

Abstract
Hexim1 is a cellular protein that associates with the positive transcription elongation factor b (P-TEFb) to regulate RNA polymerase II elongation of nascent mRNA transcripts. It directly binds to Cyclin T1 of P-TEFb and inhibits the kinase activity of Cdk9, leading to an arrest of transcription elongation. Here, we report the solution structure of the Cyclin T binding domain (TBD) of Hexim1 that forms a parallel coiled-coil homodimer composed of two segments and a preceding alpha helix that folds back onto the first coiled-coil unit. NMR titration, fluorescence, and immunoprecipitation experiments revealed the binding interface to Cyclin T1, which covers a large surface on the first coiled-coil segment. Electrostatic interactions between an acidic patch on Hexim1 and positively charged residues of Cyclin T1 drive the complex formation that is confirmed by mutagenesis data on Hexim1 mediated transcription regulation in cells. Thus, our studies provide structural insights how Hexim1 recognizes the Cyclin T1 subunit of P-TEFb, which is a key step toward the regulation of transcription elongation.
Keywords: Cyclin T1, NMR spectroscopy, transcription elongation
Transcriptional elongation by RNA polymerase II (pol II) is a highly regulated process that generates full-length RNA transcripts and coordinates transcription with pre-mRNA processing (1). The positive transcription elongation factor b (P-TEFb) is a key regulator for the transition from abortive to productive elongation of mature mRNA molecules (2). The core form of P-TEFb is a heterodimer between the cyclin-dependent kinase 9 (Cdk9) and its regulatory subunit Cyclin T1 (CycT1) or the minor forms T2 or K. Transcription activation by P-TEFb depends on the kinase activitiy of Cdk9, which phosphorylates the C-terminal domain (CTD) of the largest subunit of RNA pol II to stimulate the processivity of elongation (3). Initially, P-TEFb has been found to play a critical role in stimulating the transcription of HIV-1 genes, where the viral transactivator protein Tat recruits P-TEFb to stalled RNA pol II molecules (2). At present, P-TEFb is considered a global transcriptional elongation factor important for the expression of most RNA pol II-regulated genes (4).
Recently, it has been found that P-TEFb itself is subject to a tight regulation, because approximately half of nuclear P-TEFb in HeLa cells is sequestered in an inactive state. This inactive complex is composed of the Cdk9/CycT1 heterodimer bound to the abundant small nuclear RNA 7SK and the regulatory protein Hexim1 (5–10). Whereas 7SK is supposed to serve as a molecular scaffold to mediate the interaction of Hexim1 with P-TEFb, Hexim1 binds directly to the cyclin box repeats of CycT1 and inhibits the kinase activity of Cdk9. The other half of P-TEFb heterodimers is catalytically active and binds the bromodomain protein, Brd4 (11, 12), which has high affinity for the acetylated tails of core histones H3 and H4 and can bind the Mediator complex.
Hexim1 was initially identified as a protein whose expression is induced in vascular smooth muscle cells in response to hexamethylene bisacetamide treatment (13, 14). Human Hexim1 consists of 359 aa and is tentatively divided into four regions [supporting information (SI) Fig. 5A]: a variable N-terminal region (1–149) that is suggested to have a self inhibitory function; a central nuclear localization signal (NLS, 150–177) that interacts with the nuclear transport machinery and directly binds to 7SK snRNA; a region of highest homology (185–220), including a negatively charged cluster that might be involved in P-TEFb inhibition; and a C-terminal Cyclin T binding domain (TBD) (255–359) that leads to dimerization of Hexim molecules (7–10, 15–18). The Cyclin T binding region constitutes a stable domain and was suggested to form a coiled-coil structure.
Here, we present the solution structure of the dimeric Cyclin TBD of Hexim1 255–359 that consists of two coiled-coil segments and an adjacent helix. NMR mapping experiments and mutagenesis studies in vitro and in cells revealed the binding interface to Cyclin T1, which covers a large and highly charged surface on the first coiled-coil segment. Hexim1 recognizes Cyclin T1 through an association process that is mainly driven by electrostatic interactions. Thus, our studies provide structural insights how Hexim1 targets P-TEFb for the regulation of transcription elongation.
Results
The Hexim1-TBD Forms a Bipartite Coiled-Coil with a Preceding Helix.
The 3D structure of the TBD of human Hexim1 (residues 255–359) was determined by using heteronuclear NMR spectroscopy in solution. The structure was calculated by using 2,973 restraints and refined in a water shell, yielding a final ensemble of 20 lowest-energy structures with 96.3% of the backbone Φ-/Ψ-angle combinations in folded segments occupying the most favored region of the Ramachandran plot (SI Table 1).
The structure of the Hexim1-TBD is a parallel bipartite homodimeric coiled-coil that forms a left-handed superhelix with a length of 95 Å and a diameter of ≈20 Å (coiled-coil parameters are given in SI Table 2). The canonical α-helical fold of the coiled coil is split into two segments of approximately similar length. Helices α2/α2′ comprise residues K284 to K313 and helices α3/α3′ residues D319 to Q348, each forming eight turns of ≈40 Å length. The coiled-coil region is preceded by a short α-helix (α1) from residue T276 to N281 that docks onto the first coiled-coil segment of the mutual opposite helix α2′ (Fig. 1B and SI Fig. 6). The N-terminal 20 and the C-terminal 10 residues appeared flexible and not restrained by intermediate or long range NOEs, but truncation or extension of the N-terminal linker segment severely influenced the expression yield and solubility properties of this domain.
