

Abstract
Homologous pairing and strand exchange, which are catalyzed by Escherichia coli RecA protein, are central to homologous recombination. Homologs of this protein are found in eukaryotes; however, little has been reported on the recombinase activities of the mammalian homologs, including the human protein, denoted HsRad51. For the studies described here, we purified HsRad51 from E. coli. Although the activities of HsRad51 and RecA were qualitatively similar in the presence of ATP, there were also striking differences. The stoichiometry of binding to DNA and the rate of renaturation of complementary strands were similar for the two proteins, but rates of ATP hydrolysis, homologous pairing, and subsequent strand exchange promoted by HsRad51 were less than (null)/1;10 those of RecA. In addition, HsRad51 bound γ-thio-ATP and formed stable presynaptic complexes that promoted renaturation as rapidly as RecA, but the recombinant human protein catalyzed neither strand exchange nor homologous pairing of a single strand with duplex DNA in the presence of the ATP analog. By contrast, RecA promoted both of the latter reactions in control experiments. These observations suggest that among RecA-like proteins, HsRad51 may be a variant in which homologous pairing and strand exchange are more closely linked to the hydrolysis of ATP.
Keywords: genetic recombination, strand exchange, RecA homologs
The central events in homologous recombination are the pairing of homologous molecules and the initiation of strand exchange. Escherichia coli RecA protein is the prototype of proteins that can catalyze these reactions (1). Homologs of RecA are widely distributed in prokaryotes (1) and eukaryotes, including Saccharomyces cerevisiae (2–4), Schizosaccharomyces pombe (5), Xenopus laevis (6), Lilium longiflorum (7), Neurospora crassa (8, 9), Arabidopsis thaliana (10), mouse (11), chicken (12), and man (13, 14); however, the specific recombination events in which these proteins participate are still unclear.
In Saccharomyces cerevisiae, rad51 mutants show defects in genetic recombination and repair of damaged DNA (15, 16) and a strongly decreased yield of viable spores (3). A role in meiosis is further shown by the presence in meiotic nuclei of ScRad51 protein together with ScDmc1, another homolog of RecA that is specific to meiosis (17).
A recA-like gene in Drosophila melanogaster was found to be expressed at high level in ovary but not in testis, which correlates with the lack of meiotic recombination in male Drosophila (18). In chicken and mouse, high levels of transcription of homologs of the rad51 gene were found in lymphoid and reproductive organs (11–13). Other findings provide evidence for roles of mammalian Rad51 in meiosis and isotype switch recombination: Antibody to human Rad51 stained murine synaptonemal complexes early in meiosis (19) and stained numerous foci in nuclei of human cells exposed to DNA-damaging agents (35); and when primary murine B cells were induced to stimulate isotype switch recombination, the level of nuclear Rad51 antigen was dramatically increased (20). In the mouse, a homozygous rad51 mutation was lethal early in embryogenesis (21). The vital role of the mammalian enzyme contrasts with the viability of rad51 mutants in yeast.
The Rad51 proteins of yeast and man have been purified. The yeast protein, ScRad51, displays DNA-dependent ATPase activity, pairs single strands with homologous duplex DNA, and promotes subsequent strand exchange (22, 23). Human Rad51 protein, HsRad51, possesses DNA-dependent ATPase activity and forms nucleoprotein filaments that resemble those of RecA (24), but pairing and strand exchange activities have not been reported. In this paper, we describe experiments on highly purified HsRad51 protein in which assays of fluorescence resonance energy transfer (FRET) have helped to distinguish and characterize pairing and strand exchange.
MATERIALS AND METHODS
Enzymes and Other Reagents.
RecA protein was purified as described (25). ATP, adenosine 5′-[γ-thio]triphosphate (ATP[γS]), and phenylmethylsulfonyl fluoride were purchased from Sigma; T4 polynucleotide kinase, from New England Biolabs; and DNase I, dithiothreitol (DTT), and bovine serum albumin (BSA), from Boehringer Mannheim.
The following 83-mer oligonucleotides were synthesized on an Applied Biosystems DNA synthesizer (model 380B): A16(−), 5′-AAATGAACATAAAGTAAATAAGTATAAGGATAATACAAAATAAGTAAATGAATAAACATAGAAAATAAAGTAAAGGATATAAA; A16(+) was complementary to A16(−); W16(−), 5′-TTGATAAGAGGTCATTTTTGCGGATGGCTTAGAGCTTAATTGCTGAATCTGGTGCTGTAGCTCAACATGTTTTAAATATGCAA; and X16 was a mixture of random 83-mers.
