

Abstract
To determine whether formation of the stable complex between a serpin and a target proteinase involves a major translocation of the proteinase from its initial position in the noncovalent Michaelis complex, we have used fluorescence resonance energy transfer to measure the separation between fluorescein attached to a single cysteine on the serpin and tetramethylrhodamine conjugated to the proteinase. The interfluorophore separation was determined for the noncovalent Michaelis-like complex formed between α1-proteinase inhibitor (Pittsburgh variant) and anhydrotrypsin and for the stable complex between the same serpin and trypsin. A difference in separation between the two fluorophores of ≈21 Å was found for the two types of complex. This demonstrates a major movement of the proteinase in going from the initial noncovalent encounter complex to the kinetically stable complex. The change in interfluorophore separation is most readily understood in terms of movement of the proteinase from the reactive center end of the serpin toward the distal end, as the covalently attached reactive center loop inserts into β-sheet A of the serpin.
Keywords: α1-proteinase inhibitor Pittsburgh, proteinase inhibition, fluorescence resonance energy transfer
The inhibitory members of the serpin superfamily of proteins form complexes with target proteinases in fundamentally different ways from those of nonserpin inhibitors, such as the Kunitz and Kazal classes of protein inhibitor (1). Whereas nonserpin inhibitors form thermodynamically stable tight complexes with proteinases through extensive noncovalent interactions and do not require the participation of the active site serine of the proteinase (2), serpins form kinetically stabilized complexes that require full functioning of the catalytic apparatus of the proteinase to form a covalent complex between the active site serine and the serpin (3). Although the nature of the covalent serpin–proteinase complex has not been unequivocally established, it appears likely to be an acyl enzyme complex that represents a normal intermediate on the substrate pathway of a serine proteinase (4, 5). For this reason, serpins have been called suicide substrate inhibitors (6), since they develop their inhibitory propensity only after the initial interactions with the proteinase as a normal substrate (7).
In the absence of an x-ray structure of a serpin–proteinase complex, the nature of the kinetically trapped species has remained conjectural. Two very different types of structure have been envisioned. In one, the trapped covalent complex resembles the initially formed noncovalent Michaelis complex, with the relative positions of proteinase and serpin very similar to those in the Michaelis complex (Fig. 1). Such a complex could explain the apparent reversibility of certain serpin–proteinase complexes (8). The second type of structure involves a translocation of the proteinase from its initial position in the Michaelis complex toward the distal end of the serpin, as a result of the insertion of the reactive center loop into β-sheet A of the serpin (Fig. 1; refs. 1 and 9). Such a process is thermodynamically favorable for serpins, since their normal conformation represents a metastable state that can change to the most stable state through insertion of the reactive center loop into β-sheet A. Such a complex could explain the irreversible nature of most serpin–proteinase complexes and the need for facile insertion of the reactive center loop into β-sheet A for efficient inhibition to occur (10–13).
Figure 1.

The branched suicide substrate pathway of serpins (I) interacting with proteinase (E), showing the possible types of structure for the different intermediates and products. The initial noncovalent Michaelis-like complex (EI) is expected to resemble non-serpin proteinase–inhibitor complexes such as those of bovine pancreatic trypsin inhibitor (BPTI) and trypsin. The structure of cleaved serpin (I*; the product of the substrate branch of the pathway) is known for several serpins and has the cleaved reactive center loop completely inserted into β-sheet A. Two different types of structure are shown for the stable complex (EI+). In one, little or no movement of the proteinase has occurred and stabilization results largely from noncovalent interaction with the serpin. In the other, the P1–P1′ peptide bond has been cleaved, permitting complete loop insertion, with the intermediate trapped at the stage of the covalent acyl enzyme intermediate. The EI+ complex can decay very slowly (k5) to cleaved serpin and free proteinase.
To distinguish between such very different models, we have used fluorescence resonance energy transfer between exogenous fluorophores introduced into trypsin and the serpin α1-proteinase inhibitor Pittsburgh (14) to measure the separation between these fluorophores in the normal covalent serpin–proteinase complex and in a noncovalent complex that resembles the initial Michaelis complex. The large observed increase in separation between the fluorophores in the noncovalent and covalent complexes (Δr ≈ 21 Å) is only consistent with a large movement of the proteinase upon formation of the kinetically trapped covalent complex, as would occur upon insertion of the reactive center loop into β-sheet A of the serpin.
