

Abstract
A new method for predicting internal coding exons in genomic DNA sequences has been developed. This method is based on a prediction algorithm that uses the quadratic discriminant function for multivariate statistical pattern recognition. Substantial improvements have been made (with only 9 discriminant variables) when compared with existing methods: hexon [Solovyev, V. V., Salamov, A. A. & Lawrence, C. B. (1994) Nucleic Acids Res. 22, 5156–5163] (based on linear discriminant analysis) and grail2 [Uberbacher, E. C. & Mural, R. J. (1991) Proc. Natl. Acad. Sci. USA 88, 11261–11265] (based on neural networks). A computer program called mzef is freely available to the genome community and allows users to adjust prior probability and to output alternative overlapping exons.
Biology has entered into a new era of genomics that has far-reaching consequences in human medicine and health (1). This led to speedy identification and localization of complex disease genes (2, 3). As the Human Genome Project enters its large-scale sequencing phase, gene identification has become extremely important (4). Since human genes may span tens or hundreds of kilobases, with the protein-coding regions (exons) accounting for only a few percent of the total genomic sequence, identifying genes within large regions of uncharacterized DNA is a difficult task. The more traditional approaches to gene isolation, including identification of CpG islands and conserved sequences, as well as direct screening of cDNA libraries, are effective but very laborious. Currently, there are four basic proven robust approaches to rapid and efficient transcriptional mapping of regions of more than a few tens of kilobases of DNA (5): cDNA selection (6–8), exon trapping (9, 10), genomic sequencing with “software trapping” (11, 12), and regional assignment of randomly cloned and sequenced cDNAs (13, 14). Very soon, large-scale genomic sequencing coupled to computer prediction and experimental verification will become the major paradigm for human gene identification. Current computer methods consist basically of two types: data base similarity searches (15) and statistical pattern recognition, the latter being either rule-based or neural-network-based (see ref. 16 for a recent review). Unfortunately, systematic examination (17) of various computational methods showed that the accuracy at the nucleotide level ranges from 0.6–0.7 as measured by the correlation coefficient (see Methods) and the average fraction of actual exons identified was less than 50%.
In an effort to improve the accuracy of exon prediction and to make a new tool freely available to the genome community in a timely fashion, I chose to use quadratic discriminant analysis (QDA; see ref. 18, for example), a powerful statistical multivariate pattern-recognition method, as the basis for a new program called mzef (Michael Zhang’s Exon Finder). QDA may be thought of as a direct extension of the classical linear discriminant analysis (LDA) method pioneered by R. A. Fisher 60 years ago (19), which was used as the basis for hexon (20). One assumes that real exons and pseudoexons may be described approximately by two multinormal distributions (having different means) of some characteristic features (such as the splice site scores, etc.). Under this model (although LDA was originally formulated in a distribution-free manner) LDA differs from QDA by further assuming that the two distributions have the same covariance. Graphically, viewing the two populations as swarms of points in multidimensional (feature) space, QDA can provide a more effective curved boundary between two swarms that have different shapes and orientations than LDA, which could provide only a straight-line boundary [as shown by the simple illustration in Fig. 1 (21)].
Figure 1.

Two-variable example in which a quadratic function (solid line) separates the two groups (× and ○) completely, while the best linear function (dotted line) misclassifies an appreciable number of points.
In a recent study (22), it was shown that human internal coding exons have distinct correlation structures for different characteristic features, which implies that one should treat the two covariances (which essentially measure the correlation structures) differently. mzef uses relatively less information than hexon. hexon requires 70 bp of flanking region on each side of an exon, mzef only needs 54 bp; hexon uses up to octanucleotide compositions, mzef uses no more than hexamer compositions; and hexon measures 17 variables (7 for donor discriminant, 7 for acceptor discriminant, and 3 more for internal exon prediction), mzef measures 9 variables for discrimination. These 9 characteristic variables measure the following (see Methods for details): exon length, intron–exon transition, branch-site score, 3′ss score, exon score, strand score, frame score, 5′ss score, and exon–intron transition. The addition of branch-site information (22) is a salient feature of this program.
METHODS
In previous work (22), we extracted, classified, and characterized all human exons and their flanking regions in GenBank release 87.0 (23). The 3440 coding exons were used for computing various frequency matrices (24, 25). These matrices were computed separately for locus G+C content less than 0.48 or otherwise (22). The test set contains 43 completely sequenced genes as indicated in the tables below. They were selected randomly from the genomic DNA data that contain “complete cds” in the title (after homologs and pseudogenes were eliminated). Other sequences were used to build a training set, which had 1879 true exons and 184,217 pseudoexons [defined as an open reading frame flanked by the putative splice sites, denoted 3′ss and 5′ss (20)]. ALLSEQ data and the app gene were described in ref. 17 and *, respectively.
