

SUMMARY
Arginase is the focal enzyme of the urea cycle hydrolyzing L-arginine to urea and L-ornithine. Emerging studies have identified arginase in the vasculature and have implicated this enzyme in the regulation of NO synthesis and the development of vascular disease.
Arginase inhibits the production of nitric oxide (NO) via several potential mechanisms, including competition with NO synthase (NOS) for the substrate L-arginine, uncoupling of NOS resulting in the generation of NO scavenger, superoxide, and peroxynitrite, repression of the translation and stability of inducible NOS protein, inhibition of inducible NOS activity via the generation of urea, and by sensitization of NOS to its endogenous inhibitor, asymmetric dimethyl-L-arginine.
Upregulation of arginase inhibits endothelial NOS-mediated NO synthesis and may contribute to endothelial dysfunction in hypertension, aging, ischemia-reperfusion, and diabetes.
Arginase also redirects the metabolism of L-arginine to L-ornithine and the formation of polyamines and L-proline which are essential for smooth muscle cell growth and collagen synthesis. Induction of arginase may therefore also promote aberrant vessel wall remodeling and neointima formation.
Arginase represents a promising novel therapeutic target that may reverse endothelial and smooth muscle cell dysfunction and prevent vascular disease.
Keywords: arginine, arginase, diabetes, endothelial dysfunction, hypertension, nitric oxide synthase, smooth muscle cell proliferation
INTRODUCTION
L-arginine is a cationic, semi-essential amino acid that is involved in numerous physiological processes. It is a necessary precursor for the synthesis of L-ornithine, L-proline, polyamines, agmatine, creatine, and protein (1,2). Significantly, L-arginine is also the exclusive substrate for nitric oxide synthase (NOS) which generates the signaling molecule, nitric oxide (NO) (3) (Figure 1). NO plays a critical role in the circulation by decreasing vascular tone, platelet and leukocyte activation, smooth muscle cell proliferation, extracellular matrix deposition, and endothelial cell death (see 4,5). Decreased bioavailability of NO is a common mechanism involved in the pathogenesis of various vascular disorders, including hypertension, atherosclerosis, diabetes, and ischemia-reperfusion injury (6–8). Interestingly, clinical and experimental studies during the past decade demonstrate that L-arginine administration restores NO synthesis and vascular function in several cardiovascular diseases, suggesting that impaired L-arginine availability underlies these vascular pathologies (9–12). That L-arginine influences NO generation is clearly supported by the phenotype of a rare genetic L-arginine deficiency in humans (13). Although the precise reason for the reduction in L-arginine availability remains unknown, emerging evidence suggests that catabolism of L-arginine by arginase may be involved.
Figure 1.
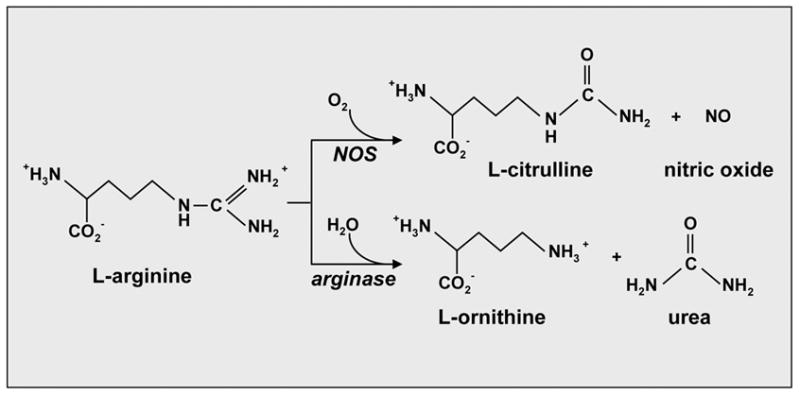
Metabolism of L-arginine by nitric oxide synthase (NOS) and arginase. NOS oxidatively degrades L-arginine into L-citrulline and nitric oxide (NO) while arginase hydrolyzes L-arginine to urea and L-ornithine.
Arginase is the central enzyme of the hepatic urea cycle and comprises the principal route for the disposal of excess nitrogen resulting from amino acid and nucleotide metabolism (14). However, recent studies indicate that arginase is also expressed in extrahepatic tissues, including blood vessels, and that it competes with NOS for the substrate L-arginine. In addition, intravenous administration of arginase has been reported to exert significant effects in the circulation, suggesting a key role for this protein in regulating vascular function (15). In this review, we will explore regulatory interactions between arginase and NOS in the vasculature and highlight recent work from our laboratory and others implicating arginase in the development of vascular disease.
ARGINASE AND NITRIC OXIDE HOMEOSTASIS
Arginase is a binuclear manganese metalloenzyme that catalyzes the hydrolysis of L-arginine to L-ornithine and urea (Figure 1). Two distinct genetic isoforms of arginase have been identified that share approximately 60% amino acid sequence homology (16,17). These isozymes differ from each other with respect to their tissue distribution, subcellular localization, and immunological reactivity (18). Aginase I is a cytosolic enzyme that is abundantly expressed in the liver and constitutes the majority of total body arginase activity. In contrast, arginase II is a mitochondrial protein that has a wide tissue distribution being most highly expressed in the kidney and prostate and poorly expressed in the liver (17,19).