MATERIALS AND METHODS
Preparation of Anhydrotrypsin and β-Trypsin.
Anhydrotrypsin was prepared from commercial crystallized trypsin (Sigma) by alkaline β-elimination of the phenylmethylsulfonyl fluoride (PMSF) adduct according to published procedures (15). Following the reaction, the solution was treated with Phe-Phe-Arg chloromethylketone (20 μM) to inhibit any remaining or regenerated active trypsin and acidified to pH 3.0. β-Anhydrotrypsin was purified from the reaction mixture by chromatography on a soybean trypsin inhibitor affinity matrix. The absence of proteolytic activity in the product was confirmed by activity assay using the chromogenic trypsin substrate S-2222 (Pharmacia Hepar, Franklin, OH). β-Trypsin was prepared from l-1-tosylamido-2-phenylethyl chloromethyl ketone (TPCK)-treated commercial trypsin (Sigma) by affinity chromatography using the same soybean trypsin inhibitor affinity matrix.
Labeling of Proteins with Fluorophores.
The Pittsburgh variant of α1-proteinase inhibitor was specifically labeled on the only free cysteine in the protein (Cys-232) by reaction with 5-iodoacetamidofluorescein (5-IAF; Molecular Probes). α1-Proteinase inhibitor was reduced with a 3-fold molar excess of dithiothreitol for 20 min at room temperature and then reacted with a 12-fold molar excess of 5-IAF overnight at 4°C in the dark. Excess reagent was removed by extensive dialysis against 20 mM phosphate buffer (pH 7.4) containing 100 mM NaCl, 0.1 mM EDTA, and 0.1% PEG 8000. The extent of labeling was determined spectrophotometrically using an extinction coefficient of 70,000 M−1·cm−1 and was found to have 0.95 labels per protein molecule.
Labeling of β-trypsin and β-anhydrotrypsin was carried out by reaction with tetramethylrhodamine isothiocyanate at pH 9.0 under identical conditions for both proteins. Although the reaction was expected to be relatively nonspecific, it was found to result in incorporation of less than one tetramethylrhodamine, even with extended reaction times, suggesting a specific labeling of one lysine side chain. To prevent autolysis of the trypsin under the conditions of labeling, the reactions were carried out on complexes between soybean trypsin inhibitor and either β-trypsin or β-anhydrotrypsin. The protein was immobilized on soybean trypsin inhibitor-Sepharose beads at pH 4.0 and any unbound material removed by several washes. The pH of the proteinase complex with soybean trypsin inhibitor was adjusted to pH 9.1 with several washes of 0.1 M Na2CO3 buffer. A 10-fold molar excess of tetramethylrhodamine isothiocyanate was added from a stock solution in dimethylformamide. The reaction was allowed to proceed for 3 hr at room temperature with gentle rocking. The beads were then washed with 0.1 M Na2CO3 buffer to remove any unreacted probe and any unbound protein. Labeled β-trypsin or β-anhydrotrypsin was eluted from the soybean trypsin inhibitor beads by resuspending the washed beads in 0.1 M citrate buffer (pH 2.9) and incubating for 5 min at room temperature. The supernatant was collected and dialyzed against 1 mM HCl/10 mM CaCl2 to remove any remaining free probe. The extent of labeling was determined spectrophotometrically using an extinction coefficient for the tetramethylrhodamine–protein adduct of 62,000 M−1·cm−1. A labeling ratio of 0.79:1 was obtained for β-anhydrotrypsin and a ratio of 1.01:1 was obtained for β-trypsin. The labeled β-trypsin was diluted with unlabeled β-trypsin to give equivalent degrees of labeling (0.79:1) for both active and inactive proteinase species for subsequent titrations. Measured efficiencies of resonance energy transfer were subsequently normalized to a label:protein ratio of 1:1 for calculation of interfluorophore separations.
The ability of both labeled α1-proteinase inhibitor and labeled trypsin to form stable covalent complex was determined by SDS/PAGE. A reaction mixture of the two components showed a high molecular weight band corresponding to the SDS-stable covalent complex, which exhibited fluorescence from both fluorescein and rhodamine fluorophores. The stoichiometry of inhibition was determined to be close to the expected 1:1.
Gel Electrophoresis.
Polyacrylamide gel electrophoresis was carried out on 10% slab gels according to the procedure of Laemmli (16) for SDS/PAGE or according to the procedure of Davis (17) for gels run under nondenaturing conditions.
