

Abstract
Misprocessing of β-amyloid precursor protein (APP) leading to the formation of elevated quantities of β-amyloid peptide (Aβ), derived by a cleavage at the β-secretase site (N-671/673aa) and by a cleavage at the γ-secretase site (C-711/713aa) of APP, is considered a key event in the pathogenesis of Alzheimer disease (AD). Point mutations near the β-secretase site in the human gene for APP, such as in the Swedish mutation-KM670/671NL lead to a form of dominantly inherited AD. These mutations are known to promote β-site cleavage and to increase levels of Aβ. Aβ has been shown previously to increase AChE activity in vitro. We wished to test whether or not translational blocking of APP-mRNA at the mutated β-site by antisense (AS) oligodeoxynucleotides (ODNs) directed to the mutated site will reduce cerebral amyloid in the Swedish transgenic mouse model (Tg2576). Mice were injected intracerebroventricularly (ICV) with AS-ODNs directed at the mutated β-site (AS-β site) or with AS-ODNs directed at the normal γ-site (AS-γ site) of human APP-mRNA, and compared with procedural controls that received ICV injections of sense ODNs at the β-site (S-β site), sense ODNs at the γ-site (S-γ site) or mismatched ODNs, and with untreated littermates (Lt) and untreated transgenic mice (Tgs). ODNs were injected into the 3rd ventricle once a week for 4 weeks. Brains were processed for ELISA analysis of sAβ40, sAβ42 and sAPPα. The physiological relevance of antisense ODNs was tested by evaluating the cerebral distribution of acetyl cholinesterase (AChE) before and after the treatment. AChE was found increased about 5-fold in Tg cortex as compared to control brain. Results show that compared to untreated and procedural controls, AS-β increased cerebral levels of sAPPα by 43% and reduced sAβ40/42 by ~39%; while simultaneously reducing the cortical density of AChE by ~4-fold in the treated Tg animals, almost to the level found in the control brain (all values p<0.0001, ANOVA, unpaired 2-tailed Student t-test), while AS-γ did not have any effect. These results indicate that antisense directed to the mutated β-site may be an effective approach to treat familial AD.
Keywords: Alzheimer disease, Intracerebroventricular, Antisense oligodeoxynucleotides, sAβ40/42, sAPPα, mRNA, AChE
Introduction
The efficacy of synthetic antisense (AS) oligodeoxynucleotides (ODNs) in inhibiting the synthesis of selected targeted proteins was recognized more than 20 years ago (Zamecnik and Stephenson, 1978). Since then, mounting evidence suggests the use of AS-ODNs for various purposes, including reducing arterial blood pressure in spontaneously hypertensive rats by inhibiting thyrotropin-releasing hormone (Garcia et al., 2001), promoting anti-tumor activity by antisense clusterin (Zellweger et al., 2001), attenuating apoptosis and cholestatic liver disease by Bid-antisense (Higuchi et al., 2001), and reduction of tumor cell viability by inhibiting bcl-2 expression (Olie et al., 2002).
Amyloidogenic processing of β-amyloid precursor protein (APP) leading to the excessive formation of β-amyloid (Aβ) is considered a key event underlying the biochemistry of Alzheimer disease (AD). Aβ is derived by a cleavage of APP at the β-secretase site (N-671/673aa) followed by a cleavage at the γ-secretase site (C-711/713aa) (Selkoe and Lansbury, 1999; Chauhan and Siegel, 2004). Point mutations in the human APP gene, such as in the Swedish double mutation-KM670/671NL, are known to promote β-site cleavage and to cause one form of dominantly inherited AD. Other mutations causing dominantly inherited AD involve presenilins 1 and 2, components of the γ-secretase complex (Selkoe and Kopan, 2003). We wished to test whether or not translational blocking of APP-mRNA at the mutated β-site by antisense (AS) oligodeoxynucleotides (ODNs) will reduce cerebral amyloid load in the Swedish mutant murine model-Tg2576. If so, this could be a significant means of preventing and treating AD pathology caused by this mutation without affecting expression of the non-mutated allele in this form of inherited AD. In addition, this approach would represent a model for treating the other dominantly inherited AD forms as well as other genetic diseases expressing a dominant mutated allele.
Coulson and co-workers reported that APP in murine neuron culture could be down regulated by AS-ODNs to initiating methionine, exon 2 or exon 4 (Coulson et al., 1997). While effects of AS-ODNs directed to native APP in senescence-accelerated mice (SAM8) have been studied (Kumar et al., 2000; Banks et al., 2001; Poon et al., 2004, 2005), there have been no published attempts to down regulate translation of APP in transgenic animal models or of specifically mutated APP associated with inherited AD.
To test our hypothesis, we designed ODNs directed to the mutated β-cleavage site of mhAPP mRNA and to the normal γ-cleavage site. These ODNs included 2’-O-(Methyl) Ethyl (2’MOE) ribosyl modification at 2’-sugar moiety capped at 5’- and 3’-ends retaining phosphorothioated (PS) bases in between. Such compounds have been demonstrated to be stable and to have high affinity and selectivity for the specific targeted mRNA as well as a high degree of resistance against extra- and intracellular nucleases (Temsamani et al., 1993; Whitesell et al., 1993; Higuchi et al., 2001; Olie et al., 2002). Early studies showed that PSODNs could be micro-infused into rat striatum resulting in uniform distribution in brain over 47 h with exponential decay over the ensuing 11 h without obvious toxicity (Broaddus et al., 1998). It was demonstrated that AS-ODNs infused into 3rd ventricle could down regulate dopamine (DA) receptors (Liang and Pan, 2002) and N-Methyl-D-Aspartate (NMDA) receptors (Murata et al., 2002). On the presumption that as a potential therapy, intraventricular infusions ultimately could be more safely performed and more precisely regulated with respect to quantity and rate of ODN administration than intraparenchymal injections, it was elected to try injection of AS-ODNs into 3rd ventricle. Accordingly, the time course of diffusion and distribution in mouse brain of 2’MOE modified ODNs directed to the β-cleavage site of APP mRNA was mapped, showing gradual spread throughout brain from 15 min to 3 h and complete clearance by 8 h after a single intracerebroventricular (ICV) injection into 3rd ventricle (Chauhan, 2002).
In the current study, we wished to obtain fundamental data on the optimal parameters for ICV administration of ODNs and a comparison of effects of ODNs directed to the β- versus the γ-cleavage sites. We report the first available data showing that ICV injection of AS-ODN directed to the β-cleavage site of mutated human (mhu) APP mRNA reduces Aβ and increases sAPPα in vivo while AS-ODN to the non-mutated hAPP γ-site has no effect on the Aβ load in this transgenic mouse model of AD.
Furthermore, since Aβ is known to increase AChE in cultured embryonal P19 carcinoma cells (Sberna, et al, 1997), in cultured cortical neurons (Fodero, et al, 2004) and in mice administered Aβ intraventricularly (Fu et al., 2006, 16872586), and since increased Aβ is associated with increased AChE in Tg mice overexpressing the CT-100 APP and Aβ (Sberna, et al, 1998) and in the Tg2576 mouse model (Fodero, et al, 2002), we wished to determine whether intraventricular ODN directed to the β- cleavage site would alter cerebral AChE distribution in Tg2576.