If fA is some frequency found in group A, we define the preference for A vs. B (say, exons vs. pseudoexons) as the ratio fA/(fA + fB) (20). The 9 feature variables are defined as follows [where 〈 · 〉 means an average, and splice site boundary is defined as (−1, 1)]: 1, exon length, x1 = log10(actual length in bp); 2, exon–intron transition, x2 = 〈(intron hexamer frequency preference in the 54-bp window to the left of the 3′ss)〉 − 〈(exon hexamer frequency preference in the 54-bp window to the right of the 3′ss)〉; 3, branch-site score, x3 = maximum branch score [measured by the log-likelihood score (22)] in the window (−54, −3); 4, 3′ss score, x4 = position-dependent triplet frequency preference for true 3′ss vs. pseudo-3′ss in the window (−24, 3); 5, exon score, x5 = 〈(hexamer frequency preference for exon vs. intron)〉; 6, strand score, x6 = 〈(hexamer frequency preference for the forward strand vs. the reverse strand)〉; 7, frame score, x7 = max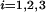 (frame-specific hexamer frequency preference for exon vs. intron in frame i); 8, 5′ss score, x8 = positional-dependent triplet frequency preference for true 5′ss vs. pseudo-5′ss in the window (−3, 8); and 9, intron–exon transition, x9 = 〈(exon hexamer frequency preference in the 54-bp window to the left of the 5′ss)〉 − 〈(intron hexamer frequency preference in the 54-bp window to the right of the 5′ss)〉.
(frame-specific hexamer frequency preference for exon vs. intron in frame i); 8, 5′ss score, x8 = positional-dependent triplet frequency preference for true 5′ss vs. pseudo-5′ss in the window (−3, 8); and 9, intron–exon transition, x9 = 〈(exon hexamer frequency preference in the 54-bp window to the left of the 5′ss)〉 − 〈(intron hexamer frequency preference in the 54-bp window to the right of the 5′ss)〉.
We applied the technique of QDA to relate the given region to one of the two alternative groups, G1 (exons) or G2 (pseudoexons) (18). The log-ratio of posterior probabilities that a sequence with feature x belongs to group G1 is given by
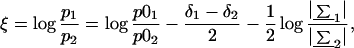 |
where p0i denotes the prior probability for the group Gi, δi = (x − μi)′Σi−1(x − μi) is the squared Mahalanobis distance between x and μi with respect to Σi, |Σ| is the determinant of Σ, and μi and Σi are the group mean and covariance matrix, respectively (computed from the training set). The optimal, or Bayes rule assigns the sequence with feature x = (x1, x2, … , x9) to exons if ξ > 0.
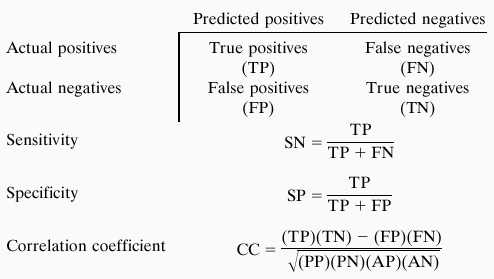
The performance measures are the standard (see ref. 17 or ref. 26, for example):
 |
To test for internal coding exons, only the genomic region between the end of the first coding exon and the beginning of the last coding exon in each gene was considered.
RESULTS AND DISCUSSION
The detailed results on a test set of 43 genes (containing 332 internal coding exons) of known organization are shown in Table 1, where mzef is compared with grail2 (27) and hexon (20) programs for internal coding exon predictions.
Table 1.
