absence of fragmentation is still consistent with formation of the analogous oxazolinone to 2: in this case, the fragmentation rates are anticipated to be significantly slower than for penicillins because the lone pair of the N in the six-membered heterocycle is conjugated with a C–C double bond, and thus less reactive. Also, with cephalosporins, the substrate may not be held in a conformation leading to cyclization. Thus, the breakdown of the cephalosporin-derived analog of 2 occurs solely by hydrolysis.
What, specifically, enables the N170L mutant to catalyze the d-Ala-d-Ala-peptidase-like fragmentation remains unknown, but the process clearly involves structural features not present in the other N170 mutants. A nonenzyme-mediated process, slow enough to be significant only with a long-lived acyl-enzyme, cannot explain the altered product specificity. Neither the N170H nor the N170D mutant, both of which form long-lived acyl-enzyme intermediates, catalyze the d-Ala-d-Ala-peptidase-like fragmentation of benzylpenicillin. The d-Ala-d-Ala-peptidase/benzylpenicillin acyl-enzyme, after denaturation with 6 M guanidine-HCl, did not release cleavage products (24). The methyl ester of benzylpenicillin is not reported to undergo this cleavage under similar buffer conditions [although cleavage does occur in trifluoroacetic acid at room temperature (25)], further supporting the notion that specific structural features of N170L β-lactamase (and the d-Ala-d-Ala-peptidases) and are required for fragmentation to occur.
We believe it is a combination of the more hydrophobic environment in the mutant active site, displacement of the hydrolytic water and E166 carboxylate by the leucine (which provides more room for the C6 side-chain to form the oxazolinone), and the conformation of the substrate being favorable for cyclization, which led to the thiazolidine–oxazolinone formation. The fact that acylation occurs with N170L β-lactamase (although probably much more slowly than with wild type) is not consistent with a mechanism involving E166 acting as a general base to activate Ser-70 via the hydrolytic water molecule (26). Since acylation is observed with mutations at E166 (5, 10, 27, 28), it is clear that if this residue is normally involved in acylation there must be alternate pathways.
Not unexpectedly, we find that N170L β-lactamase has no catalytic activity toward d-Ala-d-Ala-carboxypeptidase substrates such as diacetyl-Lys-d-Ala-d-Ala. However, our observations clearly support the evolutionary relatedness of the d-Ala-d-Ala-carboxypeptidases and the β-lactamases. Frère and coworkers have documented the reverse phenomenon with d-Ala-d-Ala-carboxypeptidase point mutants, namely increased ratios of hydrolysis to fragmentation in certain point mutations, which presumably allow greater access of water to the acyl-enzyme (29–31).
Acknowledgments
We thank Dr. Ling Chen for her help in acquiring and interpreting the LC/MS data. This work was supported by a grant from the National Science Foundation. E.R.L. was supported in part by a grant from the University of California Systemwide Biotechnology Research and Education Program.
Footnotes
Abbreviation: LC/MS, electrospray ionization mass spectrometry
References
- 1.Neu H C. Science. 1992;257:1064–1073. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- 2.Dideberg O, Charlier P, Wery J P, Dehottay P, Dursart J, Erpicum T, Frere J M, Ghuysen J M. Biochem J. 1987;245:911–913. doi: 10.1042/bj2450911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herzberg O, Moult J. Science. 1987;236:694–701. doi: 10.1126/science.3107125. [DOI] [PubMed] [Google Scholar]
- 4.Moews P C, Knox J R, Dideberg O, Charlier P, Frere J M. Protein Struct Funct Genet. 1990;7:156–171. doi: 10.1002/prot.340070205. [DOI] [PubMed] [Google Scholar]
