

Abstract
Accumulating evidence implicates a role for altered iron and copper metabolism in the pathogenesis of neurodegenerative disorders such as Alzheimer’s disease (AD). However, imbalances in the levels of the various forms of iron at different stages of AD have not been examined. In this pilot study we extracted and measured the levels of loosely bound, non-heme and total iron and copper in the frontal cortex and hippocampus of patients with mild-moderate AD (n = 3), severe AD (n = 8) and dementia with Lewy bodies (DLB, n = 6), using graphite furnace atomic absorption spectrometry (GFAAS). Additionally, the expression of iron regulatory protein 2 (IRP2) was examined in relation to the pathological hallmarks of AD and DLB, amyloid plaques, neurofibrillary tangles (NFT), and Lewy bodies, by immunohistochemistry. We found significantly decreased loosely bound iron in the hippocampal white matter of mild-moderate and severe AD patients and a trend towards increased non-heme iron in the hippocampal gray matter of severe AD patients. Furthermore, decreased levels of total copper were seen in severe AD and DLB frontal cortex compared to controls, suggesting an imbalance in brain metal levels in both AD and DLB. The decrease in loosely bound iron in mild-moderate AD patients may be associated with myelin breakdown seen in the beginning stages of AD and implicates that iron dysregulation is an early event in AD pathogenesis.
Keywords: Alzheimer’s disease, dementia with Lewy bodies, loosely bound iron, non-heme iron
A dysregulation of brain metals, especially iron and copper, has been implicated in the pathogenesis of Alzheimer’s disease (AD) [15,22,27]. Brain iron is important in neural development and function, as neurons and glial cells require iron for electron transport, myelination of axons, and as a cofactor for enzymes involved in the synthesis of neurotransmitters [14]. However, while iron deficiency impairs cell growth, iron overload can cause cellular damage. Thus, the maintenance of iron homeostasis is critical for the cell. Intracellular iron is tightly regulated by the iron regulatory proteins, IRP1 and IRP2, which post-transcriptionally regulate the expression of proteins involved in iron homeostasis, such as the transferrin receptor and ferritin, in response to intracellular “free” iron concentrations [29].
Redox active iron and copper, capable of generating reactive oxygen species (ROS), have been found associated with amyloid plaques and neurofibrillary tangles (NFT), pathological hallmarks of AD [19,27]. These and other transition metals were also demonstrated to mediate β-amyloid aggregation and neurotoxicity [15,24]. Furthermore, alterations in the localization of IRP2, but not IRP1, have been reported in AD [26]. However, little is known about the relative distribution of the different pools of iron, copper, and IRP2 expression at different stages of the disease. To address this question, we measured the levels of 1) loosely bound, non-heme, and total iron, 2) copper, and 3) examined IRP2 expression in the brains of patients with mild-moderate AD and severe AD. In addition, we evaluated cases of dementia with Lewy bodies (DLB) to determine if changes in iron levels and IRP2 expression are specific to AD or involved in both AD and DLB.
Postmortem tissue samples from the frontal cortex and hippocampus of non-demented elderly controls (n = 6, mean age = 72 ± 11 (SD), range = 56 to 86 years, M/F = 4/2, postmortem interval (PMI) = 21 ± 7), clinically and histopathologically confirmed cases of mild-moderate AD (n = 3, mean age = 83 ± 10, range = 73 to 92 years, M/F = 1/2, PMI = 11 ± 11), severe AD (n = 8, mean age = 78 ± 12, range = 59 to 91 years, M/F = 2/6, PMI = 21 ± 6), using the CERAD/NIA and Braak/Braak staging criteria, and DLB (n = 6, mean age = 78 ± 5, range = 71 to 85, M/F = 2/4, PMI = 16 ± 9) were obtained from the Neuropathology & Molecular Genetics Core of the UCLA Alzheimer’s Disease Research Center.