Fluorescence Measurements.
Fluorescence measurements were performed on an SLM8000 fluorimeter (SLM–Aminco; Urbana, IL). Emission spectra were recorded with excitation and emission slits of 4 nm, with excitation at 340 nm. Spectra were scanned at 2-nm intervals with a 5-sec integration time. For donor/acceptor titrations monitored at constant wavelength, excitation was at 340 nm and emission was at 515 nm (at which the contribution from tetramethylrhodamine is negligible).
Calculation of Interfluorophore Separation.
The separation, R, between the fluorescein/tetramethylrhodamine donor/acceptor pair was calculated from the reduction in emission intensity of the donor fluorophore as a result of nonradiative energy transfer to the acceptor, using Eq. 1:
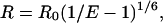 |
1 |
where E is the measured efficiency of energy transfer expressed as the fractional reduction in donor emission intensity and R0 is the separation for 50% efficiency of energy transfer (18). R0 depends on the spectral properties of the donor and acceptor fluorophores in the system under study and is given by Eq. 2:
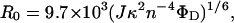 |
2 |
where J is the overlap integral, κ2 is the orientation factor, n is the refractive index, and ΦD is the donor (fluorescein) quantum yield. The overlap integral was calculated from the fluorescence emission spectrum of fluorescein-labeled α1-proteinase inhibitor Pittsburgh and the absorption spectrum of the tetramethylrhodamine-labeled trypsin sample. The quantum yield of the donor was calculated from the integrated emission spectrum of fluorescein-labeled α1-proteinase inhibitor Pittsburgh referenced to the emission spectrum of quinine sulfate in 1 M sulfuric acid, which has a known quantum yield of 0.55 (19). A value of 0.144 was calculated. A value of 1.4 was used for the refractive index (20). In the absence of other information on the orientation factor, a value of 2/3, which corresponds to isotropic rotation of both fluorophores, is frequently used and was used here.
Binding of Wild-Type and Pittsburgh Variant α1-Proteinase Inhibitor to Anhydrotrypsin.
Complex formation between α1-proteinase inhibitors and anhydrotrypsin was monitored by the change in fluorescence of p-aminobenzamidine upon displacement from anhydrotrypsin that occurred upon interaction with the serpin (21). α1-Proteinase inhibitor was titrated into a cuvette containing 4.5 μM anhydrotrypsin and 100 μM p-aminobenzamidine. Change in fluorescence of the bound probe was monitored at 345 nm, with excitation at 325 nm.
Molecular Modeling.
Possible complexes between trypsin or anhydrotrypsin and α1-proteinase inhibitor were modeled using Biopolymer (Tripos Associates, St. Louis) and the structures of cleaved α1-proteinase inhibitor (22), native antithrombin heterodimer (23), and β-trypsin (24). The heterodimer structure of antithrombin has one monomer in the latent conformation and the other with the reactive center exposed and extended. The monomer with the exposed and extended reactive center loop was used to model the noncovalent Michaelis-like complex between α1-proteinase inhibitor and anhydrotrypsin, whereas the loop-inserted monomer was used to construct models of the covalent complex. We are aware that there is likely to be some structural differences between these starting structures and the equivalent species in the complex. However, for the purposes of obtaining reasonable separations between the fluorophores in the different types of complex, this approximation to rigid bodies seems appropriate.
Materials.
5-IAF and tetramethylrhodamine isothiocyanate were from Molecular Probes. Wild-type α1-proteinase inhibitor was a recombinant protein previously expressed in baby hamster kidney cells (11). The P1 M → R variant of α1-proteinase inhibitor was a recombinant protein expressed in Escherichia coli (25) and was a gift from Philip Patston (University of Illinois, Chicago).
RESULTS AND DISCUSSION
α1-Proteinase Inhibitor Pittsburgh Forms a Tight Noncovalent Complex with Anhydrotrypsin.