Experimental Procedures
Animals
10-month-old transgenic mice harboring double Swedish mutation (KM670/671NL) (Tg2576) were obtained from Taconic Farms (Germantown, NY). The transgene-negative animals served as littermate controls (Lt), in addition to wild type controls (Wt) of different genetic background (C57BL/6J) (Jackson Laboratories, Inc.).
Animals were divided into 6 treatment-groups (N=10/group; n=5 to be used for AChE histochemistry and n=5 to be used for ELISA): [i] Tgs injected with saline vehicle; [ii] Tgs injected with AS-ODNs directed at the mutated β-site (AS-β site); [iii] Tgs injected with sense ODNs directed at the mutated β-site (S-β site); [iv] Tgs injected with AS-ODNs directed at the normal γ-site (AS-γ site); [v] Tgs injected with sense ODNs directed at the normal γ-site (S-γ site); [vi] Tgs injected with mismatched ODNs with proportional G-C content with AS- and Stest-ODNs. These treatment groups were compared with untreated Wts, Lts and Tgs to obtain the base line values.
Treatment group animals were stereotaxically implanted with stainless steel guide cannula in the 3rd ventricle and cemented to the skull with dental acrylic. Coordinates for the tip of cannula were 0.25 cm posterior to the bregma along the midline and 3.00 mm below the surface of the skull as determined in our laboratory (Chauhan et al., 2001). The guide cannula was kept screw-capped until the treatment was over. Animals were injected with test materials once a week for 4 weeks and allowed to survive 1-week post treatment.
Oligodeoxynucleotides
All test oligodeoxynucleotides (ODNs) were synthesized using standard phosphoramidite chemistry with 2’-O-(Methyl) Ethyl (2’MOE) ribosyl modification capped at 5’- and 3’-ends, and purified by Ion-Exchange Reverse Phase HPLC on 1.0 μmol scale (Qiagen-Operon, Inc., Alameda, CA). Antisense ODNs complementary to the mutated β-cleavage site targeted at 666-674aa, and complimentary to the normal γ-cleavage site targeted at 709-718aa (Fig. 1) on hAPP-mRNA (Accession No. NM_000484) were synthesized in which the underlined bases represent 2’MOE-residues with phosphorothioate-residues in between (Table 1). Respective sense ODNs and mismatched ODNs with proportional G-C content were also prepared that served as procedural controls. Lyophilized preparations of these ODNs were reconstituted with sterile saline to obtain the final concentration of 1 nmol/1μl.
Fig. 1.
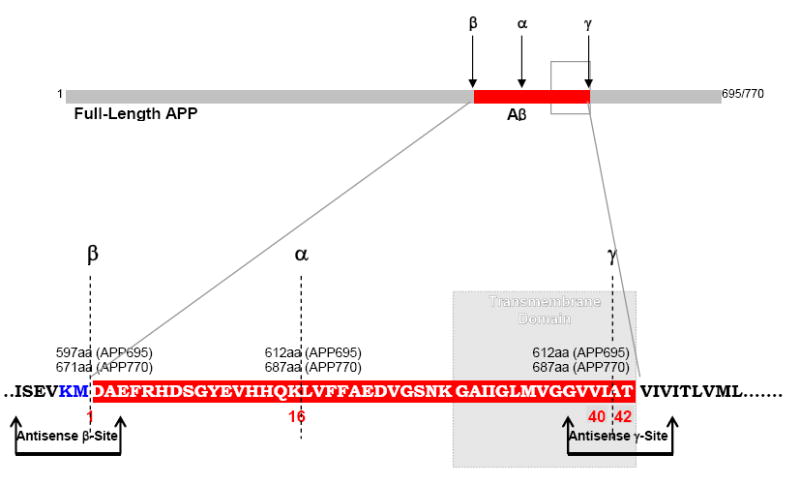
Proteolytic cleavage sites on APP molecule showing the location of Swedish mutation (Blue letters). Arrows indicate antisense oligonucleotide constructs designed to target ISEVKMDAE amino acids (aa) (β-site) and GVVIATVIV aa (γ-site) on APP.
Table 1.
Antisense and control oligodeoxynucleotides sequences
| Test ODNs | Targeted bases on hAPP-mRNA* | GC Content | |
|---|---|---|---|
| Antisense β-site | 5’-A TTC TGC ATCCAT CTT CAC TTC AGAGAT CG-3’ | 666-674aa | 44% |
| Sense β-site | 5’-CG ATC TCT GAA GTG AAG ATG GAT GCAGAA T-3’ | 666-674aa | 44% |
| Antisense γ-site | 5’-A GATGAC GAT CAC TGT CGC TAT GAC AAC ACC CG-3’ | 709-718aa | 49% |
| Sense γ-site | 5’-CG GGT GTT GTC ATA GCG ACA GTG ATC GTCATC A-3’ | 709-718aa | 49% |
| Random | 5’-A AGGACAATGAGTCTTCCTGGAACAAGTG C-3’ | Random | 49% |
Double Underlined bases are additional bases attached by the manufacturer; Underlined bases represent 2’-MOE modification [*], Swedish Mutation; Bold Italicized represent bases targeted to mutated β-site
I.C.V. Injections
Animals were anesthetized with ketamine (100mg/kg, i.p.) and xylazine (10mg/kg, i.p.) and positioned on a stereotaxic frame with ear bars plugged in and jaws fixed to a biting plate. A region over the skull was prepped with Betadine and an incision was made in the skin along the midline. The bregma point was identified and a burr hole was drilled 0.25 cm posterior to the bregma. A stainless steel guide cannula was implanted through the burr hole 3 mm below the surface of the skull into the 3rd ventricle according to the stereotaxic coordinated determined earlier (Chauhan et al., 2001). The guide cannula was cemented to the skull with dental acrylic and the cannula was tightly screw-capped.
At the time of injecting ODNs, region over the skull around implanted cannula was sterilized with Betadine and alcohol. Teflon screw cap over the guide cannula was unscrewed. A volume of 10μl containing 10 nmols of respective test ODNs was slowly injected with the use of Hamilton syringe over 10 min. Hamilton syringe was left in place for additional ~2 min to ensure proper and complete administration of test material. After intracerebral delivery of ODNs, the guide cannula was screw-capped tightly to ensure proper closure. The test material was injected once/week for 4 weeks. Animals were allowed to survive 1-week post treatment.
ELISA quantitation of sAPPα, sAβ40 and sAβ42 in Tg2576 brain
Animals were decapitated and brains were snap-frozen in liquid nitrogen until further use. Brains were homogenized in 6 volumes of chilled homogenization buffer [20 mM Tris-HCl, pH 7.8, 5 mM EDTA, 2 mM phenylmethylsulfonylfluoride (PMSF), 0.5 μg/ml leupeptin, 0.7 μg/ml pepstatin, 0.1 mg/ml phenanthroline, 0.1 mg/ml benzamidine] containing 0.2% sodium dodesyl sulphate (SDS), and centrifuged at 100,000g at 4°C for 1h. Pellets were discarded and supernatant portions were analyzed. Brain protein content was determined by Bradford’s protein assay and the samples were diluted in Tris buffered saline, pH 7.4 (TBS) to obtain total protein concentration of 100μg/100μl. An aliquot containing 100μg/100μl of total protein was analyzed by sandwich ELISA (Enzyme Linked ImmunoSorbent Assay) for quantitating SDS-soluble α-secretase cleaved APP (sAPPα) and soluble Aβ peptides (sAβ40, sAβ420).