We dissected frozen brain samples into gray and white matter and extracted loosely bound and non-heme iron according to the method of Nelson et al. [21] with slight modifications. The hippocampus consisted of a 6-8 mm tissue block from its mid-portion, sampled at approximately the level (coronal) of the lateral geniculate nucleus, and included the hippocampus proper, parahippocampal gyrus and entorhinal cortex. Small wedges of white matter were dissected out from areas around the temporal horn and within the parahippocampal gyrus in a caudo-rostral direction. Regions of the frontal cortex that were sampled correspond to Brodmann areas 10 and 11. For loosely bound iron, tissues were homogenized in 180 μl of 0.5 mM EDTA, and samples were centrifuged at 13,000 × g for 10 min, after which 34 μL of 20% tricholoroacetic acid (TCA)/0.5 mM EDTA was added to 120 μL of the supernatant. Then samples were vortexed, centrifuged again at 13,000 × g for 10 min, and the supernatant collected and stored at -20 °C. For non-heme iron, tissues were homogenized in 360 μL of 6% TCA/0.5 mM EDTA and incubated at 90 °C for 30 min. Then 0.7 ml of 0.5 mM EDTA was added, samples were centrifuged at 13,000 × g for 10 min, and the supernatant was collected and stored at −20 °C. For total iron and copper, tissues were wet ashed according to the method of Maynard et al. [20]. All metals were measured in duplicate by graphite furnace atomic absorption spectrometry (GFAAS) with a SpectrAA 220Z (Varian, Victoria, Australia).
Immunohistochemistry was done using the BioGenex Autostainer system (San Ramon, CA) with 6 μm paraffin-embedded tissue sections from the hippocampus and frontal cortex. Tissue sections were deparaffinized by two 15 min incubations in EZ-DeWax solution followed by antigen retrieval using the Antigen Retrieval Citra Plus solution, both from BioGenex, according to the manufacturer’s instructions. Endogenous peroxidase activity was inhibited by two 15 min incubations in 3% hydrogen peroxide in 10% methanol, and non-specific protein binding was blocked with Power Block (Biogenex). Antibodies used are listed in Table 1. The anti-IRP2 antibody is now available from Santa Cruz Biotechnology (Santa Cruz, CA). After 30 min incubations with the respective antibodies, sections were washed with PBS and incubated for 20 min with biotinylated secondary antibodies, followed by HRP-conjugated streptavidin. Immunoreactivity was revealed using AEC as the chromogen.
Table 1.
Antibodies used for immunohistochemistry.
| antibody | antigen | species | type | clone | source | dilution |
|---|---|---|---|---|---|---|
| IRP2 | 138-200aa of IRP2 | mouse | monoclonal | 4G11 | this study | 1:100 |
| PHF-tau | PHF-tau | mouse | monoclonal | AT8 | Pierce | 1:40 |
| Aβ40 | C-terminal 7aa from Aβ40 | rabbit | polyclonal | NA* | Chemicon | 1:100 |
| Aβ42 | C-terminal 6aa from Aβ42 | rabbit | polyclonal | NA* | Chemicon | 1:100 |
| α-synuclein | Lewy bodies from patients with DLB | mouse | monoclonal | LB509 | Zymed | 1:50 |
Not-applicable
Data are presented as means ± SEM. Statistical analysis was performed using one-way ANOVA followed by the Tukey post hoc test with SPSS 12.0.1 software. Corrections for age were done using ANCOVA analysis. P < 0.05 was considered significant.
Levels of loosely bound iron were similar between the frontal cortex and hippocampus although concentrations in the white matter tended to be higher than in the gray matter in both regions in all groups (Fig. 1). We found no differences in the levels of loosely bound iron in the frontal cortex between elderly controls, mild-moderate AD, severe AD, and DLB. However, there was a significant decrease of loosely bound iron in the hippocampal white matter in mild-moderate and severe AD brains compared to controls (3.7 ± 0.4 and 3.7 ± 0.3, respectively, vs. 5.2 ± 0.6 μg Fe/g wet weight) which remained significant when corrected for age in severe AD but was reduced to a trend in mild-moderate AD.
Fig. 1.