Recent studies have documented the presence of arginase in the vasculature. Studies in our laboratory were the first to demonstrate that vascular smooth muscle cells possess arginase activity (20). In particular, we found that cultured rat aortic smooth muscle cells exhibit significant arginase activity, and that specific growth factors and hemodynamic forces are capable of upregulating arginase activity by selectively inducing the expression of arginase I (21,22). The selective expression of arginase I in vascular smooth muscle cells has also confirmed by other investigators (23) and contrast with results obtained in endothelial cells where both isoforms of arginase are found (24–26). Arginase I and II have also been detected in several blood vessels, including the aorta, carotid and pulmonary artery, and coronary and gracilis muscle arterioles (27–32). Presently, little is known regarding the signaling pathways that regulate arginase expression in vascular cells. However, recent studies have implicated the JakStat pathway and cAMP in the induction of arginase I in vascular smooth muscle (23). In addition, the Rho/ROCK pathway appears to be involved in the elevation of endothelial arginase II activity by thrombin, whereas the epidermal growth factor receptor has been linked to the cytokine-mediated induction of arginase I and II in endothelial cells (33,34).
While arginase activity had been demonstrated in vascular cells, the lack of potent and selective pharmacological inhibitors hampered early attempts to establish a role for arginase. However, the recent development of N-hydroxy-guanidinium or boronic acid derivatives, such as NG-hydroxy-nor-L-arginine, 2(S)-amino-6-boronohexanoic acid, and S-(2-boronoethyl)-L-cysteine (BEC), that can bridge the binuclear manganese cluster of arginase has yielded highly effective and specific inhibitors that can now be used to probe arginase function (35). In addition, the increasing refinement of molecular and genetic approaches targeting gene expression offers another highly effective approach by which the role of arginase may be delineated.
Consideration of the biochemical properties of arginase and NOS supports the notion that arginase may inhibit NO synthesis by competing with NOS for L-arginine. Although the affinity of L-arginine is much higher for purified NOS (Km ~ 2–20 μM) than for arginase (Km ~ 1–5 mM), the maximum activity of arginase is more than 1,000 times that of NOS suggesting similar rates of substrate utilization at physiologic L-arginine concentrations (2). In fact, early studies of arginine metabolism by activated macrophages found that the majority of L-arginine was consumed for the production of urea rather than NO, and that inhibition of arginase or supplementation of culture media with L-arginine results in augmented NO production (36–38). More recently, inhibition of arginase has been shown to stimulate NO synthesis in endothelial cells (26). In addition, overexpression of arginase I or arginase II suppresses NO generation in endothelial cells and this is associated with a significant decrease in intracellular L-arginine content (39). Interestingly, constitutive expression of arginase in microvascular endothelial cells counteracts NO-mediated dilation, suggesting that arginase subserves a tonic vasoconstrictor function (31,32).
Genetic studies also support a role for arginase in regulating L-arginine availability. A deficiency in arginase I results in hyperargininemia in both humans and mice (40,41). However, the phenotype in arginase I-deficient mice is more acute resulting in severe symptoms of hyperammonemia and death shortly after birth. Similarly, the arginase II null mouse exhibits an approximate two-fold increase in plasma L-arginine concentration (42). However, unlike the arginase I-deficient animal, the phenotype of the arginase II-deleted animal is indistinguishable from wild type mice. These findings suggest that both isoforms of arginase play an essential role in regulating circulating L-arginine levels but that they exert disparate biological actions.
Aside from blocking NO synthesis by depleting the cell of substrate for NOS, the arginase-mediated removal of L-arginine inhibits the expression of inducible NOS (iNOS) by repressing the translation as well as the stability of iNOS protein (43,44). Furthermore, arginase may inhibit iNOS-mediated NO production through the generation of urea (45). These findings suggest that arginase downregulates NO formation via multiple mechanisms. Interestingly, NG-hydroxy-L-arginine, an intermediate formed during the catalysis of L-arginine by NOS, is a potent inhibitor of arginase, suggesting that NOS may also influence arginase activity (46). The nature and significance of these reciprocal regulatory interactions between these two enzyme systems in the circulation remain to be fully determined.
ARGINASE AND ENDOTHELIAL DYSFUNCTION
The release of NO by endothelial NOS (eNOS) plays a crucial role in preserving vascular homeostasis. In response to changes in shear stress or receptor stimulation, NO is released from the vascular endothelium to promote blood flow by inhibiting vascular tone and platelet aggregation. Endothelial dysfunction resulting from impaired NO synthesis is a central feature in many vascular disorders. Emerging evidence from numerous laboratories implicates arginase as a crucial mediator of endothelial dysfunction in several pathologic states. Several reports have linked increases in arginase activity to the development of endothelial malfunction and high blood pressure. In a recently completed study, we found that skeletal muscle arterioles isolated from hypertensive salt-loaded salt-sensitive Dahl rats exhibit higher levels of arginase I and II and blunted vasodilatory responses to the endothelium-dependent dilator, acetylcholine, compared to normotensive low salt controls (32). In addition, flow-induced dilation is abolished in high salt-treated animals. However, pretreatment of vessels with the arginase inhibitor, BEC, enhances arteriolar responses to acetylcholine and flow in both high and low salt groups. We also observed that BEC restores acetylcholine and flow-mediated dilation in high salt arterioles to levels observed in low salt vessels. Interestingly, pretreatment of vessels with L-arginine also abolishes the difference in arteriolar dilation between high and low salt groups. These results suggest that arginase activity contributes to arteriolar NO dysfunction in salt-loaded salt-sensitive Dahl rats by depleting endothelial cells of L-arginine.