Quantitation of sAPPα was performed with the use of a commercial kit (Sigma) according to the manufacturer’s directions. The capture antibody recognizes the portion of APP downstream of the β-site and therefore excludes sAPPβ. Assays of sAβ40 and sAβ42 were performed with the use of specific antibodies in a commercial kit (Signet) according to manufacturer’s directions.
The 96-well plates (Nunc) were pre-coated with the respective capture antibody (1:1000) for 24 h at 4°C. The plates were incubated overnight with 100 μl of sample containing 100 μg of total protein or standards [100-1000 pg of purified synthetic peptides (provided in the kit) at 4°C. The samples were further incubated with the respective reporter antibody for 90 min at 37°C. After 3 washes, 100 μl of anti-mouse IgG-HRP or anti-rabbit IgG-HRP conjugate (1:200) (provided in the kit) were added to each well and incubated for 90 min at room temperature. Samples were incubated with chromogenic solution (provided in the kit) for 30 min. The reaction was stopped by the addition of 100 μl of 0.5N H2SO4. The absorbance was read at 450 nm with the use of an ELISA micro titer plate reader (Molecular Devices). All samples were analyzed in triplicates.
AChE Histochemistry in Tg2576 brain
AChE histochemistry was performed according the established histochemical technique (Hedreen et al., 1985) modified from the original method of Karnovsky and Roots, (1964). Animals were perfused with 10% buffered paraformaldehyde containing 1% CaCl2. The perfused brain were preserved in 0.1M sucrose solubilized in 0.01M phosphate buffered saline (PBS), until the brain tissue were saturated and sunk at the bottom. So preserved brains were sectioned at 20μm thickness in cryostat and mounted on coverslips, and incubated in a petri dish containing incubation medium prepared as follows:
5.0 mg Substrate………(Acetyl Choline Iodide-Sigma), dissolved in 6.5 ml of 0.1M sodium hydrogen maleate buffer, pH 6.0. Then, following ingredients were sequentially added with constant stirring between each additions in order to avoid crystalization.
0.5 ml………0.1M sodium citrate
1.0 ml………30mM CuSO4
1.0 ml………H2O
1.0 ml………5mM potassium ferricyanide
5.0 mg………Silver nitrate, to be added after all above ingredients are completely dissolved.
The final incubation medium should be clear greenish/light yellow. This incubation medium is stable for ~3h. 20μm thick cryo-sections fixed on coverslips were incubated in this medium for 30 min at 37°C. Sections were washed and mounted on slides using anti-fade mounting medium (Fisher), and images captured immediately within an hour. Captured images were later analyzed with the use of computer-assisted imaging.
AChE activity was assessed by densitometric measurements of product on digitized images, performed with the use of the BioQuant Image Analyzer (R & M Biometrics, Nuhsbaum Inc., Itasca, IL) as previously described (Chauhan and Siegel, 2002). Images were captured with a CCD camera attached to a Leica microscope to generate a digitized image resolved into 255 × 255 pixels. Regions of interest (ROI) were identified under dark-field illumination for product of AChE activity. Background level was determined from the area outside the brain section. First level threshold was determined after subtracting the background level. The second level threshold was determined after subtracting non-specific binding in omit and serum controls. Thus, the net integrated optical density (IOD) representing specific immunoreactive signal was determined after subtracting first and second level thresholds. Each sample was digitized under identical illumination, threshold and camera settings.
Net IODs for AChE were quantitated within the defined high power fields (hpfs) (200◻m2/hpf) of cerebral cortex. These hpfs were precisely located between the 3rd to 5th cortical pyramidal cell layers 50μm deep from the pial surface and 50μm distant from the outer surface of corpus callosum distributed parallel to the hippocampal curvature. These hpfs were, 100μm apart, first hpf placed 1200μm away from the olfactory lobe-cortex gyrus. IODs/μm2 for 5 hpfs were obtained bilaterally in 2 sections per animal. The mean value for each animal was derived from 20 hpf-measurements. Group means were derived from 5 animals per group.
Statistical Analysis
Group means and standard deviation of means (SD) were derived from average of individual values (n=3) for each group (N=5/group) for ELISA; and from average of individual values (n=20) for each group (N=5/group) for AChE histochemistry. Data were statistically analyzed by ANOVA, unpaired 2-tailed Studentt-test. A value of p<0.05 was considered significant. The data are presented as mean ± SD (Fig. 2, Table 2; Fig. 3, Table 3).
Fig. 2.
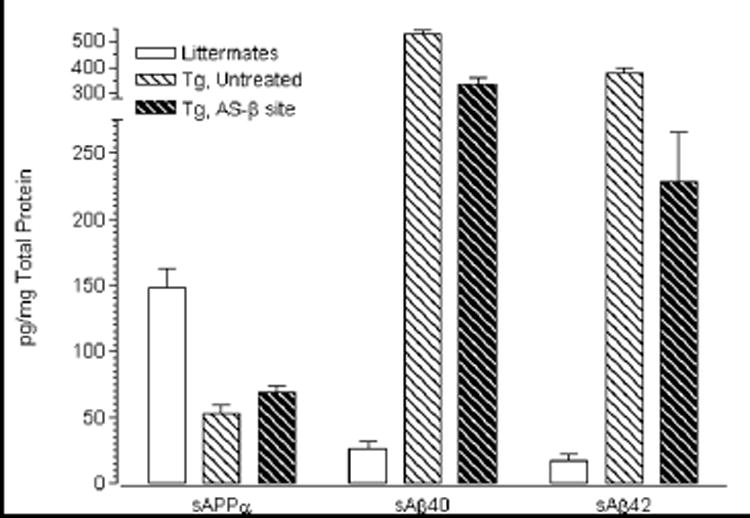
Effect of β-secretase-site directed antisense inhibition of human APP-mRNA on the levels of sAPPα, sAβ40 and sAβ42 as measured by ELISA (pg/mg protein) in Tg2576 brain. Data are statistically analyzed by ANOVA, unpaired two-tailed Student t-test, and are presented as group means and standard deviation (SD) derived from the average of individual values (n=3) for each group (N=5).
Table 2.