Fig. 1.

NMR structure of the TBD of human Hexim1. (A) Ribbon representation of the dimeric TBD (255–359). Secondary structure elements of both chains are labeled red and blue, respectively. The stammer (316–318) between the two coiled-coil segments α2 and α3 leads to an overwinding of the supercoil. (B) (Left) Backbone atoms of the 20 lowest energy structures were superimposed for the first coiled-coil segment (276–313; rmsd, 0.38 Å; see also SI Table 1). (Right) Superposition of those for the second (319–348; rmsd, 0.32 Å), including a rotation by 90° compared with Left. Side chains of residues at the coiled-coil heptad repeat positions a and d are depicted in dark and light green, respectively. (C) Electrostatic surface charge of the TBD dimer (255–359). Note the large negatively charged surface patch (red) in the first coiled-coil segment that is confined by positive charges (blue). (D) Helical wheel representation of the coiled-coil segments from 284–311 (α2/α2′) (Left) and from 319–348 (α3/α3′) (Right). Positions a to g of residues within the heptad repeat pattern are indicated. Salt bridges between g and e′ positions are depicted by dashed lines.
The TBD Coiled-Coil Dimer Interface.
Left-handed coiled-coil structures are characterized by a repeating pattern of seven residues, denoted (abcdefg)n. Residues at positions a and d are typically nonpolar and form the oligomerization interface of the helices, whereas residues at e and g are largely solvent exposed, polar residues that guide the oligomerization specificity through electrostatic interactions (19). Parallel coiled-coil structures display a distinctive “knobs-into-holes” packing, in which the side chains of residues at the heptad repeat a and d positions form successive layers. Although residues 284–348 of Hexim1 adopt the structure of a typical coiled-coil domain (Fig. 1 A–C), the sequence of the interacting dimer surface varies from the strict consensus (Fig. 1D). In the first coiled-coil segment (α2/α2′), one a position is occupied by leucine, and one by asparagine. Both residues are often observed at this position in dimeric coiled-coils (20). However, none of the four amino acids occupying an a position is β-branched (Ile, Val, Thr) as favored for a tight packing in a coiled-coil dimer (19). Instead, one a position is occupied by a charged residue (K284), and the following one is occupied by a bulky polar aromatic residue (Y291), and both are highly evolutionary conserved (SI Fig. 5B). The observed sequence of residues at the a position (K284, Y291, L298, N305) might therefore confer dimerization specificity and define the start of the coiled-coil at the expense of a lower stability because of less tight α-helical packing. The d positions in this segment are occupied by leucines and methione, which are both favored at this position. Most of the interface of the second coiled-coil segment (α3/α3′) shows good conformity with the heptad consensus sequence. The first three repeats contain leucines at the d position and valine, leucine, and asparagine at the a position. The last heptad repeat d position is occupied by histidine, which is commonly not found at this position and might thereby promote the termination of the dimeric coiled-coil.
The e and g positions are typically occupied with charged residues and influence the pairing specificity. Attractive ionic interactions between heptad repeat position g of one strand and e′ of the opposite one (denoted as i → i′ + 5) stabilize the coiled-coil dimer, whereas repelling ones destabilize it. Although E290 and E295 in the N-terminal coiled-coil form a repulsive pair, only E304 and R309 strengthen the binding interface (Fig. 1D). The situation is more uniform on the second coiled-coil segment where three favorable ionic interactions are formed (R321–E326, E328–R333, E342–R347).
Backbone Dynamics and Structural Integrity.
The flexibility of the TBD backbone was assayed by the analysis of 15N relaxation data (SI Fig. 7A). A fit of T2 and T1 relaxation times and [1H]15N NOEs by the program TENSOR2 (21) showed that rotational diffusion of the TBD is significantly better described by an axially symmetric diffusion tensor than with an isotropic model [χ2(ani)/ χ2(iso) = 0.3]. A fully anisotropic model did not further improve the fit. The long axis of the axially symmetric diffusion tensor is parallel to the long axis of the molecule with rotational correlation times τ‖ = 7.2 ns and t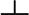 = 21.4 ns (SI Table 3). These calculations are in good agreement with values expected for the size and shape of the TBD dimer. Large amplitude backbone motions on the subnanosecond time scale are evident from decreased [1H]15N NOEs and increased 15N T2 for the N-terminal 20 and the C-terminal 10 residues and the stammer (316–318) in between the two coiled-coil segments.
= 21.4 ns (SI Table 3). These calculations are in good agreement with values expected for the size and shape of the TBD dimer. Large amplitude backbone motions on the subnanosecond time scale are evident from decreased [1H]15N NOEs and increased 15N T2 for the N-terminal 20 and the C-terminal 10 residues and the stammer (316–318) in between the two coiled-coil segments.
A superposition of the 1H/15N HSQC spectra of fragments 242–359, 255–359, and 255–316 confirmed the presence of the coiled-coil structure and the preceding α1-helix in all three fragments (SI Fig. 7B), including the construct lacking the second coiled-coil segment. The N-terminally extended fragment 242–359 did not exhibit additional folded conformation, because all additional 13 residues showed chemical shift values in the random coil range (≈8.20–8.52 ppm for 1H).