Test results
| Acc. no. | Total real exons |
grail2
|
hexon
|
mzef
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ex | lt | rt | ol | fp | ex | lt | rt | ol | fp | ex | lt | rt | ol | fp | ||
| J02843J02843 | 7 | 5 | 5 | 6 | 6 | 4 | 6 | 6 | 6 | 6 | 0 | 4 | 4 | 4 | 4 | 0 |
| J02846J02846 | 4 | 2 | 2 | 3 | 3 | 1 | 2 | 2 | 4 | 4 | 3 | 4 | 4 | 4 | 4 | 0 |
| J02933J02933 | 7 | 1 | 2 | 3 | 4 | 5 | 6 | 6 | 6 | 6 | 4 | 4 | 4 | 4 | 4 | 0 |
| J03059J03059 | 9 | 8 | 8 | 8 | 8 | 0 | 9 | 9 | 9 | 9 | 1 | 8 | 8 | 8 | 8 | 0 |
| J03930J03930 | 9 | 7 | 8 | 8 | 9 | 2 | 8 | 8 | 8 | 8 | 0 | 8 | 8 | 8 | 8 | 0 |
| J04038J04038 | 6 | 4 | 5 | 5 | 6 | 2 | 4 | 5 | 5 | 6 | 1 | 6 | 6 | 6 | 6 | 1 |
| J04617J04617 | 5 | 2 | 2 | 4 | 5 | 3 | 5 | 5 | 5 | 5 | 0 | 3 | 4 | 4 | 5 | 1 |
| J04988J04988 | 9 | 8 | 8 | 8 | 8 | 1 | 8 | 9 | 8 | 9 | 1 | 8 | 8 | 8 | 8 | 0 |
| J05096J05096 | 21 | 15 | 18 | 18 | 21 | 8 | 0 | 0 | 0 | 21 | 22 | 21 | 21 | 21 | 21 | 0 |
| J05451J05451 | 20 | 0 | 1 | 0 | 20 | 20 | 18 | 19 | 18 | 19 | 1 | 20 | 20 | 20 | 20 | 0 |
| K00650K00650 | 2 | 1 | 1 | 1 | 1 | 0 | 2 | 2 | 2 | 2 | 0 | 1 | 1 | 1 | 1 | 0 |
| K03021K03021 | 11 | 9 | 9 | 9 | 9 | 1 | 11 | 11 | 11 | 11 | 4 | 8 | 8 | 8 | 8 | 1 |
| L05072L05072 | 7 | 3 | 5 | 3 | 6 | 3 | 6 | 6 | 7 | 7 | 1 | 4 | 4 | 4 | 4 | 1 |
| L10615L10615 | 4 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 2 | 2 | 2 | 3 | 3 | 3 | 3 | 0 |
| L10641L10641 | 10 | 1 | 2 | 3 | 5 | 5 | 6 | 6 | 7 | 7 | 4 | 10 | 10 | 10 | 10 | 0 |
| L11910L11910 | 25 | 4 | 4 | 4 | 4 | 8 | 17 | 17 | 18 | 18 | 15 | 21 | 21 | 21 | 21 | 8 |
| L13470L13470 | 2 | 0 | 0 | 1 | 1 | 2 | 1 | 1 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 0 |
| L14565L14565 | 7 | 5 | 5 | 5 | 5 | 0 | 6 | 6 | 6 | 7 | 1 | 6 | 6 | 6 | 6 | 0 |
| L14927L14927 | 4 | 2 | 3 | 3 | 4 | 2 | 3 | 3 | 4 | 4 | 1 | 4 | 4 | 4 | 4 | 0 |
| M10612M10612 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 0 |
| M11228M11228 | 6 | 3 | 4 | 4 | 6 | 5 | 2 | 3 | 3 | 4 | 4 | 5 | 5 | 5 | 5 | 0 |
| M12523M12523 | 12 | 6 | 8 | 6 | 8 | 2 | 10 | 10 | 10 | 10 | 1 | 12 | 12 | 12 | 12 | 1 |
| M13792M13792 | 10 | 9 | 10 | 9 | 10 | 4 | 10 | 10 | 10 | 10 | 3 | 8 | 8 | 8 | 8 | 0 |
| M15205M15205 | 5 | 3 | 3 | 3 | 4 | 4 | 3 | 3 | 3 | 5 | 8 | 4 | 4 | 4 | 4 | 0 |
| M15840M15840 | 4 | 3 | 3 | 4 | 4 | 1 | 0 | 0 | 0 | 4 | 4 | 3 | 3 | 3 | 3 | 1 |
| M16110M16110 | 12 | 4 | 5 | 5 | 6 | 2 | 8 | 9 | 8 | 9 | 1 | 11 | 11 | 11 | 11 | 2 |
| M17262M17262 | 12 | 5 | 7 | 6 | 9 | 5 | 8 | 11 | 9 | 12 | 7 | 8 | 8 | 8 | 10 | 2 |
| M19645M19645 | 6 | 2 | 4 | 3 | 5 | 3 | 6 | 6 | 6 | 6 | 0 | 5 | 5 | 5 | 5 | 0 |
| M20543M20543 | 4 | 1 | 3 | 2 | 4 | 3 | 4 | 4 | 4 | 4 | 0 | 4 | 4 | 4 | 4 | 1 |
| M24461M24461 | 8 | 5 | 6 | 5 | 6 | 1 | 6 | 6 | 7 | 7 | 2 | 6 | 6 | 6 | 6 | 0 |
| M24842M24842 | 5 | 4 | 5 | 4 | 5 | 1 | 4 | 5 | 4 | 5 | 2 | 4 | 4 | 4 | 4 | 1 |
| M26434M26434 | 7 | 1 | 1 | 1 | 1 | 1 | 6 | 6 | 6 | 6 | 11 | 6 | 6 | 6 | 6 | 3 |
| M31061M31061 | 8 | 8 | 8 | 8 | 8 | 0 | 8 | 8 | 8 | 8 | 0 | 8 | 8 | 8 | 8 | 0 |
| M34482M34482 | 6 | 3 | 4 | 3 | 5 | 3 | 5 | 5 | 6 | 6 | 1 | 5 | 5 | 5 | 5 | 0 |
| M63391M63391 | 7 | 4 | 5 | 6 | 7 | 3 | 6 | 6 | 7 | 7 | 2 | 7 | 7 | 7 | 7 | 1 |
| M69197M69197 | 5 | 5 | 5 | 5 | 5 | 1 | 5 | 5 | 5 | 5 | 0 | 4 | 4 | 4 | 4 | 0 |