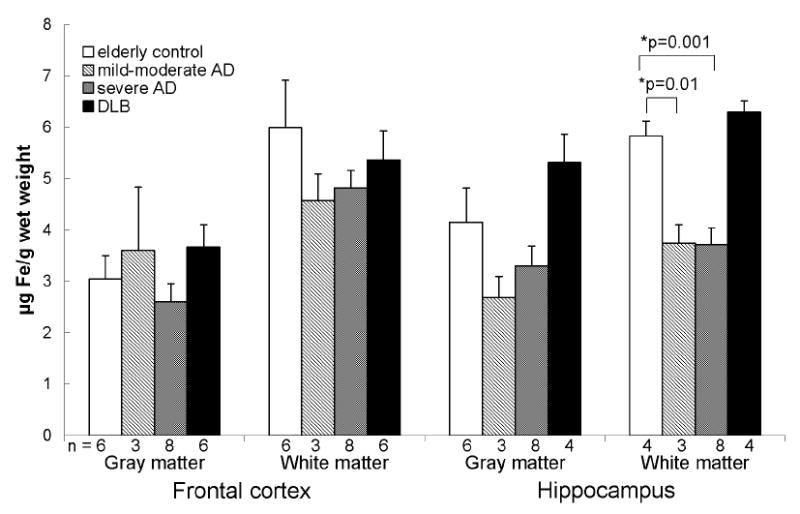
Loosely bound iron was decreased in the hippocampal white matter of mild-moderate and severe AD brains compared to controls. Data are presented as mean (μg Fe/g wet weight) ± SEM and n values for each group are shown below the bars.
*When corrected for age differences between groups, the decrease in loosely bound iron in the hippocampal white matter in severe AD remained significant (p = 0.033) whereas that for mild-moderate AD was reduced to a trend (p = 0.052).
Non-heme and total iron were also consistently higher in the white matter compared to the gray matter with slightly higher levels in the frontal white matter compared to that of the hippocampus in all groups (Fig. 2 and 3). No significant difference in non-heme and total iron was found between the groups in both regions except for higher total iron in the frontal gray matter in DLB compared to controls when corrected for age.
Fig. 2.
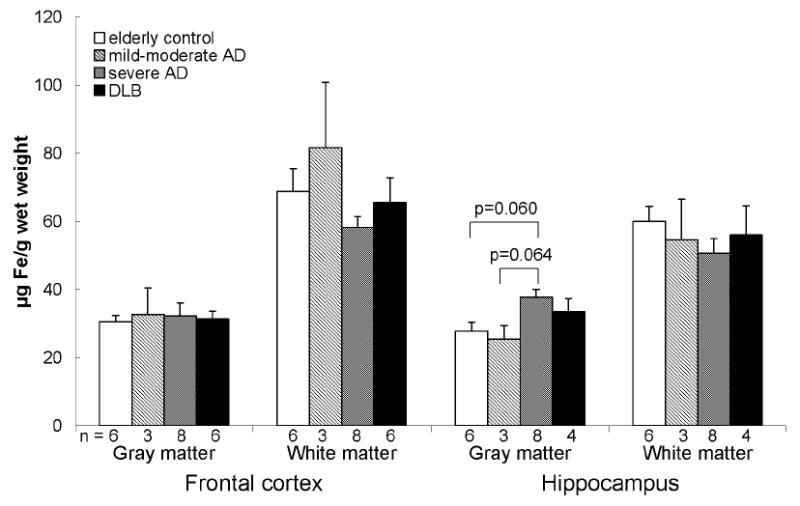
There was no statistically significant difference in non-heme iron between in the frontal cortex and hippocampus of elderly controls, mild-moderate AD, severe AD, and DLB. Data are presented as mean (μg Fe/g wet weight) ± SEM and n values for each group are shown below the bars.
*When corrected for age differences between groups, the trend toward increased non-heme iron in the hippocampal gray matter in severe AD disappeared.
Fig. 3.
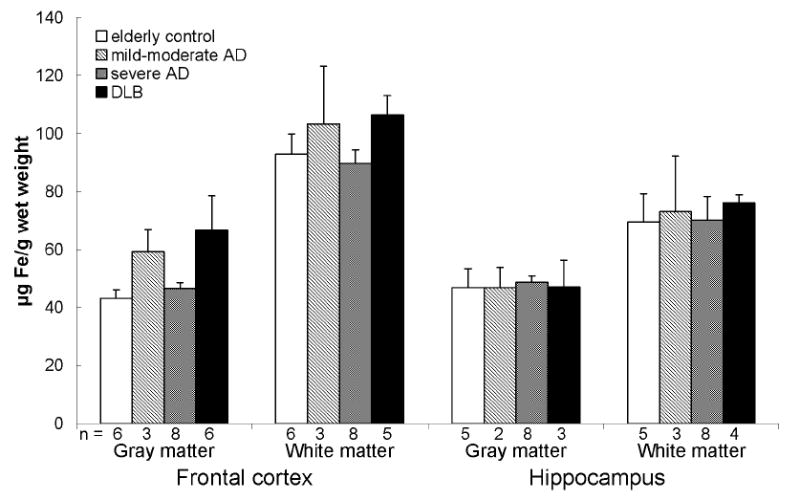
Total iron in the frontal cortex and hippocampus of elderly controls, mild-moderate AD, severe AD, and DLB. Data are presented as mean (μg Fe/g wet weight) ± SEM and n values for each group are shown below the bars.