Mapping of the Cyclin T1 Binding Interface by NMR Titration Experiments.
Upon CycT1 binding, resonances of the free Hexim1-TBD weakened continuously, indicating intermediate exchange with the bound form on the NMR timescale (SI Fig. 8). New resonances that would represent the TBD in the CycT1–TBD complex did not appear, most likely because of a low stability of CycT1 and unspecific aggregation of the 60-kDa protein complex, which would broaden the signals beyond detection. Other factors that could have influenced the observed loss in signal intensity are local motions and the degree of separation between the chemical shifts of the free and the bound form. For a quantitative evaluation, the ratio of the TBD resonance peak intensities [I(TBDfree)/I(TBDbound)] was calculated at a molar ratio of 0.5 CycT1 to TBD (Fig. 2A). Because of the overlap of resonances, some peak intensities could not be determined and were omitted in the diagram. The largest intensity changes were observed for residues within the first two heptad repeats (KEYL, 289–292) of the first coiled-coil segment. In addition, large intensity changes were observed for the backbone amides of residues E295, K296, S299, E302, and N306. All these residues are located on the first coiled-coil segment of the TBD, suggesting that this structure forms the recognition site for CycT1. Two other resonances that showed increased intensity changes are L279 and S283, located on the preceding helix α1 and the connecting loop to the coiled-coil section.
Fig. 2.

The binding interface of Cylin T1 on the Hexim1-TBD. (A) Loss of 1H/15N HSQC peak intensity upon titration of unlabeled CycT1 (1–292) to 15N-labeled TBD (255–359) at a molar ratio of 0.5. The stronger the decrease of the peak intensity compared with the spectrum without CycT1, the higher the plotted intensity ratio. (B) Display of the CycT1 binding surface on the Hexim1-TBD structure as identified by NMR perturbation measurements. The color coding corresponds to an intensity ratio for low (intensity ratio 4–10, yellow), medium (>10–15, orange), and large changes (>15, red). (C) Surface and ribbon representation of the sequence conservation of Hexim-TBDs. Sequence identities based on the evaluation of 18 Hexim genes are colored according to the degree of conservation for pink (variable, 17%), gray (average, 66%), and blue (conserved, 100%).
The results of the NMR titration were mapped on the surface of the TBD structure (Fig. 2B). Of the 12 most affected residues, 3 are located at heptad repeat b positions (L292, S299, N306), and 4 additional residues (E295, E302, K289, K296) are on the adjacent e and f positions. Y291 was the only amino acid at a core forming a position that was perturbed upon binding, inline with the suggestion that large aromatic and hydrophilic amino acids at these positions destabilize the coiled-coil interface. Mapping the identity conservation onto the structure of the TBD revealed that residues on the surface of the first coiled-coil segment are more conserved than those of the second, whereas residues in the coiled-coil interface at a and d positions are overall well conserved (Fig. 2C and SI Text). A continuous surface of high sequence conservation that is formed by residues on helices α1′ and α2 overlaps well with residues that were found to interact with CycT1 based on the NMR titration data.
Mutational Analysis of the Binding Interface Between Hexim1-TBD and Cyclin T1.
The binding interface between TBD and CycT1 was further characterized by mutagenesis studies. Based on the NMR titration data, the proximity of CycT1 binding residues on the surface of the TBD structure and on sequence conservation, 16 individual mutants of the TBD ranging from positions E277 to N311 were constructed. For direct comparisons and to minimize the influence on the structural integrity of the TBD, only alanine mutations were generated. Mutations at core forming positions that would possibly destabilize the coiled-coil dimer were omitted. In addition, a triple mutant E286A/E290A/E293A (named 3E-A) that destroys the negatively charged surface patch on the first coiled-coil segment was generated. First, GST-fused CycT1 coupled to GSH-Sepharose beads was used to pull down wild-type or mutant TBD protein that had been added in equal amounts (SI Fig. 9).
For a quantitative analysis of the mutant binding a fluorescence competition assay (22) was developed that is based on the displacement of wild-type TBD from its binding site on CycT1 by the addition of excess mutant TBD (Fig. 3A). Dissociation with wild-type TBD or buffer solution decreased the fluorescence signal intensity by 23% and 4%, respectively. Several of the TBD mutant proteins showed a significantly diminished ability to displace TBD-EDANS from CycT1. For eight of the mutant proteins, the dissociation constant Kd was >4-fold higher than for wild-type TBD (SI Table 4). Knowing the association and dissociation rates of wild-type TBD to CycT1 from previous (15) and present experiments, the kinetic parameter for the binding of mutant Hexim1 could be calculated from the slope of the curve. L292A showed a 4-fold higher koff rate but a similar kon rate, which is typical for the loss of a hydrophobic interaction upon mutation. K289A, K296A, and 3E-A all showed reduced kon rates indicating the efficacy of electrostatic interactions for the association of the two molecules.
Fig. 3.