Since nonserpin protein inhibitors of serine proteinases do not require the active site serine for formation of tight noncovalent complexes with target proteinases, the conversion of the active site serine to a dehydroalanine does not greatly reduce the strength of the highly complementary noncovalent interactions (2). In contrast, the same conversion of serine to dehydroalanine greatly affects the interaction between the proteinase and a serpin inhibitor, since the serpin is no longer able to bind covalently to the proteinase and, consequently, the strength of the interaction is determined by much less effective noncovalent interactions. It has previously been shown for α1-proteinase inhibitor that the noncovalent complex it forms with anhydrotrypsin has a KD of only ≈20 μM (3). This was considered too weak to enable saturation of the serpin with anhydrotrypsin under optimal experimental conditions. We therefore determined the strength of the noncovalent complex between anhydrotrypsin and a variant of α1-proteinase inhibitor in which the P1 residue had been mutated from methionine to arginine (Pittsburgh variant), to see if this variant could be used to give a more suitable tight binding interaction. The replacement of arginine for methionine greatly increased the strength of the interaction, decreasing KD from ≈20 μM to ≈5 nM (Fig. 2; see Fig. 4). This increase in affinity was great enough to permit saturation of the serpin with anhydroproteinase in subsequent fluorescence resonance energy transfer measurements.
Figure 2.

Ability of inactivated (anhydro-) trypsin to form tight complex with α1-proteinase inhibitor Pittsburgh but not with wild-type α1-proteinase inhibitor. Titration of α1-proteinase inhibitor Pittsburgh (○) into a solution of anhydrotrypsin (4.5 μM) in the presence of p-aminobenzamidine (100 μM) resulted in stoichiometric displacement of the noncovalently bound fluorescent probe, indicating tight 1:1 noncovalent complex formation. The solid straight lines are for visual aid only. Similar titration of wild-type α1-proteinase inhibitor (□) under the same conditions led to negligible complex formation and consequently little change in probe fluorescence.
Figure 4.

Quantitiation of the efficiency of fluorescence resonance energy transfer in covalent and noncovalent α1-proteinase inhibitor Pittsburgh–trypsin complexes. (A) Decrease in fluorescein emission of fluorescein–α1-proteinase inhibitor Pittsburgh (80 nM) as a function of added tetramethylrhodamine–anhydrotrypsin. The solid line represents the nonlinear least-squares fit of the data to a simple binding equation and gives KD of ≈5 nM. (B) Time course of decay in fluorescein emission intensity of fluorescein–α1-proteinase inhibitor Pittsburgh after mixing with two equivalents of tetramethylrhodamine-trypsin in the presence of 10 mM benzamidine to slow the reaction. The solid line is a fit of the data to a bimolecular reaction and gave a second-order rate constant, when corrected for the competitive inhibitor benzamidine, of 1.5 × 107 M−1·sec−1.
Both wild-type and P1 variant α1-proteinase inhibitors were able to form the expected SDS-stable covalent 1:1 complexes with β-trypsin, as shown by the appearance of a band on SDS/PAGE with lower mobility than the serpin at the position expected for complex (data not shown). Such experiments were carried out both with labeled and unlabeled serpin and proteinase and confirmed that the limited covalent modification had not reduced the effectiveness of either proteinase or serpin to form complex.
Differential Fluorescence Resonance Energy Transfer in the Two Types of Complex.
Stoichiometric covalent and noncovalent complexes between tetramethylrhodamine-labeled proteinase and the fluorescein-labeled serpin α1-proteinase inhibitor Pittsburgh were formed in situ and the properties of their fluorescence emission spectra examined. Compared with the emission spectrum of an equivalent complex formed between labeled α1-proteinase inhibitor Pittsburgh and unlabeled anhydrotrypsin, the intensity of the fluorescein emission spectrum of the noncovalent complex of fluorescein-labeled serpin with tetramethylrhodamine-labeled anhydrotrypsin at a molar ratio of 1:1 was reduced by ≈61% (77% when normalized) and with a corresponding increase in intensity of the acceptor fluorophore (tetramethylrhodamine; Fig. 3A). In contrast, the intensity of the donor fluorescence of the 1:1 covalent complex between comparably labeled trypsin and serpin was reduced by only ≈16% (20% when normalized; Fig. 3B).
Figure 3.