Effect of β- and γ-site directed antisense oligonucleotides on cerebral Aβ levels in Tg2576
| Wts | Lts | Tgs
Untreated |
Tgs
Vehicle |
Tgs
As-β site |
Tgs
S-β site |
Tgs
AS-γ site |
Tgs
S-γ site |
Tgs
Random |
|
|---|---|---|---|---|---|---|---|---|---|
| SAPPα | 146.3±24.7 | 147.7±25.0 | 52.53±18.5
↓ 64% [#] |
52.45±11.1
↓ 64% [#] |
59.44±10.85
↑ 13% [@] |
53.47±12.6
NS [@] |
53.6±21.4
NS [@] |
52.0±12.7
NS [@] |
51.7±17.8
NS [@] |
| sAβ40 | 25.27±3.3 | 24.61±4.9 | 527.3±18.0
↑ ~21-fold [#] |
542.9±32.7
↑ ~21-fold [#] |
330.0±29.6
↓ 37% [@] |
508.2±43.6
NS [@] |
534.9±36.2
NS [@] |
523.9±25.7
NS [@] |
538.6±33.6
NS [@] |
| sAβ42 | 17.27±2.3 | 17.13±4.6 | 374.1±23.9
↑ ~22-fold [#] |
371.5±25.6
↑ ~22-fold [#] |
227.8±38.6
↓39% [@] |
342.9±31.6
NS [@] |
374.2±24.14
NS [@] |
365.3±13.6
NS [@] |
371.6±36.3
NS [@] |
Wts, Wild type controls; Lts, Littermate controls; Tgs, Transgenic animals; AS, Antisense; S, Sense; NS, Not Significant #, Change compared to Wts/Lts; @, Change compared to Untreated or Vehicle treated Tgs All values expressed as (pg/mg protein).
Fig. 3.
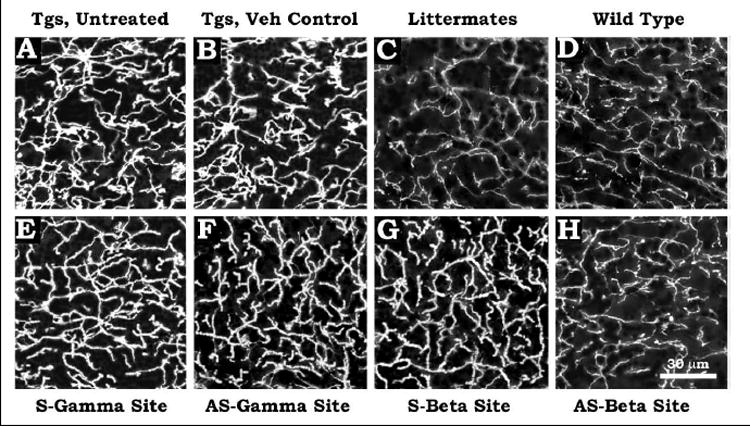
Distribution of AChE within the dendritic and axonal neuropil of transgenic Tg2576, wild type and non-transgenic littermate brains. Untreated Tgs (Fig. 3A), vehicle-injected Tgs (Fig. 3B), littermate (Fig. 3C), wild type (Fig. 3D), Tgs injected with sense γ-site ODNs (Fig. 3E), Tgs injected with antisense γ-site ODNs (Fig. 3F), Tgs injected with sense β-site ODNs (Fig. 3G), Tgs injected with antisense β-site ODNs (Fig. 3H). Scale bar=30 μm.
Table 3.
Effect of β- and γ-site directed antisense oligonucleotides on cerebral AChE Density in Tg2576
| Wts | Lts | Tgs
Untreated |
Tgs
Vehicle |
Tgs
As-β site |
Tgs
S-β site |
Tgs
AS-γ site |
Tgs
S-γ site |
Tgs
Random |
|
|---|---|---|---|---|---|---|---|---|---|
| AchE (IODs/μm2) | 13.3±1.7 | 12.7±1.0 | 63.5±8.2
↑ ~5-fold [#] |
62.6±9.1
↑ ~5-fold [#] |
14.4±2.5
↓ ~4-fold [@] |
64.1±11.1
NS [@] |
63.6±12.4
NS [@] |
62.9±8.7
NS [@] |
61.7±7.8
NS [@] |
Wts, Wild type controls; Lts, Littermate controls; Tgs, Transgenic animals; AS, Antisense; S, Sense; NS, Not Significant #, Change compared to Wts/Lts; @, Change compared to Untreated or Vehicle treated Tgs; AChE, Acetyl Cholinesterase All values expressed as (IODs/ μm2).
Results
All data for brain sAPPα and sAβ40/42 from brains of all control and experimental groups (Table 2) show that in the wild type and littermate controls, cerebral levels of sAPPα are ~147pg/mg protein, while cerebral levels of sAβ40 and sAβ42 are about ~25pg and ~17pg/mg protein respectively, with no significant difference in these values between wild type and littermate controls groups.
In all untreated and experimental transgenic mice, cerebral levels of sAPPα are reduced to 52-59pg/mg protein with no significant differences among these transgenic groups. Since our assay does not differentiate between mouse and human sAPPα, the measured sAPPα is a mixture of both. The reduction in total sAPPα in Tgs may be due to repression of mouse APP-mRNA translation by the over-expressed human APP-mRNA. The residual sAPPα of about 55pg/mg protein probably represents the human gene product, which is low because most of the human mutant APP undergoes amyloidogenic processing in this transgenic model.
Consistent with our previous reports in Tg2576 model (Chauhan and Siegel, 2003; Chauhan et al., 2005), current results show that in untreated and vehicle injected Tg2576 brain, the base levels of Aβ40 (527-540pg/mg protein) are ~1.4-fold greater than the levels of Aβ42 (371-374pg/mg protein).
Fig. 2 compares the results of injecting antisense ODNs directed to the β-cleavage site of APP (AS-β site) in Tg animals to those of littermates and untreated Tg controls, whileTable 2 shows the results of injecting antisense and sense ODNs directed to the β- (AS-β site and S-β site) and γ- (AS-γ site and S-γ site) cleavage sites of APP (AS-β site) compared to untreated Lts, Wts, Tgs and to vehicle-injected and random ODNs injected Tgs before and after the treatment.
The data show that the sum of sAβ40 and sAβ42 in the untreated Tg animals is ~900pg/mg protein, which is 18.2 times the amount of total sAPPα (Table 2). Compared to untreated or vehicle treated Tgs, antisense inhibition at the β-secretase cleavage site (AS-β site) increased cerebral sAPPα by 13% (p<0.0001, ANOVA, unpaired 2-tailed Student t-test), and reduced cerebral sAβ40/42 by 39 and 38% respectively (P<0.0001, ANOVA, unpaired 2-tailed Student t-test), as measured by sandwich ELISA (Table 2).
Thus, about 60% of APP synthesis goes on to complete APP molecules from which Aβ may be derived. The ratio of overall β-processing to α-processing is reduced to 11.2 (from 18.2 original value), which is a 40% reduction in the AS-β site injected Tgs compared to untreated Tg mice. By contrast, antisense inhibition at γ-site did not change the levels of cerebral sAβ40/42 (Table 2).
Fig. 3 shows the distribution of AChE within the dendritic and axonal neuropil of cerebral cortex of untreated Tgs (Fig. 3A), vehicle injected Tgs (Fig. 3B), littermate (Fig. 3C) and wild type (Fig. 3D) controls, Tgs injected with sense γ-site ODNs (Fig. 3E), Tgs injected with antisense γ-site ODNs (Fig. 3F), Tgs injected with sense β-site ODNs (Fig. 3G), Tgs injected with antisense β-site ODNs (Fig. 3H). It is evident that compared to wild type and littermate controls, the untreated and vehicle-injected Tgs showed abundant distribution of AChE, which did not change after sense ODNs or antisense γ-site ODNs or mismatched ODNs (Not shown). However, treatment with antisense β-site ODNs drastically reduced AChE distribution in Tg2576 brain to a level almost that of wild type and littermate brains.