Mutational analysis of the Hexim1-TBD/Cyclin T1 interaction. (A) Stopped flow dissociation competition experiments among CycT1, fluorescent-labeled TBD, and excess mutant TBDs. The preformed equimolar complex of CycT1 and TBD-EDANS was mixed with a 20-fold excess of nonlabeled TBD or buffer solution. The dissociation course was monitored for 0.5 s. The dilution and the competitive dissociation of TBD-EDANS from CycT1 by nonlabeled TBD molecules led to a characteristic decrease of the fluorescence signal. Binding constants and kinetic parameters are listed in SI Table 4. (B) Binding kinetics between TBD and wild-type CycT1 (1–292) or mutant CycT1(KR4A) (Left) as recorded by stopped flow measurements. The binding affinity to CycT1(KR4A) is ≈20-fold reduced compared with the wild-type protein, mainly because of a significantly decreased association rate kon (Right).
The loss of electrostatic attraction upon mutation of a negatively charged cluster in the binding interface of the TBD prompted us to test for a positively charged region in the cyclin box repeats of CycT1. Residues K247, R251, K253, and R254 of CycT1 (1–281) were mutated to alanines, termed KR4A, and the binding kinetics for the interaction with the TBD were determined by stopped flow measurements (Fig. 3B). The association rate kon was reduced >6-fold and the dissociation rate koff increased ≈3-fold, resulting in an apparent Kd of 68 μM for the mutant protein. These results strongly support the electrostatic nature in the association of CycT1 and TBD and help to explain previous observations of mutually exclusive binding of either Hexim1-TBD or HIV-1 Tat to CycT1 (15) by mapping the interaction to a similar region.
Binding of Mutant Hexim1 to P-TEFb in Cells.
The importance of the identified residues of Hexim1 to bind, and thus inhibit P-TEFb was further investigated by expressing selected full-length FLAG epitope-tagged Hexim1 (f.Hxm1) mutants in HeLa cells. Because the interaction between TBD and CycT1 is most likely of electrostatic nature, we mutated residues E290, E295, and E302, which form part of a negatively charged patch on the surface of the TBD (Fig. 1C). Moreover, we mutated L292 and N306 within the second and fourth heptad repeat, respectively. The selected residues were substituted by alanines individually or in combination.
Mutated and wild-type f.Hxm1 proteins were expressed in HeLa cells, immunoprecipitated from total cell lysates, and tested for binding to endogenous P-TEFb by Western blot analysis with antibodies directed against CycT1. As expected, wild-type f.Hxm1 bound CycT1 in cells (Fig. 4A, lane 2). None of the tested single amino acid substitutions abrogated binding completely (Fig. 4A, lanes 3–7), but mutation of the acidic residues E295A or E302A decreased the binding affinity (lanes 5–6). The combined mutation of E290 and E295 reduced the binding further (Fig. 4B, lane 4), whereas the double mutation E302A/N306A did not show a significant effect (lane 6). When three or four residues were substituted together, binding to CycT1 was in general strongly reduced (Fig. 4B, lanes 7–10). Levels of f.Hxm1 proteins in the immunoprecipitations were comparable (Fig. 4 A and B Lower).
Fig. 4.

Functional analysis of mutant Hexim1 proteins in cells. (A and B) Multiple amino acid substitutions in the Hexim1-TBD decrease the ability of Hexim1 to bind P-TEFb in cells. f.Hxm1 proteins that were expressed in HeLa cells from corresponding plasmid effectors (6 μg) and immunoprecipitated by anti-FLAG M2 beads are indicated above the Western blots. Arrows to the left indicate the bound P-TEFb and the amounts of immunoprecipitated f.Hxm1 proteins, respectively. (C–E) Multiple amino acid substitutions in the TBD disable the inhibitory function of Hexim1 in cells. HeLa cells expressed plasmid reporter pG6TAR (0.2 μg). Proteins that were coexpressed from corresponding plasmid effectors (Gal4.CycT1, 0.5 μg; f.Hxm1, 0.8 μg) with the plasmid reporter are presented below the CAT data. (Lower) Shown is the expression of f.Hxm1 proteins as indicated by the arrow.
P-TEFb Inhibition Requires Residues in the N-Terminal TBD Coiled-Coil Segment.
The contribution of the identified residues on the inhibitory property of Hexim1 was addressed by transcriptional assays in HeLa cells, using a system consisting of chimeric Gal4.CycT1 protein and the plasmid reporter pG6TAR, which contains six Gal4 DNA binding sites positioned upstream of the HIV-1 long terminal repeat (HIV LTR), followed by the chloramphenicol acetyltransferase (CAT) reporter gene. The recruitment of Gal4.CycT1 to the pG6TAR promoter activates transcription that depends on the kinase activity of P-TEFb (23). When we expressed the Gal4.CycT1 chimera together with pG6TAR in HeLa cells, the levels of CAT activity increased 32-fold over the basal levels, whereas coexpression of the wild-type f.Hxm1 protein decreased this activity to 4-fold (Fig. 4C, bars 1 and 2). Alanine substitutions of E290, L292, E295, and E302 reduced the ability of f.Hxm1 to inhibit P-TEFb in cells, whereas the substitution of N306 had no effect (Fig. 4C, bars 3–7). When compared with the inhibition by the wild-type f.Hxm1, this effect ranged from 1.6-fold (L292) to 5-fold (E295). The mutations K289A or K296A only slightly affected transcription inhibition, but effects were more pronounced by using lower amounts of transfected plasmids (SI Fig. 10). Based on circular dichroism data the mutant K289A has about the same amount of α-helical secondary structure as wild-type TBD indicating that the mutation did not disrupt the coiled-coil fold (SI Fig. 11).