Similar findings have also been reported in other animal models of hypertension. Aortic arginase activity is elevated in deoxycorticosterone acetate-salt hypertensive rats and it may accelerate the development of hypertension in these animals (47). In addition, arginase I activity and expression is augmented in endothelial cells of coronary arterioles isolated from renovascular hypertensive pigs (48). Moreover, coronary arterioles from these hypertensive animals demonstrate diminished NO release and NO-mediated dilation to adenosine that are partially restored by arginase inhibition or by incubation with L-arginine (48). Furthermore, aortic arginase activity and arginase I and II expression is increased in spontaneously hypertensive rats and this is associated with attenuated relaxation responses to acetylcholine (28). Significantly, chronic oral treatment with the non-selective arginase inhibitor, α-difluoromethylornithine, improves the vasorelaxant response to acetylcholine and prevents the development of hypertension in spontaneously hypertensive rats. Consistent with a role for arginase in promoting endothelial dysfunction in humans, increased arginase II expression and decreased NO synthesis has also been reported in endothelial cells of patients with pulmonary hypertension (30).
Arginase mediated endothelial dysfunction has also been discovered in aging blood vessels (27). Arginase inhibition results in significantly greater relaxation of preconstricted aortic rings obtained from old compared to young rats and this is associated with elevated expression of both arginase I and II. In addition, L-arginine results in a significant relaxation of preconstricted rings derived from young animals whereas relaxation was absent in the old rats. However, pharmacological inhibition of arginase completely restores vasorelaxation to L-arginine in old vessels such that the response is similar to that seen in young aortic rings. More recently, knockdown of arginase I expression using an antisense oligonucleotide approach has also been demonstrated to restore NO signaling in the vasculature of old animals (49). Arginase has also been shown to counteract NO-mediated dilation of coronary arterioles following ischemia-reperfusion in pigs (50). Ischemia-reperfusion of coronary vessels inhibits arteriolar production of NO and endothelium-dependent dilation, which is associated with a pronounced elevation in vascular arginase expression and activity. Moreover, arginase inhibition or L-arginine supplementation restores NO synthesis and vasodilatory function following ischemia-reperfusion, suggesting an important pathophysiological role for arginase in this setting. Interestingly, preliminary studies from our laboratory indicate that high glucose levels elevate vascular arginase activity and that arginase may also contribute to endothelial dysfunction in obesity and diabetes (51).
In addition to limiting substrate for eNOS, the arginase-mediated reduction of L-arginine may also serve to sensitize endothelial cells to the endogenous NOS inhibitor, NG-asymmetric dimethyl-L-arginine, which is elevated in various pathological conditions associated with endothelial dysfunction (52). Prior reports suggest that limited L-arginine availability also promotes uncoupling of eNOS resulting in oxygen free radical formation NO (53). Because reactive oxygen can directly inactivate NO, increased arginase activity and the resulting intracellular depletion of L-arginine may also decrease NO bioavailability. Moreover, the simultaneous generation of NO and superoxide by eNOS may increase the formation of peroxynitrite leading to increased vascular reactivity and cell damage. Thus, the catabolism of L-arginine by arginase may promote endothelial dysfunction via several distinct pathways.
The mechanism(s) involved in the upregulation of arginase in vascular disease is not known but may involve multiple mediators. In particular, increased production of inflammatory cytokines and/or growth factors observed in vascular disorders may be involved since they inhibit endothelium-dependent NO-mediated relaxation and stimulate arginase activity in cultured endothelial cells (5,24,54). Enhanced formation of reactive oxygen species is another common finding in many vascular pathologies that may also influence arginase activity since potential redox-sensitive responsive elements have been identified in the arginase I promoter region (55). In addition, changes in hemodynamics associated with hypertension may also be involved in the upregulation of arginase. In this respect, we found that cyclic strain is a potent inducer of arginase I mRNA expression in vascular cells (22). Thus, both humoral and hemodynamic stimuli may induce the expression of arginase in the vasculature.
Finally, the role of arginase in modulating NO production is not limited to the vascular endothelium. Recently, it was found that the corpus cavernosa from diabetic patients with erectile dysfunction exhibits higher arginase activity, diminished NO synthesis, and reduced smooth muscle relaxation (56). Interestingly, pharmacological inhibition of arginase activity corrects the defect in NO production and relaxation in diabetic tissue. Increased arginase expression also contributes to impaired NO synthesis and wound healing in diabetes, suggesting that arginase may contribute to many of the clinical complications associated with diabetes (57). More recently, arginase I expression was identified in cardiac myoctes and found to reduce myocardial NO production (58). Furthermore, in a model of compensated left ventricular hypertrophy, arginase expression is decreased facilitating NO synthesis (58). It is provocative to speculate that elevated arginase activity may lead to a decompensated form of heart failure. Arginase-mediated impairment in NO synthesis has also been implicated in other pathological conditions, including asthma, cystic fibrosis, glumerulonephritis, psoriasis, arthritis, and sickle cell disease (59–64).
ARGINASE AND VASCULAR SMOOTH MUSCLE CELL DYSFUNCTION
Accumulating evidence suggests that arginase may also promote vascular smooth muscle cell proliferation and extracellular matrix deposition by generating biologically important molecules. While the arginase product urea is readily secreted through the kidney, the other arginase product L-ornithine is further metabolized by ornithine decarboxylase to the polyamine, putrescine, which forms the successive polyamines spermine and spermidine via the sequential transfer of a propylamine group from S-adenosylmethionine (65). Polyamines play an integral role in the mitogenic response of cells. Vascular smooth muscle cell proliferation is preceded by increases in ornithine decarboxylase activity and polyamine synthesis, and inhibition of polyamine formation inhibits smooth muscle cell growth (66–68). L-ornithine is also converted by ornithine aminotransferase to pyrroline-5-carboxylate, which is further metabolized to L-proline, which is essential for the synthesis of many structural proteins, including collagen (5). Indeed, we recently found that production of collagen by transforming growth factor-β1 and cyclic strain is dependent on L-proline formation (21,22).