Densitometric measurements showed that compared to wild type and littermate controls, the untreated and vehicle-injected Tgs showed ~5-fold increased levels of AChE. Similarly increased levels of AChE densities were found in Tgs after administration of sense ODNs, antisense γ-site ODNs or mismatched ODNs. However, treatment with antisense β-site ODNs reduced AChE densities by ~4-fold in Tg2576 brain, almost to the levels observed in wild type and littermate brains (Table 3).
Discussion
These results indicate that ICV administration of AS-ODN directed to the β-cleavage site of APP mRNA is quite effective in improving the proportion of α-secretase to β-amyloidogenic processing. The preliminary protocol tested in this study showed not only 40% reductions in the levels of Aβ, but 40% increases in sAPPα as well and an overall 40% reduction in the ratio of β-processing to α-processing in the treated Tg as compared to untreated Tg animals. These results were obtained 1 week after 4 weekly injections. It is possible that continuous infusion or injections carried out over longer periods would have greater beneficial effects.
This reduction in Aβ peptides is due to net reduction in the synthesis of full length APP past the β-site due to interference by the AS-ODN. One would expect that since the effect of AS-ODN is to block synthesis of (mhu) APP at the β-site, then (mhu) sAPPα would also be reduced as would be Aβ. However, since almost all of the synthesized (mhu) APP in this Tg model is converted to Aβ, with very little available to form (mhu) sAPPα, the effect of reducing APP synthesis on the total measured sAPPα is probably not significant in this assay. The fact that sAPPα is actually increased in some proportion to the reduction in Aβ was surprising. Although the reason is not known, one plausible explanation is that the Aβ itself, among its other toxic actions, reduces α-secretase processing of the APP molecules whose synthesis is not blocked by the AS-ODN, which is about 60 % of potential APP synthesis in this experiment. In vitro data also show that synthetic amyloid reduces secretion of soluble APP (Takashima et al., 1995). If this hypothesis is correct, then reductions in Aβ by any means should potentiate α-processing. Furthermore, increases in Aβ past some threshold would lead to a feed-forward vicious cycle for uncontrolled Aβ production. There is considerable evidence for Aβ promoting amyloidogenesis at the expense of α-secretase processing through effects on cholesterol levels and ordering within membranes (Chauhan, 2003; Siegel et al., 2006).
Hypothetically, antisense blocking both at the β- and γ-sites on APP-mRNA was expected to reduce cerebral Aβ. However, we found that antisense inhibition only at the β-site was successful in reducing cerebral Aβ. To our surprise, antisense inhibition at the γ-site did not have any effect. The one and only hypothetical explanation to this could be the assumption that before γ-site antisense hits APP-mRNA, APP-mRNA up to the γ-site was already transcribed and hence antisense blocking at the γ-site did not work.
The fact that the AS to the β-site resulted in decreased AChE almost to the level seen in controls in parallel with reductions in the Aβ content indicates an important physiologic effect. It is possible that the neurotoxicity of Aβ in young Tg mice or early in AD brain involves AChE induction effects through action on cholinergic nerve terminals. Sberna et al (1997) found that Aβ increases Ca influx and AChE in cultured P19 cells and Hu et al. (2003) reported that Aβ increases AChE by reducing enzyme degradation. Fodero, et al (2004) observed that Aβ -induction of AChE in cultured neurons was associated with an agonist effect of Aβ at α7-nicotinic receptors and could be antagonized by inhibitors of α7 nicotinic receptors or of L- and N-type calcium channels. An anomalous glycoform of AChE, similar to an isoform seen in AD brain and CSF and which does not bind to Con A has been reported in Tg2576 mice at 4 and 8 months of age before plaque formation. (Fodero, et al. (2002); Ulrich, et al. (1990); and Mimori et al. (1997) reported that increased AChE was associated with amyloid plaques in human brain. Altered isoforms of AChE have been found increased in AD brain and CSF and co-localized with amyloid plaques (Saez-Valero et al., 2000; Talesa, 2001). Pathologic studies of AD brain usually show increased AChE activity around amyloid plaques although a general decrease in whole brain (reviewed in Frohlich, 2002). These new findings that AS to mutated β -site on APP results in decreased Aβ and AChE in parallel in transgenic mice support the suggestion that very early treatment with AChE inhibitors in at-risk individuals may be beneficial in retarding the disease process.
This method of targeting the mutated allele is a potential approach for treating familial AD or other genetic diseases with dominant inheritance. Allowing the normal APP allele to be expressed is advantageous since it has been shown that APP null mice exhibit subtle locomotor dysfunction and forelimb weakness (Heber et al., 2000) and that blocking APP by AS-ODNs to initiator methionine codon, exons 2 or 4 in primary neuronal cultures decreases adhesiveness of neurons on collagen and laminin suggesting APP is involved in cell membrane interactions with extracellular matrix (Coulson et al., 1997). APP exhibits both neuroprotective effects at lower levels and toxic effects at higher levels, depending also on the isoforms expressed (Mucke et al., 1996).
Blocking Aβ processing might also be approached by down regulating or inhibiting BACE1, which is the enzyme responsible for the β-site cleavage (Vassar et al., 1999) or presenilins which are components of the γ-cleavage complex (Iwatsubo, 2004). BACE down regulation by inhibitory nucleic acids (iNA/iNAs) or inhibitors does reduce β-secretase activity and Aβ loads effectively. However, while no obvious phenotype was discerned in early studies of BACE null mice (Kao et al., 2004; Nawrot, 2004), while a recent report indicates serious morbid effects in BACE knockout mice (Dominguez et al., 2005). Treatment and prevention of AD will demand therapy for years. Since there is about 30% sequence identity with BACE2, designing highly specific iNAs will be problematic and since there exist other substrates for BACE1 there may be unknown long-term consequences of blocking BACE1 Blocking human presenilin1 (PS1) by siRNA with resulting decreases in Aβ42 was demonstrated in CHO/PS1/APP cells (Luo et al., 2004). An siRNA specific for the mutated PS allele found in certain forms of inherited AD (Selkoe and Lansbury, 1999) may be useful in inherited disease but down regulating both native alleles of PS1 for sporadic AD is probably not a viable strategy since γ-secretase is important for other substrates including Notch and knock-outs of presenilins produce morbidity and lethality in mice (Selkoe and Kopan, 2003). However, iNA silencing of the gene for X11, an APP adaptor protein required for the γ-cleavage of APP, has been found to reduce Aβ levels in human neuroglioma culture by inhibiting γ-cleavage of APP and may have therapeutic potential (Xie et al., 2005).
Considerable research is now going into designing second and third generation iNAs, including peptide nucleic acids directed against RNA or DNA for APP (Adlerz et al., 2003; McMahon et al., 2003; Boules et al., 2004). The subject of iNAs has been comprehensively reviewed (Miller et al., 2004; Trulzsch and Wood, 2004).