Primary amines on a C6 linker (Glen Research) were added to these oligonucleotides at either the 3′ end, for A16(−) and X16, or at the 5′ end, for A16(+), for subsequent labeling with fluorescein or rhodamine by procedures that will be described elsewhere (L.R.B., M.T., and C.M.R., unpublished work). Oligonucleotides were purified on a 12% denaturing polyacrylamide gel as described (26). The absorbance at 260, 496, and 558 nm was used, respectively, to calculate the concentration of DNA, fluorescein, and rhodamine. Duplex oligonucleotides were prepared by annealing as described (26). Duplex oligonucleotides with conjugated fluorophores were labeled at 5′ ends with 32P by reaction with T4 polynucleotide kinase and examined by electrophoresis on nondenaturing 12% polyacrylamide gels to confirm complete annealing. All DNA concentrations refer to moles of nucleotide residues, except for stoichiometric ratios for duplex DNA, which are expressed as moles of protein per mole of base pairs.
Cloning of the Human rad51 Gene.
We previously cloned the coding sequence for HsRad51 with a hexahistidine tag (35); the present procedure was designed to clone the coding sequence without the tag. The HsRad51 coding sequence was amplified by PCR from a thymus cDNA library in λ Charon BS phage (28), by use of the following primers: (i) CATGCCATGGCAATGCAGATGCAGCTTG and (ii) CGCGGATCCTCAGTCTTTGGCATCTCCCACT. The PCR fragment was inserted into expression vector pTrcHisB (Invitrogen) at the NcoI and BamHI restriction sites to make plasmid pEG932. The choice of restriction enzymes enabled us to clone the Hsrad51 gene without a histidine tag, which is otherwise encoded by pTrcHisB. The identity of the inserted sequence to the published coding sequence of the Hsrad51 gene (13) was confirmed by DNA sequencing.
Purification of HsRad51.
The protein was purified from E. coli DH10B (GIBCO/BRL) carrying plasmid pEG932 (see above). Purification, details of which will be published elsewhere, involved chromatography through Q Sepharose, Bio-Gel-HTP, Mono Q, and native DNA-cellulose. By this procedure, we purified HsRad51 to apparent homogeneity: only one band of 37 kDa was seen on SDS/PAGE analysis of DNA-cellulose fractions that contained about 1.8 mg of protein per ml. The following activities were undetectable (<2% activity after incubation for 1 hr): exonuclease or endonuclease on single- or double-stranded DNA (ssDNA and dsDNA, respectively), endonuclease or topoisomerase on superhelical DNA, helicase on partial duplex substrates.
Binding of HsRad51 to Oligonucleotides.
The standard reaction mixture contained 5 μM oligonucleotide, 25 mM Hepes (pH 7.4), 1 mM MgCl2, 1 mM DTT, 2 mM ATP, and 100 μg of BSA per ml.
Binding of HsRad51 to 83-mer W16(−) was determined by a nitrocellulose filter assay as described (29), and the following fluorometric assay: W16(−) labeled with Cy3 succinimidyl ester fluor (from Amersham) at its 3′ end was incubated with different concentrations of HsRad51 at 37°C for 4 min. Enhancement of fluorescence emission from Cy3 at 565 nm was measured upon excitation at 545 nm. The Cy3 emission was plotted against the protein-to-nucleotide ratio to assess the stoichiometric requirement for saturation of the DNA.
DNase I Protection.
HsRad51 or RecA (1.2 μM) was incubated with 32P-labeled A16(−) DNA (3 μM) in the reaction mixture described below under Renaturation in the presence of ATP or ATP[γS], or in the absence of nucleotide cofactor, at 37°C for 4 min. DNase I was then added to the reaction mixture and incubation was continued at 37°C for 2 min. The reaction was immediately quenched by adding a solution so that the final concentrations of added reagents were 0.5% SDS and 25 mM EDTA. DNA was precipitated by adding cold trichloroacetic acid at a final concentration of 20%, and radioactivities of acid-soluble material in the supernatant were measured.
Renaturation.