Mutant f.Hxm1 proteins with double substitutions mostly lost their inhibitory abilities (Fig. 4D, bars 3–7). In agreement with the binding studies, the biggest effect was seen when E290 and E295 were mutated together, which resulted in the complete inactivation of f.Hxm1 (lane 4). The combination of double substitutions (Fig. 4E) or the triple mutant E286/E290/E293 (SI Fig. 10) rendered f.Hxm1 proteins nonfunctional. In summary, CycT1 recognizes a large surface composed of mostly acidic residues that are scattered along the first coiled-coil segment of the Hexim1-TBD structure.
Discussion
Our studies showed that the C-terminal region of Hexim1 forms a bipartite dimeric coiled-coil of which the first segment is required for the interaction with the CycT1 subunit of P-TEFb. The residue composition of this coiled-coil segment diverges significantly from that of an ideal parallel coiled-coil, exhibiting evolutionary conserved charged and bulky polar residues at the heptad a positions (K284, Y291) and hydrophobic residues at e and b positions (I288, L292). Moreover, conserved charged residues at positions g and e contribute to a negative surface patch (E290, E295) instead of forming a stabilizing salt bridge between the dimer chains. The second segment matches almost perfectly the preferred canonical consensus, suggesting a stable association of both strands. Thus, several features that distinguish the two coiled-coil segments provide a foundation for their functional differences.
In vitro binding assays and functional in vivo experiments delineated a large and acidic CycT1 binding interface on the first coiled-coil segment of the TBD. The observed small differences in the contribution of individual residues to the binding affinity and the inhibition of transcription activity can be accounted for by the different experimental set-up for the in vitro and in vivo experiments. In GST-pull down and fluorescence competition displacement experiments, individually purified domains were used, whereas the immunoprecipitation and in vivo transactivation analysis were performed with full-length proteins in cells that harbor the cofactors Cdk9 and 7SK snRNA. Previous studies showed that the cyclin box repeat region of CycT1 (1–292) is sufficient for binding to Hexim1 and that residues outside this domain do not contribute to the interaction with Hexim1 (9).
Despite conserved differences from the heptad repeat consensus sequence, the structure of the first coiled-coil segment and the preceding α-helix is preserved in a TBD construct that lacks the second coiled-coil segment (Hexim1 255–316, SI Fig. 7B). A functional consequence of this is that C-terminally truncated TBD still binds CycT1 and inhibits transcription activity of P-TEFb (9, 15). In contrast, full length Hexim1 with two mutated heptad repeat d positions (L287R and L294R), showed a markedly reduced ability to inhibit P-TEFb in the presence of 7SK snRNA (16). Because these mutations are at the dimerization interface, the structural integrity of the first TBD coiled-coil segment seems to be required for P-TEFb recognition. A cooperative interaction between Hexim1 and P-TEFb for two individual binding sites at the 5′ and 3′ hairpin loops of 7SK snRNA was suggested recently (24). 7SK snRNA might thus act as a stabilizing factor that gains additional specificity for the CycT1–Hexim1 interaction.
But how might Hexim1, bound to the CycT1 subunit of P-TEFb, repress the Cdk9 kinase activity? The presence of the TBD within Hexim1 was found to be required for inhibition of P-TEFb in cells (15) and an N-terminal truncation mutant of Hexim1 (181–359) inhibited P-TEFb in vitro independently of 7SK snRNA (10, 16). In analogy to the inhibition of the Cdk2/CycA complex by p27Cip2 (25), it has been suggested that tyrosines N-terminal of the determined CycT1 binding site (Y203, Y271) might block the ATP binding site in Cdk9 (10, 16). As shown here, the TBD of Hexim1 binds in close vicinity to the TRM region of the C-terminal helices of the CycT1 cyclin box repeats. Therefore, a catalytically repressive motif in Hexim1 is expected to be rather distant from the CycT1 binding site to cover the distance to the nucleotide binding site of the kinase that is supposed to reside on the opposite side of the cyclin molecule. p27Cip2 inhibits Cdk2 by binding and subsequent folding of an elongated region of ≈70 residues that covers a combined Cdk2/CycA surface (25). Moreover, studies with the Cdk2/CycA substrate CDC6 showed that recognition and discrimination against different substrates are achieved by a recruitment site on the cyclin and a second interaction site on the kinase that are connected by a linker of defined length and composition (26). For Hexim1, bridging between a Cdk9-inactivating tyrosine and the CycT1 interface would be consistent with our observation that the region N terminus of the first TBD α-helix (242–276) that is rich in conserved residues is unstructured and flexible. Alternatively, Hexim1 could bind the Cdk9/CycT1 heterodimer and alter the shape of the Cdk9 catalytic cleft by allosteric interactions, thus interfering with kinase ATP binding. Finally, binding of the Hexim1 dimer might displace the C-terminal helix of the cyclin boxes, which is known to determine the function and specificity of the respective Cyclin. Future studies will be directed to analyze the structure of the ternary complex formation between Cdk9, CycT1, and Hexim1 to further enlighten the mechanism of P-TEFb inhibition.