Several lines of evidence support a role for arginase in smooth muscle cell growth. There is increased expression of arginase and formation of polyamines in proliferating cells (69). Moreover, stable overexpression of arginase I in rat aortic smooth muscle cells is associated with increased production of polyamines and higher rates of cell proliferation (70). In addition, pharmacological inhibition of arginase activity decreases the production of polyamines and inhibits the growth of smooth muscle cells. The ability of arginase to directly stimulate smooth muscle cell proliferation by elevating polyamines synthesis may also be further amplified by the arginase-mediated suppression of NO, which is an established inhibitor of smooth muscle cell growth (see 4,5). Interestingly, excessive production of NO by iNOS also promotes smooth muscle cell apoptosis (71), providing another potential mechanism by which arginase may augment cell growth. Indeed, recent work suggests that arginase may regulate vascular remodeling. Intimal hyperplasia in premenopausal human uterine arteries is associated with enhanced arginase activity in both endothelial cells and the smooth muscle layer (72). Similarly, in a preliminary study (73), we found that arginase expression is increased following arterial injury in rats and that it contributes to the remodeling response. Interestingly, arginase has also been identified as a possible candidate gene that influences atherosusceptibility (74).
CONCLUSION
Arginase has recently emerged a critical regulator of NO synthesis that may contribute to the development of numerous pathologies, including vascular disease (Table 1). Several potential mechanisms account for the adverse effects of arginase in the vasculature (Figure 2). In the circulation, the release of NO by eNOS plays a critical role in preserving vascular homeostasis by inhibiting vascular tone, platelet aggregation, and inflammation (Figure 2). In addition, the iNOS-mediated formation of NO functions in an autocrine fashion to limit collagen synthesis and the medial expansion of smooth muscle cells by blocking cell growth and stimulating apoptosis. However, arginase may promote both endothelial and vascular smooth muscle cell dysfunction by modulating the intracellular metabolism of L-arginine. Specifically, the ability of arginase to compete with eNOS for the substrate L-arginine may impair the synthesis of NO and lead to endothelial dysfunction. In addition, arginase can redirect the metabolism of L-arginine in smooth muscle cells from NO to L-ornithine and the formation of polyamines and L-proline, which can induce vascular lesion formation by stimulating smooth muscle cell proliferation and collagen deposition. These actions of arginase are further magnified by the suppression of NO release, which serves as a well-recognized inhibitor of smooth muscle cell growth and collagen synthesis.
Table 1.
Pathological conditions associated with arginase-mediated impairment in NO synthesis.
Figure 2.
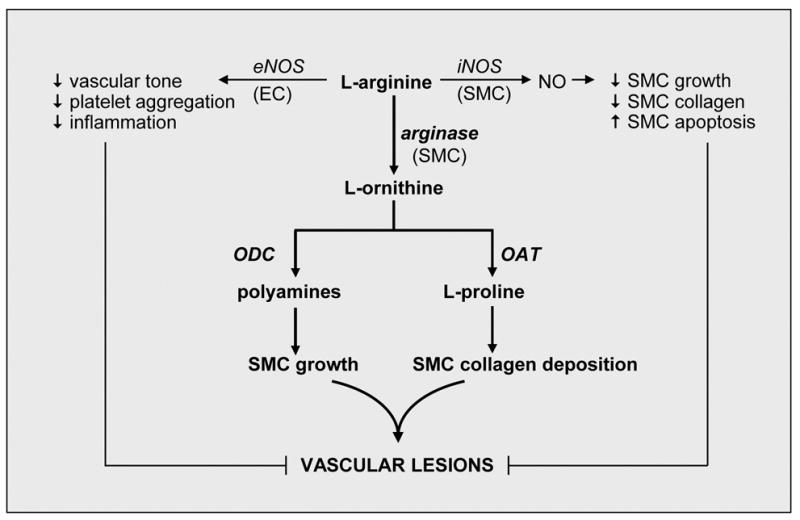
Model for the induction of vascular disease by arginase. In the circulation, the generation of NO by eNOS in endothelial cells (EC) plays a fundamental role in maintaining vascular homeostasis by inhibiting vascular tone, platelet aggregation, and inflammation. In addition, the iNOS-mediated formation of NO by vascular smooth muscle cells (SMC) functions in a negative feedback manner to inhibit collagen synthesis and SMC growth, and stimulate SMC apoptosis. However, arginase may promote both EC and SMC dysfunction by modulating the metabolism of L-arginine. In particular, the ability of arginase to compete with eNOS for the substrate L-arginine impairs NO synthesis and leads to endothelial dysfunction. In addition, arginase redirects the metabolism of L-arginine from NO to L-ornithine and the formation of polyamines and L-proline by the action of ornithine decarboxylase (ODC) and ornithine aminotransferase (OAT), respectively. The formation of polyamines and L-proline play an essential role in SMC proliferation and collagen deposition promoting the development of vascular lesions. These actions of arginase are further amplified by the repression of NO release, which serves as a well-established inhibitor of SMC growth and collagen synthesis.