HsRad51 or RecA (1.2 μM) was preincubated at 37°C for 4 min with 3 μM 83-mer, A16(−), in a reaction mixture (50 μl) containing 1 mM MgCl2, 25 mM Hepes (pH 7.4), 1 mM DTT, 2 mM ATP or ATP[γS], and 100 μg of BSA per ml. After the reaction mixture was transferred from 37°C to room temperature for 1 min, 5′-32P-labeled complementary plus strand oligomer, A16(+), was added, bringing its final concentration to 3 μM, and incubation was continued at room temperature. At various time intervals, 8-μl aliquots were taken and mixed with 2 μl of a solution to stop the reaction; final concentrations in these mixtures were 0.5% SDS, 25 mM EDTA, 0.04% bromophenol blue, 0.04% xylene cyanol, and 100 μM oligonucleotide A16(+) as an unlabeled competitor to block spontaneous renaturation. Samples were loaded on a nondenaturing 12% polyacrylamide gel after incubation for 5 min on ice, and the gel was run at 8 V/cm for 2.5 hr at room temperature in buffer containing 45 mM Tris borate (pH 8.3) and 1 mM EDTA. The gel was dried and the reaction was quantitated by PhosphorImager analysis (Molecular Dynamics).
Strand Exchange Assessed by Electrophoresis.
Preincubation of HsRad51 or RecA with 83-mer A16(−) was done as described above under Renaturation. After 4 min, the concentration of MgCl2 was increased to 30 mM, followed by addition of homologous duplex oligonucleotide 5′-32P-A16(−)/A16(+). The final concentration of the duplex oligonucleotide was 2.5 μM. To provide a heterologous control, we formed the filament on the randomized 83-mer, X16. Incubation was continued at 37°C for 1 hr for HsRad51 or 10 min for RecA. The reaction was stopped and analyzed as described above except that competitor oligonucleotide A16(+) was omitted.
Homologous Pairing and Strand Exchange Assayed by FRET.
To measure the homologous pairing of a single-stranded oligonucleotide with homologous duplex oligonucleotide, 3′-F-A16(−), an 83-mer minus strand labeled at its 3′ end with fluorescein, was preincubated with 1.2 μM HsRad51 or RecA for 4 min as described above under Renaturation. The concentration of MgCl2 was increased to 30 mM, duplex 83-mer was added at 3 μM, and incubation was continued at 37°C. The duplex was labeled with rhodamine at the 5′ end of the plus strand, 5′-R-A16(+)/A16(−).
To measure strand exchange by the fluorescence assay, the Rad51 or RecA filament was formed as described above on unlabeled A16(−) for 4 min (volume 20 μl) followed by the addition of filament to a reaction mixture (110 μl) containing 30 mM MgCl2 and 3 μM duplex oligonucleotide labeled on the 3′ end of the minus strand with fluorescein and on the 5′ end of the plus strand with rhodamine, 3′-F-A16(−)/5′-R-A16(+). The final concentrations of ssDNA and protein were 3 μM and 1.2 μM, respectively.
Fluorescence emission spectra were taken from 502 to 620 nm upon excitation at 493 nm on an SLM 8000C spectrofluorimeter (SLM Aminco, Urbana, IL). To determine the spectrum of sensitized emission (se) from rhodamine—i.e., emission due to FRET—we subtracted the summed spectra both for reaction mixtures containing only fluorescein-labeled DNA and for reaction mixtures containing only rhodamine-labeled DNA from spectra of reaction mixtures containing DNA labeled with both fluorophores. Corrections for small differences in concentration of the two fluorophores between samples were made by normalization based on reference measurements of emission from fluorescein at 520 nm and emission from rhodamine at 585 nm. A reaction mixture containing only rhodamine-DNA was used to generate a spectrum of nonsensitized emission (nse) from rhodamine following excitation at 493 nm. The nonsensitized emission from rhodamine provides the denominator of the expression that quantitates sensitized emission and permits comparison between experiments:
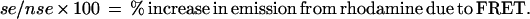 |
RESULTS
Complexes Made by HsRad51 with Oligonucleotides.
The binding of HsRad51 to 83-mer oligonucleotides in the presence of ATP was assessed by a filter assay, and by an assay based on enhancement of fluorescence from the conjugated fluorophore Cy3. According to both assays, HsRad51 bound cooperatively to a single-stranded 83-mer, and, according to the filter assay, HsRad51 also bound cooperatively to a double-stranded 83-mer (data not shown).