Differential efficiencies of fluorescence resonance energy transfer between fluorescein and tetramethylrhodamine in covalent and noncovalent serpin–proteinase complexes. (A) Effect of formation of non-covalent complex between fluorescein–α1-proteinase inhibitor Pittsburgh and tetramethylrhodamine–anhydrotrypsin. Solid line, fluorescein–α1-proteinase inhibitor Pittsburgh (82 nM) plus 250 nM unlabeled anhydrotrypsin; dashed line, tetramethylrhodamine–anhydrotrypsin; and dotted line, mixture of fluorescein–α1-proteinase inhibitor Pittsburgh (82 nM) and saturating levels (250 nM) of tetramethylrhodamine–anhydrotrypsin, showing reduction in fluorescein emission at 517 nm and increase in tetramethylrhodamine fluorescence at 575 nm. (B) Effect of formation of covalent complex between fluorescein–α1-proteinase inhibitor Pittsburgh and tetramethylrhodamine–trypsin. Solid line, fluorescein–α1-proteinase inhibitor Pittsburgh (82 nM) plus one equivalent unlabeled trypsin; dashed line, 82 nM tetramethylrhodamine–trypsin; and dotted line, 1:1 mixture of fluorescein–α1-proteinase inhibitor Pittsburgh and tetramethylrhodamine–trypsin, showing smaller reduction in fluorescein emission and smaller increase in tetramethylrhodamine emission at 575 nm in the complex.
To demonstrate both that the observed reduction in fluorescence intensity was a consequence of fluorescence resonance energy transfer upon complex formation and to more accurately determine the efficiency of energy transfer upon complete saturation of the serpin with proteinase, two separate experiments were carried out. For the noncovalent complex, a compete titration of labeled anhydrotrypsin into labeled serpin was performed. This showed that the effect was saturable and could be well fitted to a simple binding equation (Fig. 4A). The fit provided both a KD for the interaction (≈5 nM) as well as the reduction in fluorescence intensity in the doubly labeled 1:1 complex. This tight KD is in agreement with the almost linear displacement of p-aminobenzamidine by anhydrotrypsin seen at higher protein concentrations (Fig. 2). The reduction in intensity was 69%, which corresponded to an efficiency of transfer of 77%, when normalized to one acceptor label per anhydrotrypsin and when corrected for the decrease in intensity (10%) due solely to complex formation, as determined from a separate titration of labeled serpin with unlabeled anhydroproteinase. For the covalent complex, a complication arose in demonstrating that the efficiency of fluorescence resonance energy transfer was saturable, since an excess of free trypsin is known to degrade the covalent serpin–proteinase complex. We also found here that such degradation affected the fluorescence spectra. Instead, we examined the time dependence of the change in fluorescein emission intensity upon reacting two equivalents of labeled trypsin with one equivalent of labeled serpin (Fig. 4B). The curve could be well fitted to a bimolecular reaction with second-order rate constant of 1.5 × 107 M−1·sec−1, when corrected for the benzamidine competitive inhibitor (21). This is very fast compared with the second-order rate constant reported for trypsin and wild-type α1-proteinase inhibitor (1.3 × 105 M−1·sec−1; ref. 26), but in line with the large increases in rate of inhibition of arginine-specific proteinases seen for the Pittsburgh variant when compared with wild-type α1-proteinase inhibitor (25, 27). This time course thus appeared to report the formation of the covalent serpin–proteinase complex and gave a reduction in fluorescence intensity of ≈13% at completion of the reaction, corresponding to complete complexation of the serpin. This corresponded to an efficiency of resonance energy transfer of 18% when normalized to one acceptor label per trypsin and when corrected for the small increase (2%) in intensity due solely to complex formation, as determined from a separate titration between labeled serpin and unlabeled trypsin.
Large-Scale Movement of the Proteinase upon Formation of the Covalent Serpin–Proteinase Complex.
The separations between the tetramethylrhodamine on the trypsin and the fluorescein on the serpin calculated from the experimental fluorescence parameters (Table 1) were very different for the two types of complex. For the noncovalent complex between anhydrotrypsin and α1-proteinase inhibitor, an interfluorophore separation of 35 Å was calculated. For the covalent complex, the equivalent separation was calculated to be 56 Å, showing unequivocally that the proteinase in the covalent complex was in a very different position relative to Cys-232 than in the noncovalent complex. Since the noncovalent complex between anhydrotrypsin and the serpin is expected to be very similar in structure to the noncovalent Michaelis complex formed between trypsin and the serpin, these results imply that in progressing from the initial Michaelis complex to the kinetically trapped covalent complex, trypsin must undergo a large-scale movement of at least 21 Å. It should be realized, however, that the movement could be very much greater than this, since the two separations determined of 35 Å and 56 Å (R1 and R2) represent the radii of spheres centered on fluorescein covalently bound to Cys-232 (Fig. 5). An upper limit for the change in separation would be R1 + R2, which is equal to 91 Å.