Advantages and disadvantages of ICV administration as compared to IV or direct intraparenchymal infusion need to be determined for each of the compounds tested. Whitesell has shown that continuous infusion of ODN into 3rd ventricle by mini osmotic pump can maintain micromolar concentrations of PS-ODN in CSF for at least 1 week without “obvious neurologic or systemic toxicity” in rats (Whitesell et al., 1993). In SAM8, ODN to APP is transported into brain after IV injection but the IV route requires 100 times more than ICV injection to obtain the same level in brain (Banks et al., 2001). Ventricular infusions have been reported effective with regard to ODN to noggin and effects on learning in rats (Fan et al., 2003) and ODN to intercellular adhesion molecule-1 (ICAM) (Vemuganti et al., 2004). However, appropriate parameters of administration need to be determined for large brains. One report indicated ventricular AS-ODN to angiotensinogen mRNA did not diffuse throughout sheep brain as it did through rat brain (McKinley et al., 2000). The possibility of toxic effects such as inflammatory responses to the infused compounds bears testing with each substance (Elepfandt et al., 2002). Stereotaxic introduction of catheters into bilateral parenchyma has the disadvantages of hemorrhage risk and the probable requirement for operative room technology and general anesthesia if ultimately considered for human application. ICV catheter placement, on the other hand, may be performed at the bedside under local anesthesia.
Additional studies are needed to assess the effects of the AS-ODN to the mutated β-site on expression and α-secretase processing of the normal human APP protein in cultured heterozygous cells, the possible production of brain inflammatory responses to the injected ODNs, the permeation of ODNs through larger brains, optimal concentrations and frequency of administration, and finally effects in primate brains. This method is not expected to be useful in sporadic AD or in Down syndrome in which there is no mutation in the APP gene.
Conclusions
This is the first demonstration that AS-ODN directed to the mutated β-cleavage site of APP reduces Aβ levels and increases sAPPα levels while reversing the increases in AChE expression otherwise observed in Tg2576 mice. The antisense effects probably stem from relieving promotion of amyloidogenic- relative to α-processing by the inhibition of β -site cleavage and the consequent reduction in Aβ levels. The advantage of using AS-ODNs to the specific APP mutations in the inherited forms of AD are that the normal APP allele may be expressed and that the reduction in Aβ may allow increased sAPPα production both from the normal allele and any APP escaping the ODN block which may have additional beneficial effects in retarding the disease progression.
Acknowledgments
This work was supported in part by NIH (AT001218 & NS044538, N.B.C.). We are grateful to the Neuroscience reviewer who brought the paper by Sbena et al. (1997) to our attention.
Abbreviations
- α
Alpha
- ACh
Acetyl Choline
- AChE
Acetyl Cholinesterase
- AD
Alzheimer’s disease
- Aβ
β-amyloid
- APP
β-amyloid precursor protein
- AS
Antisense
- ANOVA
Analysis of Variance
- β
beta
- BACE
β-site APP cleaving Enzyme
- DA
Dopamine
- DNA
Deoxyribo Nucleic Acid
- EDTA
EthyleneDiamineTetraacetic Acid
- ELISA
Enzyme-Linked Immunosorbent Assay
- γ
gamma
- HRP
Horse Radish peroxidase
- hAPP
Human APP (β-amyloid precursor protein)
- ICV
Intracerebroventricular
- IgG
Immunoglobulin
- iNA(s)
Inhibitory Nucleic Acids
- ICAM
Intracellular Adhesion Molecule 1
- Lt/Lts
littermates (non-transgenic mice)
- Mg
milligram
- ml
milliliter
- μg
microgram
- μl
microliter
- Mhu
mutated human gene
- mRNA
messenger ribonucleic acid
- 2’-MOE
2’-O-(Methyl) Ethyl
- NMDA
N-Methyl-D-Aspartate
- ODN(s)
Oligodeoxynucleotides
- Pg
picograms
- PS
Phosphorothioate
- PS1
Presenilin 1
- PMSF
Phenyl Methyl Sulfonyl Fluoride
- sAPPα
α-cleaved soluble APP
- sAβ40
β- and γ-cleaved soluble Aβ40
- sAβ42
β- and γ-cleaved soluble A β42
- SD
Standard deviation
- SDS
Sodium dodecyl sulphate
- TBS
Tris buffered saline
- Tg/Tgs
Transgenic mice
Footnotes
Appropriate Section Editor for Manuscript Handling: Dr. M. Segal, Weizman Institute of Science, Department of Neurobiology, Hertzl Street, Rohovot 76100, Israel.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adlerz I, Soomets U, Holmlund I, Viirlaid S, Langel U, Iverfeldt K. Down-regulation of amyloid precursor protein by peptide nucleic acid oligomer in cultured rat primary neurons and astrocytes. Neurosci Lett. 2003;336(1):55–59. doi: 10.1016/s0304-3940(02)01219-3. [DOI] [PubMed] [Google Scholar]
- Banks WA, Farr SA, Butt W, Kumar VB, Franko MW, Morley JE. Delivery across the blood-brain barrier of antisense directed against amyloid beta: reversal of learning and memory deficits in mice overexpressing amyloid precursor protein. J Pharmacol Exp Ther. 2001;297(3):1113–1121. [PubMed] [Google Scholar]
- Boules M, Williams K, Gollatz E, Fauq A, Richelson E. Down-regulation of amyloid precursor protein by peptide nucleic acid in vivo. J Mol Neurosci. 2004;24(1):123–128. doi: 10.1385/JMN:24:1:123. [DOI] [PubMed] [Google Scholar]
- Broaddus WC, Prabhu SS, Gillies GT, Neal J, Conrad WS, Chen ZJ, Fillmore H, Young HF. Distribution and stability of antisense phosphorothioate oligonucleotides in rodent brain following direct intraparenchymal controlled-rate infusion. J Neurosurg. 1998;88(4):734–742. doi: 10.3171/jns.1998.88.4.0734. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ, Lichtor T. Distribution of intraventricularly administered antiamyloid-beta peptide (Abeta) antibody in the mouse brain. J Neurosci Res. 2001;66(2):231–235. doi: 10.1002/jnr.1215. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ. Reversal of amyloid beta toxicity in Alzheimer’s disease model Tg2576 by intraventricular antiamyloid beta antibody. J Neurosci Res. 2002;69(1):10–23. doi: 10.1002/jnr.10286. [DOI] [PubMed] [Google Scholar]
- Chauhan NB. Trafficking of intracerebroventricularly injected antisense oligonucleotides in the mouse brain. Antisense Nucleic Acid Drug Dev. 2002;12(5):353–357. doi: 10.1089/108729002761381320. [DOI] [PubMed] [Google Scholar]