The stoichiometry of the interactions of Rad51 with DNA was observed by multiple assays (Table 1). Maximal binding, strand exchange, and ATPase activity occurred when there was one molecule of protein per two to three nucleotide residues or base pairs (Table 1). This stoichiometric relationship is very similar to that for RecA protein (30) and ScRad51 (22) and for HsRad51 interacting with long single strands (24).
Table 1.
Stoichiometry of interactions of HsRad51 with 83-mer oligonucleotides
| Assay | % of maximal reaction at protein/DNA ratio
|
||
|---|---|---|---|
| 1:2 | 1:3 | 1:6 | |
| Binding to ssDNA | |||
| Filter assay | 100 | 92 | 56 |
| Fluorescence assay | 100 | 95 | 78 |
| Strand exchange | |||
| Gel assay | 100 | 96 | 32 |
| Binding to dsDNA | |||
| Filter assay | 100 | 98 | 44 |
| ATPase activity with ssDNA | 100 | 84 | 51 |
Reaction conditions were as described in the text. Binding reactions were carried out at 37°C for 4 min. The same stoichiometry was observed when the strand exchange assay was done with 33-mer or 83-mer oligonucleotides. The strand exchange assay was carried out for 60 min and reached a yield of 60% in the case the 83-mer substrates. Protein/DNA ratio refers to moles of HsRad51 per mole of nucleotide residue, or, in the case of binding to duplex oligonucleotide, mole of base pairs. ATPase was assayed as described (25).
The binding of HsRad51 to 83-mer, in the presence of either ATP or ATP[γS], conferred protection from DNase I comparable to protection afforded by RecA protein. In the absence of nucleotide cofactor, HsRad51 protected the DNA less well (Table 2).
Table 2.
DNase I protection of DNA bound to HsRad51 or RecA
| Cofactor | % DNA protected from DNase I digestion
|
|
|---|---|---|
| RecA | HsRad51 | |
| ATP | 90 | 90 |
| ATP[γS] | 95 | 79 |
| None | 91 | 30 |
The reaction was done as described in the text. The amount of DNase I used (0.067 unit/μl) was sufficient to digest all unprotected DNA molecules in 2 min. Protected DNA was the fraction that remained trichloroacetic acid-insoluble.
Homologous Pairing of a Single Strand with Duplex DNA.
We devised two assays based on fluorescence spectroscopy that measure homologous pairing and strand exchange, respectively (see below). For studies with oligonucleotides as substrates, the spectroscopic approach enables one to distinguish homologous pairing from subsequent strand exchange, and provides assays that measure pairing and strand exchange in solution, in real time.
Homologous pairing of a single-stranded oligonucleotide with a duplex oligonucleotide was monitored by FRET. An oligonucleotide labeled at its 3′ end with fluorescein, 3′-F-A16(−), was used to form a presynaptic filament with HsRad51. Another fluorophore, rhodamine, was attached to the 5′ end of the complementary strand in duplex DNA, 5′-R-A16(+)/A16(−). Homologous pairing between the two DNA molecules should juxtapose the two fluors, resulting in nonradiative energy transfer from fluorescein to rhodamine when fluorescein is excited at 493 nm, near the wavelength for maximal excitation. As a result of this energy transfer, the fluorescence emission from fluorescein is quenched and that from rhodamine is enhanced.
As expected, homologous pairing of the described substrates by HsRad51 enhanced rhodamine emission (Fig. 1A). The net increase in rhodamine emission due to energy transfer, the so-called sensitized emission, was approximately 100% above the background of nonsensitized emission from rhodamine (Fig. 1B). The use of heterologous substrates resulted in a negligible enhancement of rhodamine emission above background (Fig. 1 C and D).
Figure 1.