*When corrected for age differences between groups, there was a significant increase in total iron in the frontal gray matter in DLB compared to controls (p = 0.048).
Levels of total copper were higher in the frontal cortex compared to the hippocampus in normal brains (Fig. 4). This difference is abolished in severe AD and DLB. Compared to controls, total copper in severe AD and DLB was significantly lower in the frontal gray matter (6.9 ± 1.2 vs. 3.9 ± 0.3 and 3.6 ± 0.6 μg Cu/g wet weight, respectively) although this was reduced to a trend when corrected for age. No differences in total copper concentrations were seen between any of the groups in the hippocampus.
Fig. 4.
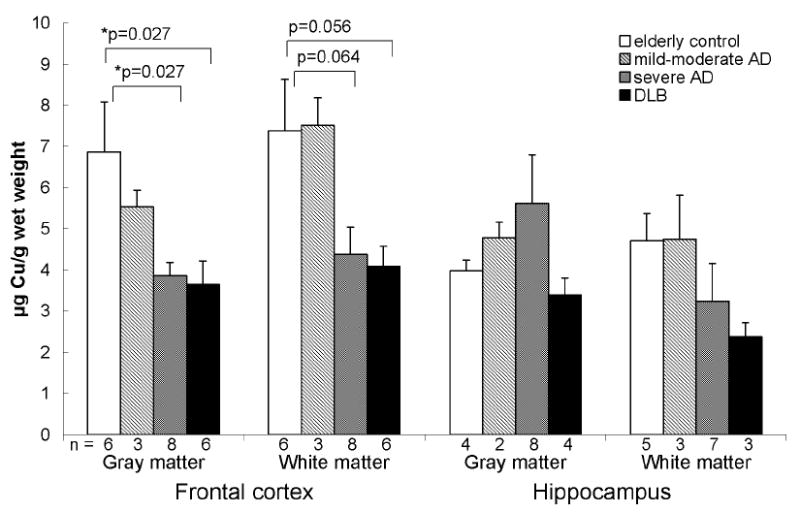
Total copper was decreased in the gray matter of the frontal cortex in severe AD and DLB compared to controls. Data are presented as mean (μg Cu/g wet weight) ± SEM and n values for each group are shown below the bars.
*When corrected for age differences between groups, the decreases in total copper in the frontal gray matter in severe AD and DLB were reduced to trends (p = 0.057 for both groups) while the trends toward decreased total copper in the frontal white matter disappeared.
IRP2 expression was found to be primarily intraneuronal with sparing of leptomeninges, oligodendrocytes, astrocytes, and white matter tracts. IRP2 expression was limited to the cytoplasm of neurons in the gray matter of the frontal cortex and pyramidal neurons in the hippocampus. The dentate gyrus was generally negative and expressed IRP2 only in rare instances. We did not find significant differences in IRP2 expression between controls, mild-moderate AD, severe AD, and DLB (Fig. 5), and IRP2 did not colocalize with senile plaques, NFT, or Lewy bodies in the cases we examined (data not shown).
Fig. 5.