Based on this model, we propose that arginase is a highly attractive therapeutic target in modifying the arterial response to injury. Since treatment modalities directed against arginase offer the possibility of correcting both vascular endothelial and smooth muscle cell dysfunction, they may prove advantageous over other treatment options that target a single cell type. In addition, targeting arginase provides a more selective approach in correcting alterations in L-arginine metabolism compared to L-arginine administration. While dietary L-arginine supplementation may restore NO synthesis, the shunting of L-arginine to alternative pathways as well as the many NO-independent actions of L-arginine may compromise the effectiveness of this nutritional strategy (75). The continued development of specific, selective molecules that target arginase activity and/or expression represents a promising area of research that may lead to novel therapeutic interventions in the treatment of vascular disease.
Acknowledgments
This work was supported by National Institutes of Health grants R01 HL59976 (WD), R01 HL74966 (WD), R01 HL76187 (RAJ), the Juvenile Diabetes Research Foundation (WD), and an Institutional Development Award (IdeA) Program P20 RR017659 (FKJ) from the National Center for Research Resources.
Abbreviations
- NO
nitric oxide
- NOS
nitric oxide synthase
- eNOS
endothelial nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- BEC
S-(2-boronoethyl)-L-cysteine
References
- 1.Barbul B. Arginine: biochemistry, physiology, and therapeutic implications. J Parenter Enteral Nutr. 1986;10:227–238. doi: 10.1177/0148607186010002227. [DOI] [PubMed] [Google Scholar]
- 2.Wu G, Morris SM., Jr Arginine metabolism: nitric oxide and beyond. Biochem J. 1998;336:1–17. doi: 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmer RMJ, Ashton AS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1998;333:664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- 4.Loscalzo J, Welch G. Nitric oxide and its role in the cardiovascular system. Prog Cardiovasc Dis. 1995;35:87–104. doi: 10.1016/s0033-0620(05)80001-5. [DOI] [PubMed] [Google Scholar]
- 5.Durante W. Regulation of L-arginine transport and metabolism in vascular smooth muscle cells. Cell Biochem Biophys. 2001;35:19–34. doi: 10.1385/CBB:35:1:19. [DOI] [PubMed] [Google Scholar]
- 6.John S, Schmeider RE. Potential mechanisms of impaired endothelial function in arterial hypertension and hypercholesterolemia. Curr Hypertens Rep. 2003;5:199–207. doi: 10.1007/s11906-003-0021-1. [DOI] [PubMed] [Google Scholar]
- 7.Soriano FG, Virag L, Szabo C. Diabetic endothelial dysfunction: role of reactive oxygen and nitrogen species production and poly(ADP-ribose) polymerase activation. J Mol Med. 2001;79:437–448. doi: 10.1007/s001090100236. [DOI] [PubMed] [Google Scholar]
- 8.Lefer Am, Lefer DJ. The role of nitric oxide and cell adhesion molecules on the microcirculation in ischemia-reperfusion. Cardiovas Res. 1996;32:743–751. [PubMed] [Google Scholar]
- 9.Drexler H, Zeiher AM, Meinzer K, Just H. Correction of endothelial dysfunction in coronary microcirculation of hypercholestrolemic patients by L-arginine. Lancet. 1991;67:1301–1308. doi: 10.1016/0140-6736(91)92372-9. [DOI] [PubMed] [Google Scholar]
- 10.Cooke JP, Singer AH, Tsao P, Zera P, Rowan RA, Billingham ME. Antiatherogenic effects of L-arginine in the hypercholestrolemic rabbit. J Clin Invest. 1992;90:1168–1172. doi: 10.1172/JCI115937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tarry WC, Markhoul RG. L-Arginine improves endothelial-dependent vasorelaxation and reduces intimal hyperplasia after balloon injury. Arterioscler Thromb. 1994;14:938–943. doi: 10.1161/01.atv.14.6.938. [DOI] [PubMed] [Google Scholar]
- 12.Tousoulis D, Antoniades C, Tentolouris C, Goumas G, Stefanadis C, Toutouzas P. L-Arginine in cardiovascular disease: dream or reality? Vasc Med. 2002;7:203–211. doi: 10.1191/1358863x02vm434ra. [DOI] [PubMed] [Google Scholar]
- 13.Loscalzo J. An experiment in nature: genetic L-arginine deficiency and NO insufficiency. J Clin Invest. 2001;108:663–664. doi: 10.1172/JCI13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meijer AJ, Lamers WH, Chamuleau AFM. Nitrogen metabolism and ornithine cycle function. Physiol Rev. 1990;70:701–748. doi: 10.1152/physrev.1990.70.3.701. [DOI] [PubMed] [Google Scholar]
- 15.Nishimura H, Rosenblum WI, Nelson GH, Boynton S. Agents that modify EDRF formation alter antiplatelet properties of brain arteriolar endothelium in vivo. Am J Physiol. 1991;261:H15–H21. doi: 10.1152/ajpheart.1991.261.1.H15. [DOI] [PubMed] [Google Scholar]
- 16.Dizikes GJ, Grody WW, Kern RM, Cederbaum SD. Isolation of human liver arginase cDNA and demonstration of nonhomology between the two human arginase genes. Biochem Biophys Res Commun. 1986;141:53–59. doi: 10.1016/s0006-291x(86)80333-3. [DOI] [PubMed] [Google Scholar]