Materials and Methods
Plasmid Cloning, Protein Expression, and Purification.
Plasmids encoding Hexim1-TBD fragments (242–359, 255–359, 255–316) were generated as described in ref. 15. Site-directed mutagenesis was performed by using the mega-primer method similarly as described in ref. 27. The DNA coding for CycT1 (1–272 or 1–292) was cloned into pGEX-4T1-tev (Amersham, Piscataway, NJ) and expressed as GST-fusion protein. The plasmid reporter pG6TAR and the plasmids coding for Gal4.CycT1 and f.Hxm1 are described in ref. 18. To construct mutant f.Hxm1 proteins, the pFLAG-CMV-2.Hxm1 plasmids were subjected to site-directed mutagenesis, using the QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA).
Hexim1 and CycT1 proteins were expressed from Escherichia coli BL21(DE3) cells (Novagen, Madison, WI) and purified as described in ref. 15. Uniformly 15N- or 13C/15N-labeled TBD was prepared in minimal medium containing 15NH4Cl as the sole nitrogen source or 13C6 d-glucose as the sole carbon source, respectively. Fractions were analyzed by SDS/PAGE, and fractions containing Hexim1-TBD proteins (≈98% pure) were concentrated (Amicon filter) up to 1.0 mM in 20 mM KPi (pH 7.4) buffer/50 mM NaCl/1 mM DTE and stored at −80°C. Fluorescence labeling of the TBD with the dye 1,5-IAEDANS (Molecular Probes, Eugene, OR) was performed as described in ref. 15. Protein concentrations were determined by Bradford assay (Bio-Rad, Hercules, CA) and extinction coefficient measurements.
NMR Spectroscopy.
NMR samples contained ≈0.6–1.2 mM Hexim1-TBD in 20 mM KPi (pH 7.2) buffer (95% H2O/5% D2O or 100% D2O) with 50 mM NaCl/10 mM TCEP·HCl/1 mM DTE. The sample for measuring residual dipolar couplings was supplemented with ≈16 mg/ml PF1 phages (ASLA Biotech, Riga, Latvia). Mapping of the binding interface was performed on 0.1 mM samples of 15N-labeled TBD titrated with 0–0.1 mM unlabeled CycT1 (1–292). NMR spectra were acquired at 308 K on Bruker (Newark, DE) DRX600 and DRX800 spectrometers. Data were processed with NMRPipe (28) and analyzed by using NMRView (29). The assignments for 13C, 15N, and 1H nuclei are described in ref. 30.
Distance restraints were obtained from 15N- and 13C-edited NOESY spectra. Interchain distance restraints were derived from a 3D 13C F1-edited, F3-filtered HMQC-NOESY spectrum (31, 32) recorded on a sample containing 13C/12C mixed Hexim1-TBD dimers. 15N-1H residual dipolar couplings were obtained from the analysis of 15N-1H IPAP-HSQC (33) and 3D Hα-coupled HN(CO)CA data (34) acquired on the sample containing PF1 phages. The maximal 1DN-H was 16 Hz. Information about the backbone dynamics was derived from the measurement of 15N-relaxation experiments, including T1, T2, and [1H]15N NOE (35).
Structure Calculations.
Structure calculations were performed with X-PLOR-NIH (36), using molecular dynamics in torsion angle and cartesian coordinate space. Distance restraints were generated by using NMRView and classified according to NOE-crosspeak intensities. Upper bounds were 2.8, 3.5, 4.5, and 5.5 Å. The lower bound was always 1.8 Å. For all NOE restraints, r−6 sum averaging was used. Initially, all distance restraints from the 3D 15N-edited NOESY spectrum were assigned as intrachain, because backbone amides in α-helices only rarely mediate interchain contacts. For the next calculation step, interchain distance restraints from the 3D F1-edited, F3-filtered 13C-NOESY spectrum of the heterolabeled dimer sample were added. Distance restraints from the 13C-edited NOESY spectrum of the uniformly 13C-labeled sample were assigned as intra- or intermonomer or ambiguous, based on the presence of the same correlations in the 3D F1-edited F3-filtered 13C-NOESY spectrum and the initial structure calculations. Backbone dihedral angle restraints for φ and ψ were derived based on 3JHNHα, using error bounds of ±25° (±30° at the helix termini) and ±40°, respectively.
For regions with α-helical secondary structure, hydrogen bonds were defined by HN-O distance bounds of 1.8–2.3 Å and N O distance bounds of 2.6–3.1 Å. Based on 3JHαHβ2/3 and 3JNHβ2/3 coupling constants and NOE data, side chain χ1 angles were restrained to one of the staggered conformations (60°, 180°, −60°) ±30°. Residual dipolar couplings were included, using the ISAC algorithm (37) by calling the TENSO statement. The alignment tensors for residues 277–315 and 319–348 were fitted independently during the calculation. The final values for the force constants during the molecular dynamics in torsion angle (cartesian coordinate) space were 150 (50) kcal mol−1Å−2 for NOE distance restraints, 50 (300) kcal mol−1 rad−2 for dihedral angle restraints, and 0.6 (1.2) kcal mol−1 Hz−2 for residual dipolar coupling restraints. The 20 best of 200 calculated structures were finally refined in a water shell (38, 39).