Homologous pairing assayed by FRET. (A) Pairing of homologous single-stranded and duplex oligonucleotides, 3′-F-A16(−) with 5′-R-A16(+)/A16(−). The dotted line represents the summed emission spectra for a reaction lacking a rhodamine-labeled strand and for a reaction lacking a fluorescein-labeled strand, after excitation at 493 nm. The solid line represents the spectrum of the complete reaction mixture containing DNA conjugated with both dyes. (B) Difference spectrum derived from A: the sensitized emission from rhodamine—i.e., the net enhancement in rhodamine emission as a consequence of energy transfer, from which the background of nonsensitized emission has been subtracted. Since the sensitized and nonsensitized emission in this experiment were of equal magnitude, the net emission from rhodamine increased 100% as a result of energy transfer. (C) Heterologous DNA, 3′-F-X16 plus 5′-R-A16(+)/A16(−), otherwise as in A. The superposition of the two curves means that there was no detectable FRET. (D) Difference spectrum derived from C. The solid line represents a FRET signal that is negligible compared with the nonsensitized emission from rhodamine, shown for comparison by the dotted line. (E) Difference spectrum for a reaction of the homologous substrates shown in A, in the presence of ATP[γS]. As in D, the solid line represents a FRET signal that is negligible compared with the nonsensitized emission from rhodamine, shown for comparison by the dotted line.
Strand Exchange.
To detect strand exchange with oligonucleotides as substrates we used both the standard electrophoretic assay (see Materials and Methods) and another variant of the fluorometric assay (see below).
HsRad51 was preincubated with 83-mer oligonucleotide A16(−), followed by addition of a homologous duplex oligonucleotide, 5′-32P-A16(−)/A16(+). After 60 min, 60% of the labeled strands were displaced from the duplex oligonucleotide in the reaction promoted by HsRad51 (Fig. 2A, lane 3), compared with 78% in a reaction promoted by RecA protein under similar conditions (lane 7). Strand exchange catalyzed by HsRad51 required homology (lane 4), ATP (lane 5), and Mg2+ (lane 6). The optimal exchange reaction was observed at a ratio of one protein monomer per two to three nucleotides, the same stoichiometric relationship as observed for DNA binding (Table 1).
Figure 2.

Strand exchange mediated by HsRad51. Strand exchange reactions were done with single-stranded 83-mer A16(−) and duplex 83-mer 5′-32P-A16(−)/A16(+) as described in the text. (A) All lanes contained reaction mixtures with the following omissions or substitutions: lane 1, no protein; lane 2, HsRad51 but no ssDNA; lane 3, HsRad51 complete reaction in presence of ATP; lane 4, HsRad51 with X16 substituted for A16(−) as a heterologous control; lane 5, HsRad51 without ATP; lane 6, HsRad51 without Mg2+; lane 7, RecA substituted for HsRad51 in a complete reaction; lane 8, HsRad51 with ATP[γS] in place of ATP. (B) Lack of strand exchange mediated by HsRad51 in presence of ATP[γS]. Lanes 1, 2, 3, and 4, complete HsRad51 reaction at 5, 15, 30, and 45 min, respectively; lanes 5, 6, 7, and 8, complete RecA reaction at 5, 10, 15, and 30 min, respectively.
Relative Rates of Homologous Pairing and Strand Exchange.
As noted above, when fluorescein and rhodamine are juxtaposed, the emission from fluorescein is quenched and that from rhodamine is enhanced as a result of energy transfer. If one starts with a duplex oligonucleotide in which the two strands are juxtaposed, as in 3′-F-A16(−)/5′-R-A16(+), strand exchange should separate the two fluors and lead to enhanced emission from fluorescein, as it does (Fig. 3). In side-by-side experiments, using substrates 3′-F-A16(−) and 5′-R-A16(+)/A16(−), we observed the quenching of emission from fluorescein as homologous pairing occurred. We thus compared the rates of homologous pairing and strand exchange for both HsRad51 and RecA protein.
Figure 3.

Time courses of homologous pairing and strand exchange. (A) HsRad51. (B) RecA. Pairing and strand exchange were measured by two FRET assays with different respective locations of the fluorescent probes in the substrates (see Materials and Methods). Solid lines are plots of quenching of fluorescence from fluorescein as a result of homologous pairing; dotted lines are plots of enhancement of fluorescence as a result of strand exchange. The intensity of fluorescein emission was observed at 520 nm upon excitation at 493 nm.
The respective rates of pairing and strand exchange were indistinguishable for HsRad51 (Fig. 3A), and, moreover, were at least an order of magnitude slower than pairing and strand exchange catalyzed by RecA protein (Fig. 3B). Pairing and strand exchange by RecA was completed in 60 sec, while the same reactions by HsRad51 were not completed even at 900 sec. A similarly slow strand exchange by HsRad51 was seen when the time course was measured by the electrophoretic assay (data not shown).