- Chauhan NB. Membrane dynamics, cholesterol homeostasis and Alzheimer’s disease. J Lipid Res. 2003;44(11):2019–2029. doi: 10.1194/jlr.R300010-JLR200. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ. Effect of PPF and ALCAR on the induction of NGF- and p75-mRNA and on APP processing in Tg2576 brain. Neurochem Int. 2003;43(3):225–233. doi: 10.1016/s0197-0186(03)00006-8. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ. Intracerebroventricular passive immunization in transgenic mouse models of Alzheimer’s disease. Expert Rev Vaccines. 2004;3(6):717–725. doi: 10.1586/14760584.3.6.717. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ, Feinstein DL. Propentofylline attenuates tau hyperphosphorylation in Alzheimer’s Swedish mutant model Tg2576. Neuropharmacology. 2005;48(1):93–104. doi: 10.1016/j.neuropharm.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Coulson EJ, Barrett GL, Storey E, Bartlett PF, Beyreuther K, Masters CL. Down-regulation of the amyloid protein precursor of Alzheimer’s disease by antisense oligonucleotides reduces neuronal adhesion to specific substrata. Brain Res. 1997;770(12):72–80. doi: 10.1016/s0006-8993(97)00757-9. [DOI] [PubMed] [Google Scholar]
- Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels I, Camacho IE, Marjaux E, Craessaerts K, Roebroek AJ, Schwake M, D’hooge R, Bach P, Kalinke U, Moechars D, Alzheimer C, Reiss K, Saftig P, de Strooper B. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem. 2005;280(35):30797–30806. doi: 10.1074/jbc.M505249200. [DOI] [PubMed] [Google Scholar]
- Elepfandt P, Rupprecht S, Schoning-burkhardt B, Volk HD, Woiciechowsky C. Oligodeoxynucleotides induce brain inflammation in rats when infused intracerebroventricularly. Neurosci Lett. 2002;322(2):107–110. doi: 10.1016/s0304-3940(02)00093-9. [DOI] [PubMed] [Google Scholar]
- Fan XT, Cai WG, Yang Z, Xu HW, Zhang JH. Effect of antisense oligonucleotide of noggin on spatial learning and memory of rats. Acta Pharmacol Sin. 2003;24(5):394–397. [PubMed] [Google Scholar]
- Fodero LR, Saez-Valero J, McLean CA, Martins RN, Beyreuther K, Masters CL, Robertson TA, Small DH. Altered glycosylation of acetylcholinesterase in APP (SW) Tg2576 transgenic mice occurs prior to amyloid plaque deposition. J Neurochem. 2002;81(3):441–8. doi: 10.1046/j.1471-4159.2002.00902.x. [DOI] [PubMed] [Google Scholar]
- Fodero LR, Mok SS, Losic D, Martin LL, Aguilar MI, Barrow CJ, Livett BG, Small DH. Alpha7-nicotinic acetylcholine receptors mediate an Abeta(1-42)-induced increase in the level of acetylcholinesterase in primary cortical neurones. J Neurochem. 2004;88(5):1186–93. doi: 10.1046/j.1471-4159.2003.02296.x. [DOI] [PubMed] [Google Scholar]
- Frohlich ED. Academic pursuits in the multispecialty clinic. Med Clin North Am. 1992;76(5):1003–6. doi: 10.1016/s0025-7125(16)30301-7. [DOI] [PubMed] [Google Scholar]
- Fu AL, Dong ZH, Sun MJ. Protective effects of N-acetyl-L-cysteine on amyloid beta-peptide-induced learning and memory deficits in mice. Brain Res. 2006;1109(1):201–206. doi: 10.1016/j.brainres.2006.06.042. [DOI] [PubMed] [Google Scholar]
- Garcia SI, Alvarez AL, Porto PI, Garfunkel VM, Finkielman S, Pirola CJ. Antisense inhibition of thyrotropin-releasing hormone reduces arterial blood pressure in spontaneously hypertensive rats. Hypertension. 2001;37(2 part 2):365–370. doi: 10.1161/01.hyp.37.2.365. [DOI] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellne J, Aguzzi A, Rulicke T, Von Kretzschmar H, Von Koch C, Sisodia S, Tremml P, Lipp HP, Wolfer DP, Muller U. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20(21):7951–7963. doi: 10.1523/JNEUROSCI.20-21-07951.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedreen JC, Bacon SJ, Price DL. A modified histochemical technique to visualize acetyl cholinesterase containing axons. J Histochem Cytochem. 1985;33(2):134–140. doi: 10.1177/33.2.2578498. [DOI] [PubMed] [Google Scholar]
- Higuchi H, Miyoshi H, Bronk SF, Zhang H, Dean N, Gores GJ. Bid antisense attenuates bile acid-induced apoptosis and cholestatic liver injury. J Pharmacol Exp Ther. 2001;299(3):866–873. [PubMed] [Google Scholar]
- Hu W, Gray NW, Brimijoin S. Amyloid-beta increases acetylcholinesterase expression in neuroblastoma cells by reducing enzyme degradation. J Neurochem. 2003;86(2):470–8. doi: 10.1046/j.1471-4159.2003.01855.x. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T. The gamma-secretase complex: machinery for intramembrane proteolysis. Curr Opin Neurobiol. 2004;14(3):379–383. doi: 10.1016/j.conb.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Kao SC, Krichevsky AM, Kosik KS, Tsai IH. BACE1 suppression by RNA interference in primary cortical neurons. J Biol Chem. 2004;279(3):1942–1949. doi: 10.1074/jbc.M309219200. [DOI] [PubMed] [Google Scholar]
- Karnovsky MJ, Roots L. A “direct coloring” thiocholine method for cholinesterase. J Histochem Cytochem. 1964;12:219–221. doi: 10.1177/12.3.219. [DOI] [PubMed] [Google Scholar]
- Kumar VB, Farr SA, Flood JF, Kamlesh V, Franko M, Banks WA, Morley JE. Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged samp8 mice. Peptides. 2000;21(12):1769–1775. doi: 10.1016/s0196-9781(00)00339-9. [DOI] [PubMed] [Google Scholar]
- Liang SL, Pan JT. Pretreatment with antisense oligodeoxynucleotide against D(2) or D(3) receptor mRNA diminished dopamine’s inhibitory effect on dorsomedial arcuate neurons in brain slices of estrogen-treated ovariectomized rats. Brain Res. 2002;926(12):156–164. doi: 10.1016/s0006-8993(01)03314-5. [DOI] [PubMed] [Google Scholar]
- Luo HM, Deng H, Xiao F, Gao Q, Weng W, Zhang PF, Li XG. Down-regulation amyloid beta-protein 42 production by interfering with transcript of presenilin 1 gene with siRNA. Acta Pharmacol Sin. 2004;25(12):1613–1618. [PubMed] [Google Scholar]
- McKinley MJ, Guzzo-Pernell N, Sinnayah P. Antisense oligonucleotide inhibition of angiotensinogen in the brains of rats and sheep. Methods. 2000;22(3):219–225. doi: 10.1006/meth.2000.1073. [DOI] [PubMed] [Google Scholar]
- McMahon BM, Stewart J, Fauq A, Younkin S, Younkin L, Richelson E. Peptide nucleic acids targeted to the amyloid precursor protein. J Mol Neurosci. 2003;20(3):261–265. doi: 10.1385/JMN:20:3:261. [DOI] [PubMed] [Google Scholar]
- Miller VM, Gouvion CM, Davidson BL, Paulson HL. Targeting alzheimer’s disease genes with RNA interference: an efficient strategy for silencing mutant alleles. Nucleic Acids Res. 2004;32(2):661–668. doi: 10.1093/nar/gkh208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori Y, Nakamura S, Yukawa M. Abnormalities of acetylcholinesterase in Alzheimer’s disease with special reference to effect of acetylcholinesterase inhibitor. Behav Brain Res. 1997;83(12):25–30. doi: 10.1016/s0166-4328(97)86041-x. [DOI] [PubMed] [Google Scholar]