Table 1.
Fluorescence parameters for tetramethylrhodamine–proteinase/fluorescein–α1-proteinase inhibitor Pittsburgh complexes
| Parameters | Values |
|---|---|
| J, M−1·nm3 | 3.06 × 10−13 |
| ΦD, fluorescein | 0.144 |
| R0 (2/3) | 43.4 Å |
| E1, noncovalent complex | 77% |
| E2, covalent complex | 18% |
| R1, noncovalent complex | 35 Å |
| R2, covalent complex | 56 Å |
Figure 5.

Models of noncovalent and covalent serpin–proteinase complexes. The serpin (solid ribbon) is shown in the center in an orientation such that the reactive center loop in the unreacted molecule is at the top of the molecule and β-sheet A is seen edge-on, running from top to bottom. The F helix is on the surface of β-sheet A at the bottom right side (distal end of β-sheet from the reactive center). The serpin shown is antithrombin in its crystal structure conformation that has an appropriate exposed and extended reactive center loop for initial noncovalent docking with proteinase. However, the structural homology between serpins is such that the location indicated for the Cys-232 residue is equivalent to that in α1-proteinase inhibitor. The proteinase (three-stranded ribbon) is shown in three different locations corresponding to three distinct types of complex. Two circles are drawn, centered on the position of Cys-232 in α1-proteinase inhibitor Pittsburgh, with radii of 35 Å and 56 Å, corresponding to the interfluorophore separations measured for the noncovalent and covalent serpin–proteinase complexes, respectively. Structure A (proteinase at top) represents the noncovalent complex of the anhydroproteinase or of the active proteinase in the initial Michaelis-like complex. Structure B (proteinase in the middle) represents a covalent partially loop-inserted (up to P9) covalent complex. Note the abutment of the proteinase against the F α-helix. Structure C (proteinase at the bottom), represents a covalent complex in which the reactive center loop has completely inserted into β-sheet A and has resulted in translocation of the proteinase from the proximal (top) end of the serpin to the distal (bottom) end.
Although some parameters in these distance measurements are not fully determined, with corresponding uncertainties in the absolute values of R1 and R2, the conclusions that R1 and R2 are very different and that the proteinase must therefore undergo large-scale movement upon formation of the covalent complex are not in doubt. Thus, the use of 2/3 for the orientation factor (κ2) may be in error if the two fluorophores are not undergoing isotropic rotation. However, we have previous labeled Cys-232 with a nitroxide spin label and obtained an EPR spectrum that was characterized by isotropic rotation of the nitroxide (P.G.W.G., unpublished work), suggesting a nonconstraining environment. In addition, both the donor and acceptor fluorophores are probably in similar environments in the two complexes, so that if the correct value of κ2 were different from 2/3, the same altered value would be appropriate for both distance calculations.
Another uncertainty concerns the homogeneity of the tetramethylrhodamine labeling. Although only ≈0.8 labels per anhydrotrypsin were incorporated, we do not know whether this represents nearly complete labeling of a single site or less complete labeling of multiple sites. In favor of uniform labeling is that, even with excess reagent present, the degree of labeling at much longer reaction times tended to a plateau of about one label per trypsin (data not shown). Thus there may be one site that is uniquely reactive, either as a result of steric constraints on the bulky tetremethylrhodamine group or of an unusually low pKa for one amino group. Importantly, the labeling of both trypsin and anhydrotrypsin was carried out under identical reaction conditions. Therefore, even if more than one site carried partial acceptor label, the label distribution is likely to be very similar in both types of proteinase. Since the most important finding in this study is that there is a large change in interfluorophore separation, even partial multiple labeling that is the same in both types of complex should not lead to a change in this conclusion.
Correlation of Interfluorophore Separations with Possible Structures of Serpin–Proteinase Complex.
Because only the location of the donor fluorophore is known with certainty and because only a single attachment site was available in the serpin, it is not possible to use the present distance measurements to unambiguously determine the location and orientation of the proteinase in each of the two types of complexes. However, since there are x-ray structures available for several serpin structures, both in native and in cleaved loop-inserted forms, as well as for trypsin, it is very useful to consider the possible structures of the complexes based on modeling the interaction of these structures, both as a test of the fluorescence resonance energy transfer method in this system and to attempt discrimination between different models for the covalent complex.