Homologous Pairing vs. Hydrolysis of ATP.
HsRad51 hydrolyzed ATP in a reaction that was completely dependent upon the presence of DNA, but was very slow: in an experiment in which we found that the kcat for hydrolysis by RecA protein was 20, the kcat for HsRad51 was less than 1 (data not shown).
In the presence of ATP[γS], RecA protein can promote homologous pairing and limited strand exchange (Fig. 2B, lanes 5–8) (31–33), whereas HsRad51, in contrast, was unable to promote either strand exchange (Fig. 2A, lane 8; Fig. 2B, lanes 1–4) or the pairing of a single strand with duplex DNA, as assessed by the FRET assay (Fig. 1E). As noted above, HsRad51 binds to single-stranded oligonucleotides in a reaction that depends on either ATP or ATP[γS] (Table 2). The binding of 35S-labeled ATP[γS] to HsRad51 in the presence of an oligonucleotide was quantitatively the same as the binding to RecA (data not shown).
In contrast to its slow rate of pairing of a single strand with duplex DNA in the presence of ATP and its inability to catalyze that reaction in the presence of ATP[γS], HsRad51 promoted the renaturation of complementary single strands as fast and efficiently as RecA protein in the presence of either ATP or ATP[γS] (Fig. 4).
Figure 4.

Renaturation promoted by HsRad51 and RecA. Renaturation reactions were carried out as described in the text. The fraction of labeled oligonucleotide in double-stranded form was determined by gel electrophoresis. ▵, No protein; •, HsRad51 and ATP; ○, HsRad51 and ATP[γS]; ▪, RecA and ATP; □, RecA and ATP[γS].
DISCUSSION
We have purified HsRad51 protein that was overproduced in E. coli, and we have characterized the basic parameters of its action, using oligonucleotides as substrates. By several criteria, we found that the stoichiometry of interaction of HsRad51 with oligonucleotides is similar to that of RecA protein with DNA. Previous experiments of Benson et al. (24) showed that similarly prepared HsRad51 forms a nucleoprotein filament that resembles the filament formed by E. coli RecA protein, but also exhibits some differences.
The recombinant human protein carries out the hallmark reactions of RecA protein, including DNA-dependent hydrolysis of ATP, renaturation of complementary strands, homologous pairing of a single strand with duplex DNA, and strand exchange. There are, however, significant quantitative differences in the activities of HsRad51 as compared with RecA protein. These include rates of ATP hydrolysis, homologous pairing, and strand exchange that are at least an order of magnitude lower than the rates of these reactions promoted by E. coli RecA protein in side-by-side comparisons.
The much slower reactions promoted by HsRad51 do not appear to be due to a lower fraction of active molecules in the preparation, since the stoichiometry of the reaction is very similar to that of its E. coli homolog. It is possible, of course, that the average specific activity of recombinant HsRad51 is lower because it is missing some modification that is normally made in human cells, or that a factor is missing in our reactions. ScRad51, the yeast homolog of RecA, has been reported to require the action of an ssDNA-binding protein (23). In our system, however, with recombinant HsRad51 acting on oligonucleotides, we were unable to detect an effect of the human ssDNA-binding protein, RPA, or E. coli ssDNA-binding protein (unpublished observations).
Another set of observations suggests that the relative catalytic inefficiency of HsRad51 is related to its slow hydrolysis of ATP. First, we note from previous reports that the yeast homolog, ScRad51, also hydrolyzes ATP slowly and appears to promote strand exchange slowly (23), and that HsRad51 appears to have a reduced capacity to unwind duplex DNA (24). In the present experiments, we found that in the presence of ATP[γS], HsRad51 was completely unable to promote strand exchange of as few as 83 nucleotide residues. Moreover, the lack of strand exchange by HsRad51 in the presence of ATP[γS] was attributable to a failure to form stable joint molecules as detected by a sensitive and nondisruptive fluorescence assay. Under the same conditions, RecA protein promoted strand exchange in the presence of ATP[γS] (Fig. 2B). The failure of HsRad51 to form joint molecules in the presence of ATP[γS] does not appear to be attributable to the more obvious trivial causes: HsRad51 bound ATP[γS] and formed nucleoprotein complexes (Table 2) that were as active as those made by RecA protein in the promotion of protein-dependent renaturation of complementary strands of DNA (Fig. 4). Since HsRad51 can bind to both ss- and dsDNA, the simultaneous binding to both in the presence of ATP[γS] might provide another uninteresting reason for the failure to form joint molecules. We found, however, that when single-stranded 83-mer was preincubated with HsRad51 and ATP[γS], the subsequent addition of a 10-fold excess of homologous duplex oligonucleotide did not detectably “steal” protein from the complex of HsRad51 with 83-mer (unpublished observations).