- Mucke L, Abraham CR, Masliah E. Neurotrophic and neuroprotective effects of hAPP in transgenic mice. Ann N Y Acad Sci. 1996;777:82–88. doi: 10.1111/j.1749-6632.1996.tb34405.x. [DOI] [PubMed] [Google Scholar]
- Murata M, Suzuki M, Tanaka K, Tajiri K, Emori K, Kurachi M. N-methyl-D-aspartate-R1 receptor antisense oligodeoxynucleotide modulates pre-and postsynaptic expression of D2 dopamine receptors in the rat. Neurosci Lett. 2002;335(1):9–12. doi: 10.1016/s0304-3940(02)01161-8. [DOI] [PubMed] [Google Scholar]
- Nawrot B. Targeting BACE with small inhibitory nucleic acids - a future for Alzheimer’s disease therapy? Acta Biochim Pol. 2004;51(2):431–444. [PubMed] [Google Scholar]
- Nijholt I, Farchi N, Kye M, Sklan EH, Shoham S, Verbeure B, Owen D, Hochner B, Spiess J, Soreq H, Blank T. Stress-induced alternative splicing of acetylcholinesterase results in enhanced fear memory and long-term potentiation. Mol Psychiatry. 2004;9(2):174–83. doi: 10.1038/sj.mp.4001446. [DOI] [PubMed] [Google Scholar]
- Olie RA, Hall J, Natt F, Stahel RA, Zangemeister-Wittke U. Analysis of Ribosyl-modified, mixed backbone analogs of a bcl-2/bcl-xl antisense oligonucleotide. Biochim Biophys Acta. 2002;1576(12):101–109. doi: 10.1016/s0167-4781(02)00300-7. [DOI] [PubMed] [Google Scholar]
- Poon HF, Joshi G, Sultana R, Farr SA, Banks WA, Morley JE, Calabrese V, Butterfield DA. Antisense directed at the Abeta region of APP decreases brain oxidative markers in aged senescence accelerated mice. Brain Res. 2004;1018(1):86–96. doi: 10.1016/j.brainres.2004.05.048. [DOI] [PubMed] [Google Scholar]
- Poon HF, Farr SA, Banks WA, Pierce WM, Klein JB, Morley JE, Butterfield DA. Proteomic identification of less oxidized brain proteins in aged senescence-accelerated mice following administration of antisense oligonucleotide directed at the Abeta region of amyloid precursor protein. Brain Res Mol Brain Res. 2005;138(1):8–16. doi: 10.1016/j.molbrainres.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Saez-Valero J, Mok SS, Small DH. An unusually glycosylated form of acetylcholinesterase is a CSF biomarker for Alzheimer’s disease. Acta Neurol Scand Suppl. 2000;176:49–52. doi: 10.1034/j.1600-0404.2000.00307.x. [DOI] [PubMed] [Google Scholar]
- Sberna G, Saez-Valero J, Beyreuther K, Masters CL, Small DH. The amyloid beta-protein of Alzheimer’s disease increases acetylcholinesterase expression by increasing intracellular calcium in embryonal carcinoma P19 cells. J Neurochem. 1997 Sep;69(3):1177–84. doi: 10.1046/j.1471-4159.1997.69031177.x. [DOI] [PubMed] [Google Scholar]
- Sberna G, Saez-Valero J, Li QX, Czech C, Beyreuther K, Masters CL, McLean CA, Small DH. Acetylcholinesterase is increased in the brains of transgenic mice expressing the C-terminal fragment (CT100) of the beta-amyloid protein precursor of Alzheimer’s disease. J Neurochem. 1998;71(2):723–31. doi: 10.1046/j.1471-4159.1998.71020723.x. [DOI] [PubMed] [Google Scholar]
- Selkoe D, Lansbury PT. Biochemistry of Alzheimer’s and Prion diseases. In: Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD, editors. Basic Neurochemistry. lippincot raven; new york: 1999. pp. 949–970. [Google Scholar]
- Selkoe D, Kopan R. Notch and Presenilin: (Regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci. 2003;26:565–597. doi: 10.1146/annurev.neuro.26.041002.131334. [DOI] [PubMed] [Google Scholar]
- Siegel GJ, Chauhan NB, Karczmar A. Linking amyloid and tau biology in Alzheimer’s disease and its Cholinergic aspects. In: Karczmar A, editor. Exploring Cholinergic Mechanisms in Brain Disease. Kluver-Academic-Plenum Press; NY: 2006. pp. 569–627. [Google Scholar]
- Takashima A, Yamaguchi H, Noguchi K, Michel G, Ishiguro K, Sato K, Hoshino T, Hoshi M, Imahori K. Amyloid beta peptide induces cytoplasmic accumulation of amyloid protein precursor via tau protein kinase I/glycogen synthase kinase-3 beta in rat hippocampal neurons. Neurosci Lett. 1995;198(2):83–86. doi: 10.1016/0304-3940(95)11964-x. [DOI] [PubMed] [Google Scholar]
- Talesa VN. Acetylcholinesterase in Alzheimer’s disease. Mech Ageing Dev. 2001;122(16):1961–9. doi: 10.1016/s0047-6374(01)00309-8. [DOI] [PubMed] [Google Scholar]
- Temsamani J, Tang JY, Padmapriya A, Kubert M, Agrawal S. Pharmacokinetics, biodistribution, and stability of capped oligodeoxynucleotide phosphorothioates in mice. Antisense Res Dev. 1993;3(3):277–284. doi: 10.1089/ard.1993.3.277. [DOI] [PubMed] [Google Scholar]
- Trulzsch B, Wood M. Applications of nucleic acid technology in the CNS. J Neurochem. 2004;88(2):257–265. doi: 10.1111/j.1471-4159.2004.02153.x. [DOI] [PubMed] [Google Scholar]
- Ulrich J, Meier-Ruge W, Probst A, Meier E, Ipsen S. Senile plaques: staining for acetylcholinesterase and A4 protein: a comparative study in the hippocampus and entorhinal cortex. Acta Neuropathol (Berl) 1990;80(6):624–8. doi: 10.1007/BF00307630. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Vemuganti R, Dempsey RJ, Bowen KK. Inhibition of intercellular adhesion molecule-1 protein expression by antisense oligonucleotides is neuroprotective after transient middle cerebral artery occlusion in rat. Stroke. 2004;35(1):179–184. doi: 10.1161/01.STR.0000106479.53235.3E. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Geselowitz D, Chavany C, Fahmy B, Walbridge S, Alger JR, Neckers LM. Stability, clearance, and disposition of intraventricularly administered oligodeoxynucleotides: implications for therapeutic application within the central nervous system. Proc Natl Acad Sci U S A. 1993;90(10):4665–4669. doi: 10.1073/pnas.90.10.4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Romano DM, Tanzi RE. RNA interference-mediated silencing of x11alpha and x11beta attenuates amyloid beta-protein levels via differential effects on beta-amyloid precursor protein processing. J Biol Chem. 2005;280(15):15413–15421. doi: 10.1074/jbc.M414353200. [DOI] [PubMed] [Google Scholar]
- Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A. 1978;75(1):280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger T, Miyake H, Cooper S, Chi K, Conklin BS, Monia BP, Gleave ME. Antitumor activity of antisense clusterin oligonucleotides is improved in vitro and in vivo by incorporation of 2′-O-(2-methoxy) ethyl chemistry. J Pharmacol Exp Ther. 2001;298(3):934–940. [PubMed] [Google Scholar]