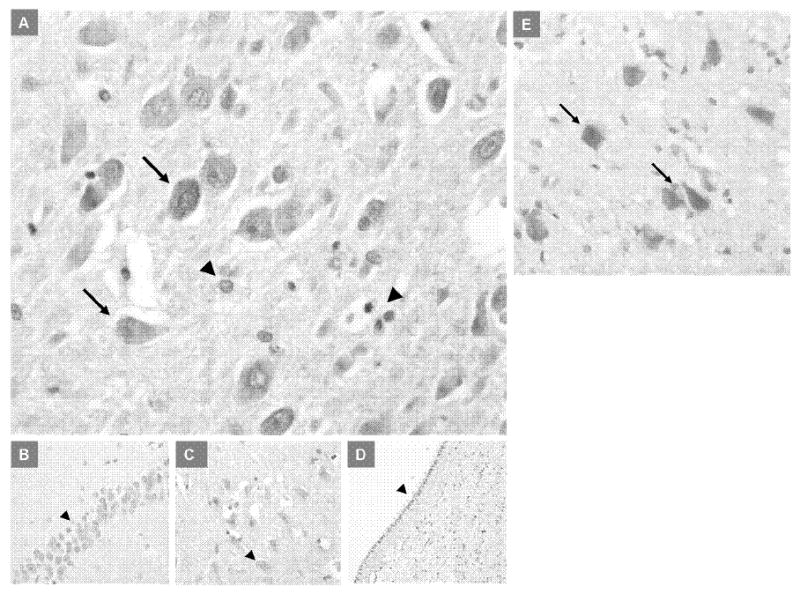
IRP2 expression in the hippocampus of elderly control and AD brains. No differences in the intensity or localization of IRP2 expression was seen in mild-moderate AD, severe AD, or DLB brains compared to controls. A) Cytoplasmic IRP2 expression in the CA1-2 hippocampal neurons in a severe AD patient (arrows); arrowheads show absence of staining in small glial cells (×200). B) Absence of IRP2 expression in the hippocampal dentate neurons (arrowhead) in a severe AD patient (×200). C) Absence of IRP2 expression in the hippocampal CA3 neurons (arrowhead) in a severe AD patient (×200). D) Absence of IRP2 expression in the ependymal cells (arrowhead) in a severe AD patient (×100). E) Cytoplasmic IRP2 expression in the CA4 hippocampal neurons (arrows) in an elderly control brain (×200).
The purpose of this study was to more closely examine alterations in iron and copper levels in AD at different stages of the disease. The decrease in loosely bound iron levels in the hippocampal white matter in cases with mild-moderate AD and severe AD but not DLB compared to elderly controls suggests that alterations in loosely bound iron may occur relatively early in AD and be specific to this disease process. Our results are consistent with a previous study that showed decreased transferrin and iron in AD white matter [8] and may be a cause or consequence of myelin breakdown and oligodendrocyte damage observed in AD [2,25]. Oligodendrocytes have high basal metabolism, which is needed for lipid synthesis, and consequently show vulnerability to oxidative stress. Not surprisingly, these cells contain the highest levels of iron out of all cell types in the brain, and up to 70% of brain iron is associated with myelin [1,7]. Loosely bound iron is a labile, chelatable pool of non-heme iron that mediates the transfer of iron from ferritin storage sites to where it is needed for enzymatic and biosynthetic activity and includes iron attached to phosphate esters and carbohydrates [11,16]. Iron deficiency causing poor myelination is associated with mental deficits in children [1], and decreased myelin basic protein has been shown to correlate with cognitive decline in AD [30]. Our findings of decreased loosely bound iron in the white matter of the hippocampus, a region generally affected in the initiating stages of the disease, are in agreement with an early loss of oligodendrocytes in AD [5].
Non-heme iron consists of iron tightly bound to proteins such as the iron transport proteins transferrin and lactoferrin and in intracellular storage proteins such as ferritin [11,13,17]. The majority of iron in the hippocampal gray matter was non-heme, in agreement with previous studies showing that most iron in the hippocampus is ferritin-like [10]. The lack of major differences in non-heme and total iron levels between the groups suggest a relocalization and sequestering of iron into plaques rather than a general increase in bulk tissue as high concentrations of iron have been consistently reported in amyloid plaques [6,19]. These results are consistent with previous reports that have found no difference in non-heme and total iron concentrations in bulk tissue from AD and control brains [12,23] but at variance with others who have reported increases in non-heme and total iron in AD brain. Total protein levels have been demonstrated to be significantly decreased in AD frontal cortex [18] which may partly explain some of the increases in iron found when measured per milligram of protein. However, further studies with larger sample sizes will be necessary to confirm changes in non-heme and total iron. IRP2 expression was confined to neurons in the gray matter and did not differ between controls, AD, and DLB, in the frontal cortex and hippocampus, which is consistent with the lack of major differences in iron levels in the gray matter of both regions.