Our working hypothesis, which rationalizes the observed behavior of HsRad51, is that the low rate of hydrolysis of ATP affects a rate-limiting step that is essential for both homologous pairing and strand exchange. There may be an energy-demanding step that is required either to effect homologous recognition or to stabilize an initial recognition complex. The role of ATP hydrolysis in the pairing and exchange reactions of the extensively studied RecA protein remains uncertain. If, as we suggest, human HsRad51 is a variant that exhibits a more direct coupling between hydrolysis of ATP on the one hand and homologous pairing and strand exchange on the other, further study of this protein may provide important clues on the underlying mechanism of homologous recognition and strand exchange.
Acknowledgments
We are grateful to Jan Zulkeski for data processing and to Zhufang Li for technical assistance. We thank Dr. Marc Wold for the generous gift of RPA and Dr. T. Jovin for helpful advice on measurement of FRET. We appreciate the careful reading of the manuscript by our colleagues Ewa Folta-Stogniew and Oleg Kovalenko. We thank Dr. Steve West for a preprint of related studies of HsRad51, which we received just prior to submission of this manuscript (34). This research was sponsored by the following grants from the National Institutes of Health: 5PO1 CA39238-09 and 5R37 GM33504-13.
Footnotes
Abbreviations: HsRad51, Homo sapiens Rad51 protein; ScRad51, Saccharomyces cerevisiae Rad51 protein; RecA, Escherichia coli RecA protein; ATP[γS], adenosine 5′-[γ-thio]triphosphate; FRET, fluorescence resonance energy transfer; ssDNA, single-stranded DNA; dsDNA, double-stranded DNA.
References
- 1.Kowalczykowski S C, Eggleston A K. Annu Rev Biochem. 1994;63:991–1043. doi: 10.1146/annurev.bi.63.070194.005015. [DOI] [PubMed] [Google Scholar]
- 2.Aboussekhra A, Chanet R, Adjiri A, Fabre F. Mol Cell Biol. 1992;12:3224–3234. doi: 10.1128/mcb.12.7.3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shinohara A, Ogawa H, Ogawa T. Cell. 1992;69:457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 4.Basile G, Aker M, Mortimer R K. Mol Cell Biol. 1992;12:3235–3246. doi: 10.1128/mcb.12.7.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muris D F R, Vreken K, Carr A M, Broughton B C, Lehmann A R, Lohman P H M, Pastink A. Nucleic Acids Res. 1993;21:4586–4591. doi: 10.1093/nar/21.19.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maeshima K, Morimatsu K, Shinohara A, Horii T. Gene. 1995;160:195–200. doi: 10.1016/0378-1119(95)00148-y. [DOI] [PubMed] [Google Scholar]
- 7.Terasawa M, Shinohara A, Hotta Y, Ogawa H, Ogawa T. Genes Dev. 1995;9:925–934. doi: 10.1101/gad.9.8.925. [DOI] [PubMed] [Google Scholar]
- 8.Cheng R, Baker T I, Cords C E, Radloff R J. Mutat Res. 1993;294:223–234. doi: 10.1016/0921-8777(93)90005-2. [DOI] [PubMed] [Google Scholar]
- 9.Hatakeyama S, Ishii C, Inoue H. Mol Gen Genet. 1995;249:439–446. doi: 10.1007/BF00287106. [DOI] [PubMed] [Google Scholar]
- 10.Sato S, Hotta Y, Tabata S. DNA Res. 1995;2:89–93. doi: 10.1093/dnares/2.2.89. [DOI] [PubMed] [Google Scholar]