O distance bounds of 2.6–3.1 Å. Based on 3JHαHβ2/3 and 3JNHβ2/3 coupling constants and NOE data, side chain χ1 angles were restrained to one of the staggered conformations (60°, 180°, −60°) ±30°. Residual dipolar couplings were included, using the ISAC algorithm (37) by calling the TENSO statement. The alignment tensors for residues 277–315 and 319–348 were fitted independently during the calculation. The final values for the force constants during the molecular dynamics in torsion angle (cartesian coordinate) space were 150 (50) kcal mol−1Å−2 for NOE distance restraints, 50 (300) kcal mol−1 rad−2 for dihedral angle restraints, and 0.6 (1.2) kcal mol−1 Hz−2 for residual dipolar coupling restraints. The 20 best of 200 calculated structures were finally refined in a water shell (38, 39).
In Vitro Binding Studies and Fluorescence Measurements.
Fluorescence experiments were performed at 20°C in 20 mM Hepes (pH 7.5) buffer containing 50 mM NaCl, 1 mM TCEP, and 1% (vol/vol) glycerol. Kinetic dissociation measurements were done by using a SX.18MV stopped flow apparatus (Applied Photophysics, Leatherhead, Surrey, U.K.) by rapid mixing of a preequilibrated solution of 5 μM CycT1 (1–292) and 5 μM dimeric TBD-EDANS with 100 μM excess of wild-type or mutant dimeric TBD. The dansyl-based fluorophore was excited at 337 nm, and the fluorescence emission was recorded through a 420-nm cutoff filter. The competition displacement curve was fitted to the fluorescence traces according to pseudo second-order kinetics. Dissociation experiments were analyzed with Scientist software, Version 1.0 (OriginLab, Northampton, MA).
Transient Transfection and CAT Reporter Gene Assay.
HeLa cells were seeded into six-well plates or 100-mm-diameter Petri dishes ≈12 h before transfection and transfected with FuGENE6 reagent (Roche Applied Science, Indianapolis, IN). CAT enzymatic assays were performed as described in ref. 23. Fold transactivation represents the ratio between the Gal4.CycT1-activated transcription and the activity of the reporter plasmid alone. Error bars give standard errors of the mean.
Supplementary Material
Acknowledgments
We thank Diana Ludwig for expert technical assistance; Roger Goody for stimulating discussions, valuable help on kinetic data evaluation, and continuous support; Q. Zhou, J.H. Yik (both at University of California, Berkeley, CA), and H. Tanaka (University at Tokyo, Tokyo, Japan) for sharing plasmid reagents; and Chris A. E. M. Spronk (Center for Molecular and Biomolecular Informatics, University of Nijmegen, Nijmegen, The Netherlands) for help by the structural refinement in a water shell. This work was supported Deutsche Forschungsgemeinschaft Grant GE-976/2 (to M.G.), Schweizer Nationalfonds Grant 31-109712 (to S.G.), National Institutes of Health Grant R01 AI49104 (to B.M.P.), a fellowship from the Treubel Fonds (Basel, Switzerland) (to S.A.D.), American Foundation for AIDS Research Grant 106584-36-RFNT (to M.B.).
Abbreviations
- CycT1/A
Cyclin T1/A
- P-TEFb
positive transcription elongation factor b
- TBD
Cyclin T binding domain.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and NMR restraints have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2GD7).
This article contains supporting information online at www.pnas.org/cgi/content/full/0701848104/DC1.
References
- 1.Sims RJ, 3rd, Belotserkovskaya R, Reinberg D. Genes Dev. 2004;18:2437–2468. doi: 10.1101/gad.1235904. [DOI] [PubMed] [Google Scholar]
- 2.Peterlin BM, Price DH. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 3.Goodrich JA, Kugel JF. Nat Rev Mol Cell Biol. 2006;7:612–616. doi: 10.1038/nrm1946. [DOI] [PubMed] [Google Scholar]
- 4.Saunders A, Core LJ, Lis JT. Nat Rev Mol Cell Biol. 2006;7:557–567. doi: 10.1038/nrm1981. [DOI] [PubMed] [Google Scholar]
- 5.Yang Z, Zhu Q, Luo K, Zhou Q. Nature. 2001;414:317–322. doi: 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen VT, Kiss T, Michels AA, Bensaude O. Nature. 2001;414:322–325. doi: 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- 7.Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q. Mol Cell. 2003;12:971–982. doi: 10.1016/s1097-2765(03)00388-5. [DOI] [PubMed] [Google Scholar]