Compared to controls, copper levels in the frontal cortex were decreased in the gray matter in severe AD and DLB. These results are in agreement with reports of decreased levels of copper in the AD brain [9] and in the brains of transgenic mice overexpressing APP [20]. The decrease in copper is not seen in mild-moderate AD but only in severe AD, suggesting that this phenomenon may occur at a later stage in disease progression. Overproduction of APP and Aβ is hypothesized to pool copper into the extracellular space from the intracellular compartment and thus cause a deficiency of intracellular copper in AD brain [4,20]. Furthermore, increased levels of copper in CSF and plasma have been found in AD patients in contrast to the decrease seen in brain [3,28]. Mechanisms similar to that seen in AD brains may also underlie lower copper levels in DLB, since both diseases share some common pathologic features. A limitation of this and other post-mortem studies is their cross sectional nature, and thus we have examined iron levels in the brains of AD patients at different stages of the disease. A related limitation is the difficulty of obtaining tissue quickly after death although there were no significant differences in the PMIs between our patient groups.
In conclusion, we found decreased iron in severe AD hippocampus and decreased copper in the frontal cortex in severe AD and DLB while finding no significant differences in IRP2 expression between the groups. These results lend further support to the hypothesis of dysregulation of brain metals in AD and related dementias while shedding light on the timeframe in which the alterations in iron and copper metabolism occur in the pathogenesis of the disease.
Acknowledgments
W.M.K. was supported in part by NIH grant AG20948. H.V.V. was supported in part by NIA grant P50 AG16570 and the Daljit S. & Elaine Sarkaria Chair in Diagnostic Medicine. We would like to thank Wayne Kelln, Waheed Baqai and Justine Pomakian for technical assistance.
References
- 1.Bartzokis G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol Aging. 2004a;25:5–18. doi: 10.1016/j.neurobiolaging.2003.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Bartzokis G, Sultzer D, Lu PH, Nuechterlein KH, Mintz J, Cummings JL. Heterogeneous age-related breakdown of white matter structural integrity: implications for cortical “disconnection” in aging and Alzheimer’s disease. Neurobiol Aging. 2004b;25:843–851. doi: 10.1016/j.neurobiolaging.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Basun H, Forssell LG, Wetterberg L, Winblad B. Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer’s disease. J Neural Transm Park Dis Dement Sect. 1991;3:231–258. [PubMed] [Google Scholar]
- 4.Bayer TA, Multhaup G. Involvement of amyloid beta precursor protein (AbetaPP) modulated copper homeostasis in Alzheimer’s disease. J Alzheimers Dis. 2005;8:201–206. doi: 10.3233/jad-2005-8212. [DOI] [PubMed] [Google Scholar]
- 5.Benes FM. A disturbance of late myelination as a trigger for Alzheimer’s disease. Neurobiol Aging. 2004;25:41–43. doi: 10.1016/j.neurobiolaging.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Bush AI. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003;26:207–214. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 7.Connor JR. Myelin breakdown in Alzheimer’s disease: a commentary. Neurobiol Aging. 2004;25:45–47. doi: 10.1016/j.neurobiolaging.2003.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Connor JR, Snyder BS, Beard JL, Fine RE, Mufson EJ. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. J Neurosci Res. 1992;31:327–335. doi: 10.1002/jnr.490310214. [DOI] [PubMed] [Google Scholar]
- 9.Deibel MA, Ehmann WD, Markesbery WR. Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: possible relation to oxidative stress. J Neurol Sci. 1996;143:137–142. doi: 10.1016/s0022-510x(96)00203-1. [DOI] [PubMed] [Google Scholar]
- 10.Friedman A, Galazka-Friedman J, Bauminger ER, Koziorowski D. Iron and ferritin in hippocampal cortex and substantia nigra in human brain - Implications for the possible role of iron in dementia. J Neurol Sci. 2006;248:31–34. doi: 10.1016/j.jns.2006.05.056. [DOI] [PubMed] [Google Scholar]