- 11.Morita T, Yoshimura Y, Yamamoto A, Murata K, Mori M, Yamamoto H, Matsushiro A. Proc Natl Acad Sci USA. 1993;90:6577–6580. doi: 10.1073/pnas.90.14.6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bezzubova O, Shinohara A, Mueller R G, Ogawa H, Buerstedde J. Nucleic Acids Res. 1993;21:1577–1580. doi: 10.1093/nar/21.7.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shinohara A, Ogawa H, Matsuda Y, Ushio N, Ikeo K, Ogawa T. Nat Genet. 1993;4:239–243. doi: 10.1038/ng0793-239. [DOI] [PubMed] [Google Scholar]
- 14.Yoshimura Y, Morita T, Yamamoto A, Matsushiro A. Nucleic Acids Res. 1993;21:1665. doi: 10.1093/nar/21.7.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petes T D, Malone R E, Symington L S. In: The Molecular and Cellular Biology of the Yeast Saccharomyces: Genome Dynamics, Protein Synthesis, and Energetics. Broach J R, Jones E W, Pringle J R, editors. Vol. 1. Plainview, NY: Cold Spring Harbor Lab. Press; 1991. pp. 407–521. [Google Scholar]
- 16.Game J C. Cancer Biol. 1993;4:73–83. [PubMed] [Google Scholar]
- 17.Bishop D K. Cell. 1994;79:1081–1092. doi: 10.1016/0092-8674(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 18.McKee B D, Ren X, Hong C. Chromosoma. 1996;104:479–488. doi: 10.1007/BF00352112. [DOI] [PubMed] [Google Scholar]
- 19.Plug A W, Xu J, Reddy G, Golub E I, Ashley T. Proc Natl Acad Sci USA. 1996;93:5920–5924. doi: 10.1073/pnas.93.12.5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li M-J, Peakman M-C, Golub E I, Reddy G, Ward D C, Radding C M, Maizels N. Proc Natl Acad Sci USA. 1996;93:10222–10227. doi: 10.1073/pnas.93.19.10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Matsushiro A, Yoshimura Y, Morita T. Proc Natl Acad Sci USA. 1996;93:6236–6240. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sung P, Robberson D L. Cell. 1995;82:453–461. doi: 10.1016/0092-8674(95)90434-4. [DOI] [PubMed] [Google Scholar]
- 23.Sung P. Science. 1994;265:1241–1243. doi: 10.1126/science.8066464. [DOI] [PubMed] [Google Scholar]
- 24.Benson F E, Stasiak A, West S C. EMBO J. 1994;13:5764–5771. doi: 10.1002/j.1460-2075.1994.tb06914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shibata T, Cunningham R P, Radding C M. J Biol Chem. 1981;256:7557–7564. [PubMed] [Google Scholar]
- 26.Rao B J, Chiu S K, Radding C M. J Mol Biol. 1993;229:328–343. doi: 10.1006/jmbi.1993.1038. [DOI] [PubMed] [Google Scholar]
- 27.Ashley T, Plug A, Xu J, Solari A J, Reddy G, Golub E I, Ward D C. Chromosoma. 1995;104:19–28. doi: 10.1007/BF00352222. [DOI] [PubMed] [Google Scholar]
- 28.Swaroop A, Weissman S M. Nucleic Acids Res. 1988;16:8739. doi: 10.1093/nar/16.17.8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rao B J, Jwang B, Radding C M. J Mol Biol. 1990;213:789–809. doi: 10.1016/S0022-2836(05)80264-5. [DOI] [PubMed] [Google Scholar]
- 30.DiCapua E, Engel A, Stasiak A, Koller T. J Mol Biol. 1982;157:87–103. doi: 10.1016/0022-2836(82)90514-9. [DOI] [PubMed] [Google Scholar]
- 31.Rosselli W, Stasiak A. J Mol Biol. 1990;216:335–352. doi: 10.1016/S0022-2836(05)80325-0. [DOI] [PubMed] [Google Scholar]
- 32.Menetski J P, Bear D G, Kowalczykowski S C. Proc Natl Acad Sci USA. 1990;87:21–25. doi: 10.1073/pnas.87.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J-I, Cox M M, Inman R B. J Biol Chem. 1992;267:16438–16443. [PubMed] [Google Scholar]
- 34.Baumann P, Benson F E, West S. Cell. 1996;87:757–766. doi: 10.1016/s0092-8674(00)81394-x. [DOI] [PubMed] [Google Scholar]
- 35.Haaf T, Golub E I, Reddy G, Radding C M, Ward D C. Proc Natl Acad Sci USA. 1995;92:2298–2302. doi: 10.1073/pnas.92.6.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]