- 17.Vockley JG, Jenkinson CP, Shukla H, Kern RM, Grody WW, Cederbaum SD. Cloning and characterization of the human type II arginase gene. Genomics. 1996;2:118–123. doi: 10.1006/geno.1996.0606. [DOI] [PubMed] [Google Scholar]
- 18.Jenkinson CP, Grody WW, Cederbaum SD. Comparative properties of arginases. Comp Biochem Physiol. 1996;114B:107–132. doi: 10.1016/0305-0491(95)02138-8. [DOI] [PubMed] [Google Scholar]
- 19.Morris SM, Jr, Bhamidipati D, Kepka-Lenhart D. Human type II arginase: sequence analysis and tissue-specific expression. Gene. 1997;193:157–161. doi: 10.1016/s0378-1119(97)00099-1. [DOI] [PubMed] [Google Scholar]
- 20.Durante W, Liao L, Peyton KJ, Schafer AI. Lysophosphatidylcholine regulates cationic amino acid transport and metabolism in vascular smooth muscle cells: role in polyamine synthesis. J Biol Chem. 1997;272:30154–30159. doi: 10.1074/jbc.272.48.30154. [DOI] [PubMed] [Google Scholar]
- 21.Durante W, Liao L, Reyna SV, Peyton KJ, Schafer AI. Transforming growth factor-β1 stimulates L-arginine transport and metabolism in vascular smooth muscle cells: role in polyamine and collagen synthesis. Circulation. 2001;103:1121–1127. doi: 10.1161/01.cir.103.8.1121. [DOI] [PubMed] [Google Scholar]
- 22.Durante W, Liao L, Reyna SV, Peyton KJ, Schafer AI. Physiologic cyclic stretch directs L-arginine transport and metabolism to collagen synthesis in vascular smooth muscle cells. FASEB J. 2000;14:1775–1783. doi: 10.1096/fj.99-0960com. [DOI] [PubMed] [Google Scholar]
- 23.Wei LH, Jacobs AT, Morris SM, Jr, Ignarro LJ. IL-4 and IL-13 upregulate arginase I expression by cAMP and JAK/STAT6 pathways in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2000;279:C248–C256. doi: 10.1152/ajpcell.2000.279.1.C248. [DOI] [PubMed] [Google Scholar]
- 24.Buga GM, Singh R, Pervin A, et al. Arginase activity in endothelial cells: inhibition by NG-hydroxy-L-arginine during high-output NO production. Am J Physiol Heart Circ Physiol. 1996;271:H1988–H1998. doi: 10.1152/ajpheart.1996.271.5.H1988. [DOI] [PubMed] [Google Scholar]
- 25.Bachetti T, Comini L, Francolini G, et al. Arginase pathway in human endothelial cells in pathophysiological conditions. J Mol Cell Cardiol. 2004;37:515–523. doi: 10.1016/j.yjmcc.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Chicoine LG, Paffet ML, Young TL, Nelin LD. Arginase inhibition increases nitric oxide production in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L60–L68. doi: 10.1152/ajplung.00194.2003. [DOI] [PubMed] [Google Scholar]
- 27.Berkowitz DE, White R, Li D, et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003;108:2000–2006. doi: 10.1161/01.CIR.0000092948.04444.C7. [DOI] [PubMed] [Google Scholar]
- 28.Demougeot C, Prigent-Tesssier A, Marie C, Berthelot A. Arginase inhibition reduced endothelial dysfunction and blood pressure rising in spontaneously hypertensive rats. J Hypertens. 2005;23:971–978. doi: 10.1097/01.hjh.0000166837.78559.93. [DOI] [PubMed] [Google Scholar]
- 29.Durante W, Tulis DA, Keswani AN, Wang H, Peyton KJ. Arginase promotes vascular smooth muscle cell proliferation and neointima formation. Cardiovasc Pathol. 2004;13:S46. [Google Scholar]
- 30.Xu W, Kaneko T, Zheng S, et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary artery hypertension. FASEB J. 2004;18:1746–1748. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- 31.Zhang C, Hein TW, Wang W, Chang C-I, Kuo L. Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. FASEB J. 2001;15:1264–1266. doi: 10.1096/fj.00-0681fje. [DOI] [PubMed] [Google Scholar]
- 32.Johnson FK, Johnson RA, Peyton KJ, Durante W. Arginase inhibition restores arteriolar endothelial function in Dahl rats with salt-induced hypertension. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1057–1062. doi: 10.1152/ajpregu.00758.2004. [DOI] [PubMed] [Google Scholar]
- 33.Ming X-F, Barandier C, Viswambharan H, et al. Thrombin stimulates human endothelial arginase enzymatic activity via the RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation. 2004;110:3708–3714. doi: 10.1161/01.CIR.0000142867.26182.32. [DOI] [PubMed] [Google Scholar]
- 34.Nelin LD, Chicoine LG, Reber KM, English BK, Young TL, Liu Y. Cytokine-induced endothelial arginase expression is dependent on epidermal growth factor receptor. Am J Respir Cell Mol Biol. 2005;33:394–401. doi: 10.1165/rcmb.2005-0039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christianson DW. Arginase: structure, mechanism, and physiological role in male and female sexual arousal. Acc Chem Res. 2005;38:191–201. doi: 10.1021/ar040183k. [DOI] [PubMed] [Google Scholar]
- 36.Chang CI, Liao JC, Kuo L. Arginase modulates nitric oxide production in activated macrophages. Am J Physiol Heart Circ Physiol. 1998;274:H342–H348. doi: 10.1152/ajpheart.1998.274.1.H342. [DOI] [PubMed] [Google Scholar]