The separation measured for the complex between anhydrotrypsin and α1-proteinase inhibitor Pittsburgh first serves as a test of the method. Since this is a relatively tightly binding but noncovalent complex that derives much of its binding energy from the P1 arginine residue (from comparison of the Kd values of the wild-type and variant complexes), a reasonable model for the complex can be obtained by appropriate docking of the S and S′ subsites on the anhydroproteinase with residues in the reactive center of the serpin. The extended ribbon reactive loop conformation seen in one monomer of the antithrombin heterodimer (23) was used and was docked in similar orientation to that for the “canonical” loop conformation seen in trypsin–bovine pancreatic trypsin inhibitor (BPTI) complexes (28). The resulting structure (Fig. 5, structure A) places the proteinase in a position such that a sphere of radius 35 Å, centered on Cys-232 of the serpin (at the end of the F2 helix), includes about two-thirds of the volume of the proteinase. There are several lysines on the surface of this sphere that, if labeled with tetramethylrhodamine, would give the observed interfluorophore separation of 35 Å. We can therefore conclude that the fluorescence energy transfer method applied to this system gives an interfluorophore separation for the noncovalent complex that is consistent with the type of structure for such a complex. In contrast, a sphere of radius 56 Å, centered at the same point, does not intersect the proteinase at all in this structure (Fig. 5), thus requiring a major movement of the proteinase to give a structure consistent with the longer interfluorophore separation measured for the covalent complex.
Structures for the covalent complex that are consistent with the longer interfluorophore separation and that maintain contact between the active site serine of the proteinase and the P1 residue of the proteinase, as required for the covalent complex, require a major movement of the proteinase both radially and circularly from Cys-232. Two representative structures were modeled. Both used clockwise rotation of the trypsin about a vertical axis centered on Cys-232 of the serpin and radial displacement consistent with the likely position of the P1 residue as the reactive center loop inserted into β-sheet A and the requirement that the P1 residue be covalently bound to the proteinase active site. In addition, only clockwise rotation over the right-hand surface of the serpin was considered, since this is the only feasible route for insertion of the reactive center loop into β-sheet A and also results in contacts between the proteinase and the serpin that may be the source of disturbance of the catalytic triad (29) and consequently of the kinetic trapping of the serpin–proteinase complex. The structure involving the largest movement (Fig. 5, structure C) represents full insertion of the reactive center loop into β-sheet A and was modeled using the location of the P1 residue in the cleaved structure of α1-proteinase inhibitor (22). A structure (Fig. 5, structure B) intermediate between this extreme structure and the starting structure was also considered. This represents the structure expected if only partial insertion of the reactive center loop into β-sheet A occurs, up to residue P9. The end point was determined by the point at which the proteinase impinges on the F α-helix. This helix covers the distal surface of the A sheet (bottom in Fig. 5) and might therefore impede further loop insertion, while the proteinase is still covalently attached to the P1 residue. Both of these structures placed lysine side chains at or close to the longer separation of 56 Å and are thus consistent with the measured interfluorophore separation for the covalent complex.
Conclusions.
The results presented here show that the method of fluorescence resonance energy transfer can be successfully applied to mapping serpin–proteinase complexes and most importantly show for the first time that the location of the proteinase in a covalent complex is very different from that in the noncovalent complex. This is consistent with much evidence that supports a mechanism of kinetic trapping of the complex in which the reactive center loop, with the proteinase covalently attached, inserts into β-sheet A and drags the proteinase with it.
Acknowledgments
We thank Dr. Philip Patston for the generous gift of recombinant α1-proteinase inhibitor Pittsburgh variant and Dr. Steven Olson for help with preparation of anhydrotrypsin, for the use of his SLM8000 fluorimeter, and for helpful discussions during the study. This work was supported by Grant HL49234 from the National Institutes of Health.