- 8.Yik JH, Chen R, Pezda AC, Samford CS, Zhou Q. Mol Cell Biol. 2004;24:5094–5105. doi: 10.1128/MCB.24.12.5094-5105.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michels AA, Nguyen VT, Fraldi A, Labas V, Edwards M, Bonnet F, Lania L, Bensaude O. Mol Cell Biol. 2003;23:4859–4869. doi: 10.1128/MCB.23.14.4859-4869.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michels AA, Fraldi A, Li Q, Adamson TE, Bonnet F, Nguyen VT, Sedore SC, Price JP, Price DH, Lania L, Bensaude O. EMBO J. 2004;23:2608–2619. doi: 10.1038/sj.emboj.7600275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 12.Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Mol Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 13.Kusuhara M, Nagasaki K, Kimura K, Maass N, Manabe T, Ishikawa S, Aikawa M, Miyazaki K, Yamaguchi K. Biomed Res. 1999;20:273–279. [Google Scholar]
- 14.Ouchida R, Kusuhara M, Shimizu N, Hisada T, Makino Y, Morimoto C, Handa H, Ohsuzu F, Tanaka H. Genes Cells. 2003;8:95–107. doi: 10.1046/j.1365-2443.2003.00618.x. [DOI] [PubMed] [Google Scholar]
- 15.Schulte A, Czudnochowski N, Barboric M, Schonichen A, Blazek D, Peterlin BM, Geyer M. J Biol Chem. 2005;280:24968–24977. doi: 10.1074/jbc.M501431200. [DOI] [PubMed] [Google Scholar]
- 16.Li Q, Price JP, Byers SA, Cheng D, Peng J, Price DH. J Biol Chem. 2005;280:28819–28826. doi: 10.1074/jbc.M502712200. [DOI] [PubMed] [Google Scholar]
- 17.Dulac C, Michels AA, Fraldi A, Bonnet F, Nguyen VT, Napolitano G, Lania L, Bensaude O. J Biol Chem. 2005;280:30619–30629. doi: 10.1074/jbc.M502471200. [DOI] [PubMed] [Google Scholar]
- 18.Barboric M, Kohoutek J, Price JP, Blazek D, Price DH, Peterlin BM. EMBO J. 2005;24:4291–4303. doi: 10.1038/sj.emboj.7600883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lupas AN, Gruber M. Adv Protein Chem. 2005;70:37–78. doi: 10.1016/S0065-3233(05)70003-6. [DOI] [PubMed] [Google Scholar]
- 20.Woolfson DN. Adv Protein Chem. 2005;70:79–112. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- 21.Dosset P, Hus JC, Blackledge M, Marion D. J Biomol NMR. 2000;16:23–28. doi: 10.1023/a:1008305808620. [DOI] [PubMed] [Google Scholar]
- 22.Goody RS. In: Kinetic Analysis of Macromolecules. A Practical Approach. Johnson KA, editor. Oxford: Oxford Univ Press; 2003. pp. 153–170. [Google Scholar]
- 23.Taube R, Lin X, Irwin D, Fujinaga K, Peterlin BM. Mol Cell Biol. 2002;22:321–331. doi: 10.1128/MCB.22.1.321-331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Egloff S, Van Herreweghe E, Kiss T. Mol Cell Biol. 2006;26:630–642. doi: 10.1128/MCB.26.2.630-642.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pavletich NP. J Mol Biol. 1999;287:821–828. doi: 10.1006/jmbi.1999.2640. [DOI] [PubMed] [Google Scholar]
- 26.Cheng KY, Noble ME, Skamnaki V, Brown NR, Lowe ED, Kontogiannis L, Shen K, Cole PA, Siligardi G, Johnson LN. J Biol Chem. 2006;281:23167–23179. doi: 10.1074/jbc.M600480200. [DOI] [PubMed] [Google Scholar]
- 27.Breuer S, Gerlach H, Kolaric B, Urbanke C, Opitz N, Geyer M. Biochemistry. 2006;45:2339–2349. doi: 10.1021/bi052052c. [DOI] [PubMed] [Google Scholar]
- 28.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 29.Johnson BR, Blevins RA. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 30.Dames SA, Schönichen A, Grzesiek S, Geyer M. J Biomol NMR. 2006;36:39. doi: 10.1007/s10858-006-9010-4. [DOI] [PubMed] [Google Scholar]
- 31.Lee W, Revington MJ, Arrowsmith C, Kay LE. FEBS Lett. 1994;350:87–90. doi: 10.1016/0014-5793(94)00740-3. [DOI] [PubMed] [Google Scholar]
- 32.Dames SA, Kammerer RA, Wiltscheck R, Engel J, Alexandrescu AT. Nat Struct Biol. 1998;5:687–691. doi: 10.1038/90444. [DOI] [PubMed] [Google Scholar]
- 33.Ottiger M, Delaglio F, Bax A. J Magn Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- 34.Bax A, Kontaxis G, Tjandra N. Methods Enzymol. 2001;339:127–174. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- 35.Grzesiek S, Bax A, Hu JS, Kaufman J, Palmer I, Stahl SJ, Tjandra N, Wingfield PT. Protein Sci. 1997;6:1248–1263. doi: 10.1002/pro.5560060613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 37.Sass H-J, Musco G, Stahl SJ, Wingfield PT, Grzesiek S. J Biomol NMR. 2001;21:275–280. doi: 10.1023/a:1012998006281. [DOI] [PubMed] [Google Scholar]
- 38.Linge JP, Williams MA, Spronk CAEM, Bonvin AMJJ, Nilges M. Proteins. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- 39.Nabuurs SB, Nederveen AJ, Vranken W, Doreleijers JF, Bonvin AMJJ, Vuister GW, Vriend G, Spronk CAEM. Proteins. 2004;55:483–486. doi: 10.1002/prot.20118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.