- 37.Hey C, Boucher JL, Vadon-Le Goff S, Ketterer G, Wessler I, Racke K. Inhibition of arginase in rat and rabbit alveolar macrophages by N -hydroxy-D,L-indospicine, effects of L-arginine utilization by nitric oxide synthase. Br J Pharmacol. 1997;121:395–400. doi: 10.1038/sj.bjp.0701143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tenu JP, Lepoivre M, Moali C, Brollo M, Mansuy D, Boucher JL. Effects of the new arginase inhibitor N -hydroxy-nor-L-arginine on NO synthase activity in murine macrophages. Nitric Oxide. 1999;3:427–438. doi: 10.1006/niox.1999.0255. [DOI] [PubMed] [Google Scholar]
- 39.Li H, Meininger CJ, Hawker JR, et al. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline synthesis in endothelial cells. Am J Physiol Endocrinol Metab. 2001;280:E75–E82. doi: 10.1152/ajpendo.2001.280.1.E75. [DOI] [PubMed] [Google Scholar]
- 40.Crombez EA, Cederbaum SD. Hyperargininemia due to liver arginase deficiency. Mol Genet Metabol. 2005;84:243–251. doi: 10.1016/j.ymgme.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 41.Iyer RK, Yoo PK, Kern RM, et al. Mouse model for human arginase deficiency. Mol Cell Biol. 2002;22:4491–4498. doi: 10.1128/MCB.22.13.4491-4498.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi O, Morris SM, Jr, Zoghbi H, Porter CW, O’Brien WE. Generation of a mouse model for arginase II deficiency by targeted disruption of the arginase II gene. Mol Cell Biol. 2001;21:811–813. doi: 10.1128/MCB.21.3.811-813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee J, Ryu H, Ferrante RJ, Morris SM, Jr, Ratan RR. Translational control of inducible nitric oxide synthase expression by arginine can explain the arginase paradox. Proc Natl Acad Sci USA. 2003;100:4843–4848. doi: 10.1073/pnas.0735876100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.El-Gayar S, Thuring-Nahler H, Pfeilschifter J, Rollinghoff M, Bogdan C. Translational control of inducible nitric oxide synthase by IL-13 and arginine availability in inflammatory macrophages. J Immunol. 2003;171:4561–4568. doi: 10.4049/jimmunol.171.9.4561. [DOI] [PubMed] [Google Scholar]
- 45.Prabhakar SS, Zeballos GA, Montoya-Zavala M, Leonard C. Urea inhibits nitric oxide synthase in macrophage cell line. Am J Physiol Cell Physiol. 1997;273:C1882–1888. doi: 10.1152/ajpcell.1997.273.6.C1882. [DOI] [PubMed] [Google Scholar]
- 46.Boucher J-L, Custot J, Vadon S, et al. N -hydroxy-L-arginine, an intermediate in the L-arginine to nitric oxide pathway is a strong inhibitor of liver and macrophage arginase. Biochem Biophys Res Commun. 1994;203:1614–1621. doi: 10.1006/bbrc.1994.2371. [DOI] [PubMed] [Google Scholar]
- 47.Rodriguez S, Richert L, Berthelot A. Increased arginase activity in aorta of mineralocorticoid-salt hypertensive rats. Clin Exp Hypertension. 2000;22:75–85. doi: 10.1081/ceh-100100063. [DOI] [PubMed] [Google Scholar]
- 48.Zhang C, Hein TW, Wang W, et al. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension. 2004;44:935–943. doi: 10.1161/01.HYP.0000146907.82869.f2. [DOI] [PubMed] [Google Scholar]
- 49.White AR, Ryoo S, Li D, et al. Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension. doi: 10.1161/01.HYP.0000198543.34502.d7. In press. [DOI] [PubMed] [Google Scholar]
- 50.Hein TW, Zhang C, Wang W, Chang C-I, Thengchaisri N, Kuo L. Ischemia-reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: counteracting role of arginase. FASEB J. 2003;17:2328–2330. doi: 10.1096/fj.03-0115fje. [DOI] [PubMed] [Google Scholar]
- 51.Durante W, Johnson FK, Johnson RA, Peyton KJ. Arginase induces endothelial dysfunction and hypertension in obese Zucker rats. Diabetes. 2005;54(Suppl 1):A51. [Google Scholar]
- 52.Boger RH, Bode-Boger SM. Asymmetric dimethylarginine, derangements of the endothelial nitric oxide synthase pathway, and cardiovascular disease. Semin Thromb Hemost. 2000;26:539–545. doi: 10.1055/s-2000-13210. [DOI] [PubMed] [Google Scholar]
- 53.Berka V, Wu G, Yeh HC, Palmer G, Tsai AL. Three different oxygen-induced radical species in endothelial nitric oxide synthase oxygenase domain under regulation by L-arginine and tetrahydrobiopterin. J Biol Chem. 2004;279:32243–32251. doi: 10.1074/jbc.M404044200. [DOI] [PubMed] [Google Scholar]
- 54.Nelin L, Nash H, Chicoine L. Cytokine treatment increases arginine metabolism and uptake in bovine pulmonary artery arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1232–L1239. doi: 10.1152/ajplung.2001.281.5.L1232. [DOI] [PubMed] [Google Scholar]
- 55.Kawamoto S, Amaya Y, Murakami K, et al. Complete nucleotide sequence of cDNA and deduced amino acid sequence of the rat liver arginase. J Biol Chem. 1987;262:6280–6283. [PubMed] [Google Scholar]