References
- 1.Gettins P G W, Patston P A, Olson S T. Serpins: Structure, Function and Biology. Austin, TX: Landes; 1996. [Google Scholar]
- 2.Ako H, Foster R J, Ryan C A. Biochemistry. 1974;13:132–139. doi: 10.1021/bi00698a021. [DOI] [PubMed] [Google Scholar]
- 3.Olson S T, Bock P E, Kvassman J, Shore J D, Lawrence D A, Ginsburg D, Björk I. J Biol Chem. 1995;270:30007–30017. doi: 10.1074/jbc.270.50.30007. [DOI] [PubMed] [Google Scholar]
- 4.Lawrence D A, Ginsburg D, Day D E, Berkenpas M B, Verhamme I M, Kvassman J-O, Shore J D. J Biol Chem. 1995;270:25309–25312. doi: 10.1074/jbc.270.43.25309. [DOI] [PubMed] [Google Scholar]
- 5.Wilczynska M, Fa M, Ohlsson P I, Ny T. J Biol Chem. 1995;270:29652–29655. doi: 10.1074/jbc.270.50.29652. [DOI] [PubMed] [Google Scholar]
- 6.Patston P A, Gettins P, Beechem J, Schapira M. Biochemistry. 1991;30:8876–8882. doi: 10.1021/bi00100a022. [DOI] [PubMed] [Google Scholar]
- 7.Waley S G. Biochem J. 1991;279:87–94. doi: 10.1042/bj2790087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shieh B-H, Potempa J, Travis J. J Biol Chem. 1989;264:13420–13423. [PubMed] [Google Scholar]
- 9.Wright H T, Scarsdale J N. Proteins. 1995;22:210–225. doi: 10.1002/prot.340220303. [DOI] [PubMed] [Google Scholar]
- 10.Hopkins P C R, Carrell R W, Stone S R. Biochemistry. 1993;32:7650–7657. doi: 10.1021/bi00081a008. [DOI] [PubMed] [Google Scholar]
- 11.Hood D B, Huntington J A, Gettins P G W. Biochemistry. 1994;33:8538–8547. doi: 10.1021/bi00194a020. [DOI] [PubMed] [Google Scholar]
- 12.Huntington J A, Patston P A, Gettins P G W. Protein Sci. 1995;4:613–621. doi: 10.1002/pro.5560040403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hopkins P C R, Stone S R. Biochemistry. 1995;34:15872–15879. doi: 10.1021/bi00048a033. [DOI] [PubMed] [Google Scholar]
- 14.Owen M C, Brennan S O, Lewis J H, Carrell R W. N Engl J Med. 1983;309:694–698. doi: 10.1056/NEJM198309223091203. [DOI] [PubMed] [Google Scholar]
- 15.Ako H, Foster R J, Ryan C A. Biochem Biophys Res Commun. 1972;47:1402–1407. doi: 10.1016/0006-291x(72)90228-8. [DOI] [PubMed] [Google Scholar]
- 16.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 17.Davis B J. Ann NY Acad Sci. 1964;121:404–427. doi: 10.1111/j.1749-6632.1964.tb14213.x. [DOI] [PubMed] [Google Scholar]
- 18.Lakowicz J R. Principles of Fluorescence Spectroscopy. New York: Plenum; 1983. [Google Scholar]
- 19.Melhuish N Z. J Opt Soc Am. 1964;54:183–188. [Google Scholar]
- 20.Beardsley K, Cantor C R. Proc Natl Acad Sci USA. 1970;65:39–46. doi: 10.1073/pnas.65.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans S A, Olson S T, Shore J D. J Biol Chem. 1982;257:3014–3017. [PubMed] [Google Scholar]
- 22.Löbermann H, Tokuoka R, Deisenhofer J, Huber R. J Mol Biol. 1984;177:731–757. [PubMed] [Google Scholar]
- 23.Carrell R W, Stein P E, Fermi G, Wardell M R. Structure (London) 1994;2:257–270. doi: 10.1016/s0969-2126(00)00028-9. [DOI] [PubMed] [Google Scholar]
- 24.Marquart M, Walter J, Deisenhofer J, Bode W, Huber R. Acta Crystallogr B. 1983;39:480–490. [Google Scholar]
- 25.Schapira M, Ramus M-A, Jallat S, Carvallo D, Courtney M. J Clin Invest. 1986;77:635–637. doi: 10.1172/JCI112347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beatty K, Bieth J, Travis J. J Biol Chem. 1980;255:3931–3934. [PubMed] [Google Scholar]
- 27.Patston P A, Roodi N, Schifferli J A, Bischoff R, Courtney M, Schapira M. J Biol Chem. 1990;265:10786–10791. [PubMed] [Google Scholar]
- 28.Perona J J, Craik C S. Protein Sci. 1995;4:337–360. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plotnick M I, Mayne L, Schechter N M, Rubin H. Biochemistry. 1996;35:7586–7590. doi: 10.1021/bi960233w. [DOI] [PubMed] [Google Scholar]