- 5.Strynadka N C J, Adachi H, Jensen S E, Johns K, Sielecki A, Betzel C, Sutoh K, James M N J. Nature (London) 1992;359:700–705. doi: 10.1038/359700a0. [DOI] [PubMed] [Google Scholar]
- 6.Knox J R, Moews P C. J Mol Biol. 1991;220:435–456. doi: 10.1016/0022-2836(91)90023-y. [DOI] [PubMed] [Google Scholar]
- 7.Joris B, Ghuysen J M, Dive G, Renard A, Dideberg O, Charlier P, Frere J M, Kelly J A, Boyington J C, Moews P C, Knox J R. Biochem J. 1988;250:313–324. doi: 10.1042/bj2500313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matagne A, Frere J M. Biochim Biophys Acta. 1995;1246:109–127. doi: 10.1016/0167-4838(94)00177-i. [DOI] [PubMed] [Google Scholar]
- 9.Fisher J, Belasco J G, Kholsa S, Knowles J R. Biochemistry. 1980;19:2895–2901. doi: 10.1021/bi00554a012. [DOI] [PubMed] [Google Scholar]
- 10.Escobar W A, Tan A K, Lewis E R, Fink A L. Biochemistry. 1994;33:7619–7626. doi: 10.1021/bi00190a015. [DOI] [PubMed] [Google Scholar]
- 11.Martin M T, Waley S G. Biochem J. 1988;254:923–925. doi: 10.1042/bj2540923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuroki R, Weaver L H, Matthews B W. Nat Struct Biol. 1995;2:1007–1011. doi: 10.1038/nsb1195-1007. [DOI] [PubMed] [Google Scholar]
- 13.Joris B, Ledent P, Dideberg O, Fonze E, Lamotte-Brasseur J, Kelly J A, Ghuysen J M, Frere J M. Antimicrob Agents Chemother. 1991;35:2294–2301. doi: 10.1128/aac.35.11.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelly J A, Kuzin A P. J Mol Biol. 1995;254:223–236. doi: 10.1006/jmbi.1995.0613. [DOI] [PubMed] [Google Scholar]
- 15.Hammarstrom S, Strominger J L. Proc Natl Acad Sci USA. 1975;72:3463–3467. doi: 10.1073/pnas.72.9.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammarstrom S, Strominger J L. J Biol Chem. 1976;251:7947–7949. [PubMed] [Google Scholar]
- 17.Waxman D J, Strominger J L. J Biol Chem. 1979;254:12056–12061. [PubMed] [Google Scholar]
- 18.Ghuysen J-M, Frere J-M. Nature (London) 1975;258:168–170. doi: 10.1038/258168a0. [DOI] [PubMed] [Google Scholar]
- 19.Frere J-M, Ghuysen J-M, Vanderhaeghe H, Adriaens P, Delgelaen J, DeGraeve J. Nature (London) 1976;260:451–454. doi: 10.1038/260451a0. [DOI] [PubMed] [Google Scholar]
- 20.Georgopapadakou N H, Liu F Y, Ryono D E, Neubeck R, Gordon E M, Pluscec J. Anal Biochem. 1984;137:125–128. doi: 10.1016/0003-2697(84)90357-9. [DOI] [PubMed] [Google Scholar]
- 21.Ellman G L. Arch Biochem Biophys. 1959;82:70–72. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 22.Adriaens P, Meesschaert B, Frere J M, Vanderhaeghe H, Degelaen J, Ghuysen J M, Eyssen H. J Biol Chem. 1978;253:3660–3665. [PubMed] [Google Scholar]
- 23.Clarke H T, Johnson J R, Robinson R, editors. The Chemistry of Penicillin. Princeton: Princeton Univ. Press; 1949. [Google Scholar]
- 24.Hammarstrom S, Strominger J L. Proc Natl Acad Sci USA. 1975;72:3463–3467. doi: 10.1073/pnas.72.9.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bell M R, Carlson J A, Oesterlin R. J Org Chem. 1972;37:2733–2735. doi: 10.1021/jo00982a023. [DOI] [PubMed] [Google Scholar]
- 26.Vijayakumar S, Ravishankar G, Pratt R F, Beveridge D L. J Am Chem Soc. 1995;117:1722–1730. [Google Scholar]
- 27.Adachi H, Ohta T, Matsuzawa H. J Biol Chem. 1991;266:3186–3191. [PubMed] [Google Scholar]
- 28.Escobar W A, Tan A K, Fink A L. Biochemistry. 1991;30:10783–10787. doi: 10.1021/bi00108a025. [DOI] [PubMed] [Google Scholar]
- 29.Bourguignon-Bellefroid C, Wilkin J-M, Joris B, Aplin R T, Houssier C, Prendergast F G, Van Beeumen J, Ghuysen J-M, Frere J-M. Biochem J. 1992;282:361–367. doi: 10.1042/bj2820361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilkin J-M, Jamin M, Joris B, Frere J-M. Biochem J. 1993;293:195–201. doi: 10.1042/bj2930195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkin J-M, Jamin M, Damblon C, Zhao G H, Joris B, Duez C, Frere J-M. Biochem J. 1993;291:537–544. doi: 10.1042/bj2910537. [DOI] [PMC free article] [PubMed] [Google Scholar]