- 56.Bivalacqua TJ, Hellstrom WJG, Kadowitz PJ, Champion HC. Increased expression of arginase II in human diabetic corpus cavernosum in diabetic-associated erectile dysfunction. Biochem Biophys Res Commun. 2001;283:923–927. doi: 10.1006/bbrc.2001.4874. [DOI] [PubMed] [Google Scholar]
- 57.Kampfer H, Pfeilschifter J, Frank S. Expression and activity of arginase isozymes during normal and diabetic-impaired skin repair. J Invest Dermatol. 2003;121:1544–1551. doi: 10.1046/j.1523-1747.2003.12610.x. [DOI] [PubMed] [Google Scholar]
- 58.Jung AS, Kubo H, Wilson RM, Houser SR, Margulies KB. Modulation of contractility by myocyte-derived arginase in normal and hypertrophied feline myocardium. Am J Physiol Heart Circ Physiol. doi: 10.1152/ajpheart.01104.2005. [DOI] [PubMed] [Google Scholar]
- 59.Meurs H, Maarsingh H, Zaagsma J. Arginase and asthma: novel insights into nitric oxide homeostasis and airway hyperresponsiveness. TIPS. 2003;24:450–454. doi: 10.1016/S0165-6147(03)00227-X. [DOI] [PubMed] [Google Scholar]
- 60.Grasemann H, Schwiertz R, Mathiesen S, Racke K, Ratjen F. Increased arginase activity in cystic fibrosis airways. Am J Respir Crit Care Med. 2005;172:1523–1528. doi: 10.1164/rccm.200502-253OC. [DOI] [PubMed] [Google Scholar]
- 61.Waddington SN, Cattell V. Arginase in glomerulonephritis. Exp Nephrol. 2000;8:128–134. doi: 10.1159/000020660. [DOI] [PubMed] [Google Scholar]
- 62.Bruch-Gerharz D, Schnorr O, Suschek C, et al. Arginase I overexpression in psoriasis: limitation of inducible nitric oxide synthase activity as a molecular mechanism for keratinocyte hyperproliferation. Am J Pathol. 2003;162:203–211. doi: 10.1016/S0002-9440(10)63811-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carraliza I, Moncada S. Increased expression of arginase II in patients with different forms of arthritis: implications of the regulation of nitric oxide. J Rheumatol. 2002;29:2261–2265. [PubMed] [Google Scholar]
- 64.Morris CR, Kato GJ, Poljakovic M, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and limited substrate availability in sickle cell disease. JAMA. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tabor CW, Tabor H. Polyamines. Annu Rev Biochem. 1984;53:749–790. doi: 10.1146/annurev.bi.53.070184.003533. [DOI] [PubMed] [Google Scholar]
- 66.Durante W, Liao L, Iftikhar I, Cheng K, Schafer AI. Platelet-derived growth factor regulates vascular smooth muscle cell proliferation by inducing cationic amino acid transporter gene expression in cultured vascular smooth muscle cells. J Biol Chem. 1996;271:11838–11843. doi: 10.1074/jbc.271.20.11838. [DOI] [PubMed] [Google Scholar]
- 67.Durante W, Liao L, Peyton KJ, Schafer AI. Thrombin stimulates vascular smooth muscle cell polyamine synthesis by inducing cationic amino acid transporter and ornithine decarboxylase gene expression. Circ Res. 1998;83:217–223. doi: 10.1161/01.res.83.2.217. [DOI] [PubMed] [Google Scholar]
- 68.Endean ED, Kispert JF, Martin KW, O’Connor W. Intimal hyperplasia is reduced by ornithine decarboxylase inhibition. J Surg Res. 1991;50:634–637. doi: 10.1016/0022-4804(91)90054-p. [DOI] [PubMed] [Google Scholar]
- 69.Ignarro LJ, Buga GM, Wei LH, Bauer PM, Wu G, Soldato P. Role of arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc Natl Acad Sci USA. 2001;98:4202–4208. doi: 10.1073/pnas.071054698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wei LH, Wu G, Morris SM, Jr, Ignarro LJ. Elevated arginase I expression in rat aortic smooth muscle cells increases cell proliferation. Proc Natl Acad Sci USA. 2001;98:9260–9264. doi: 10.1073/pnas.161294898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu X, Chapman GB, Peyton KJ, Schafer AI, Durante W. Carbon monoxide inhibits apoptosis in vascular smooth muscle cells. Cardiovasc Res. 2002;55:396–405. doi: 10.1016/s0008-6363(02)00410-8. [DOI] [PubMed] [Google Scholar]
- 72.Loyaga-Rendon R, Sakamoto S, Beppu M, et al. Accumulated endogenous nitric oxide synthase inhibitors, enhanced arginase activity, attenuated dimethylarginine dimethylaminohydrolase activity and intimal hyperplasia in premenopausal human uterine arteries. Atherosclerosis. 2005;178:231–239. doi: 10.1016/j.atherosclerosis.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 73.Durante W, Tulis DA, Kewani AN, Wang H, Peyton KJ. Arginase promotes vascular smooth muscle cell proliferation and neointima formation. Cardiovasc Pathol. 2004;13:S46. [Google Scholar]
- 74.Teupser D, Burkhardt R, Wilfert W, Haffner I, Nebandahl K, Thiery J. Identification of macrophage arginase I as a new candidate gene of atherosclerosis resistance. Arterioscler Thromb Vasc Biol. 2006;26:365–371. doi: 10.1161/01.ATV.0000195791.83380.4c. [DOI] [PubMed] [Google Scholar]
- 75.Wu G, Meininger CJ. Arginine nutrition and cardiovascular function. J Nutr. 2000;130:2626–2629. doi: 10.1093/jn/130.11.2626. [DOI] [PubMed] [Google Scholar]