- 11.Gutteridge JM. Iron and oxygen radicals in brain. Ann Neurol. 1992;32:S16–21. doi: 10.1002/ana.410320705. [DOI] [PubMed] [Google Scholar]
- 12.Hallgren B, Sourander P. The non-haemin iron in the cerebral cortex in Alzheimer’s disease. J Neurochem. 1960;5:307–310. doi: 10.1111/j.1471-4159.1960.tb13369.x. [DOI] [PubMed] [Google Scholar]
- 13.Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J, Connor JR. Demonstration and characterization of the iron regulatory protein in human brain. J Neurochem. 1996;67:838–844. doi: 10.1046/j.1471-4159.1996.67020838.x. [DOI] [PubMed] [Google Scholar]
- 15.Huang X, Atwood CS, Moir RD, Hartshorn MA, Tanzi RE, Bush AI. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer’s Abeta peptides. J Biol Inorg Chem. 2004;9:954–960. doi: 10.1007/s00775-004-0602-8. [DOI] [PubMed] [Google Scholar]
- 16.Kala SV, Hasinoff BB, Richardson JS. Brain samples from Alzheimer’s patients have elevated levels of loosely bound iron. Int J Neurosci. 1996;86:263–269. doi: 10.3109/00207459608986717. [DOI] [PubMed] [Google Scholar]
- 17.Levenson CW, Tassabehji NM. Iron and ageing: an introduction to iron regulatory mechanisms. Ageing Res Rev. 2004;3:251–263. doi: 10.1016/j.arr.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Loeffler DA, Connor JR, Juneau PL, Snyder BS, Kanaley L, DeMaggio AJ, Nguyen H, Brickman CM, LeWitt PA. Transferrin and iron in normal, Alzheimer’s disease, and Parkinson’s disease brain regions. J Neurochem. 1995;65:710–724. doi: 10.1046/j.1471-4159.1995.65020710.x. [DOI] [PubMed] [Google Scholar]
- 19.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 20.Maynard CJ, Cappai R, Volitakis I, Cherny RA, White AR, Beyreuther K, Masters CL, Bush AI, Li QX. Overexpression of Alzheimer’s disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J Biol Chem. 2002;277:44670–44676. doi: 10.1074/jbc.M204379200. [DOI] [PubMed] [Google Scholar]
- 21.Nelson SR, Pazdernik TL, Samson FE. Measurement of loosely-bound iron in brain regions using redox cycling and salicylate. Cell Mol Biol (Noisy-le-grand) 2000;46:649–655. [PubMed] [Google Scholar]
- 22.Perry G, Sayre LM, Atwood CS, Castellani RJ, Cash AD, Rottkamp CA, Smith MA. The role of iron and copper in the aetiology of neurodegenerative disorders: therapeutic implications. CNS Drugs. 2002;6:339–352. doi: 10.2165/00023210-200216050-00006. [DOI] [PubMed] [Google Scholar]
- 23.Religa D, Strozyk D, Cherny RA, Volitakis I, Haroutunian V, Winblad B, Naslund J, Bush AI. Elevated cortical zinc in Alzheimer disease. Neurology. 2006;67:69–75. doi: 10.1212/01.wnl.0000223644.08653.b5. [DOI] [PubMed] [Google Scholar]
- 24.Rottkamp CA, Raina AK, Zhu X, Gaier E, Bush AI, Atwood CS, Chevion M, Perry G, Smith MA. Redox-active iron mediates amyloid-beta toxicity. Free Radic Biol Med. 2001;30:447–450. doi: 10.1016/s0891-5849(00)00494-9. [DOI] [PubMed] [Google Scholar]
- 25.Sjobeck M, Englund E. Glial levels determine severity of white matter disease in Alzheimer’s disease: a neuropathological study of glial changes. Neuropathol Appl Neurobiol. 2003;29:159–169. doi: 10.1046/j.1365-2990.2003.00456.x. [DOI] [PubMed] [Google Scholar]
- 26.Smith MA, Wehr K, Harris PL, Siedlak SL, Connor JR, Perry G. Abnormal localization of iron regulatory protein in Alzheimer’s disease. Brain Res. 1998;788:232–236. doi: 10.1016/s0006-8993(98)00002-x. [DOI] [PubMed] [Google Scholar]
- 27.Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Squitti R, Lupoi D, Pasqualetti P, Dal Forno G, Vernieri F, Chiovenda P, Rossi L, Cortesi M, Cassetta E, Rossini PM. Elevation of serum copper levels in Alzheimer’s disease. Neurology. 2002;59:1153–1161. doi: 10.1212/wnl.59.8.1153. [DOI] [PubMed] [Google Scholar]
- 29.Wallander ML, Leibold EA, Eisenstein RS. Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim Biophys Acta. 2006;1763:668–689. doi: 10.1016/j.bbamcr.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang DS, Bennett DA, Mufson EJ, Mattila P, Cochran E, Dickson DW. Contribution of changes in ubiquitin and myelin basic protein to age-related cognitive decline. Neurosci Res. 2004;48:93–100. doi: 10.1016/j.neures.2003.10.002. [DOI] [PubMed] [Google Scholar]