| M85276M85276 | 3 | 2 | 2 | 2 | 3 | 1 | 2 | 2 | 3 | 3 | 1 | 2 | 2 | 2 | 2 | 0 |
| M91463M91463 | 9 | 7 | 8 | 7 | 9 | 2 | 3 | 5 | 5 | 8 | 5 | 7 | 7 | 7 | 8 | 1 |
| M94579M94579 | 9 | 7 | 8 | 8 | 9 | 2 | 8 | 9 | 8 | 9 | 1 | 9 | 9 | 9 | 9 | 0 |
| M96264M96264 | 9 | 2 | 4 | 2 | 6 | 4 | 5 | 5 | 6 | 6 | 2 | 5 | 5 | 5 | 5 | 0 |
| X05006X05006 | 6 | 2 | 4 | 4 | 6 | 5 | 2 | 3 | 3 | 4 | 2 | 4 | 4 | 4 | 4 | 0 |
| X63600X63600 | 4 | 1 | 3 | 1 | 3 | 2 | 3 | 3 | 3 | 3 | 0 | 3 | 3 | 3 | 3 | 0 |
| D00596D00596 | 5 | 3 | 3 | 3 | 3 | 4 | 2 | 2 | 3 | 3 | 5 | 4 | 4 | 4 | 4 | 0 |
| Total | 332 | 168 | 202 | 196 | 260 | 128 | 236 | 249 | 256 | 298 | 124 | 280 | 281 | 281 | 285 | 25 |
Acc. no, accession number; ex, matched exons; lt, matched left ends; rt, matched right ends; ol, overlap matches; fp, false exons.
For a better comparison, we increased the threshold for hexon so that its sensitivity at the nucleotide level was the same as mzef. We used the default threshold for grail2, as its sensitivity was already lower than that of mzef. The statistical summary of tests both at the exon level (sn and sp) and at the base-pair level (SN, SP, and CC) is shown in Table 2. These results show that, on average, mzef has higher accuracy measured by the correlation coefficient. Furthermore, mzef tends to have higher specificity (i.e., fewer false positives). The improvement is more dramatic at the exon level.
Table 2.
Test summary
| Program | sn | sp | SN | SP | CC |
|---|---|---|---|---|---|
| grail2 | 0.51 | 0.57 | 0.79 | 0.85 | 0.80 |
| fgeneh | 0.71 | 0.65 | 0.88 | 0.80 | 0.83 |
| mzef | 0.84 | 0.92 | 0.88 | 0.95 | 0.90 |
See Methods for definitions. Lowercase letters indicate the exon level; uppercase, the base-pair level.
As a second test, I obtained the ALLSEQ data (GenBank release 85.0) used in the recent evaluation of gene structure prediction programs (17) and their results for grail2 and fgeneh. fgeneh is hexon plus dynamic programming gene assembly, which can substantially increase the accuracy by requiring frame compatibility and distance constraint among exons (20). Since the statistics in that evaluation included the first and the last coding exons, I recomputed the statistics of the 1509 internal coding exons in 473 genes of ALLSEQ. The comparison of this test is summarized in Table 3. Again mzef performed better on average. I should point out the training set for mzef does overlap with ALLSEQ.
Table 3.
ALLSEQ summary
| Program | sn | sp | SN | SP | CC |
|---|---|---|---|---|---|
| grail2 | 0.53 | 0.60 | 0.79 | 0.92 | 0.83 |
| fgeneh | 0.73 | 0.78 | 0.83 | 0.93 | 0.85 |
| mzef | 0.78 | 0.86 | 0.87 | 0.95 | 0.89 |
As a final test, I used a new sequence (about 301 kb, kindly provided by K. Murakami before publication) which includes the amyloid precursor protein gene, app. In a recent workshop, Murakami and Tsukuni reported that hexon was more sensitive, but less specific than grail2* when used to predict the app gene structure. The statistical evaluation of internal exon predictions from grail2, hexon, and mzef is shown in Table 4. For a better comparison, the threshold for grail2 was increased so that grail2 and mzef had the same sensitivity at the nucleotide level. For hexon, I used its default threshold, as its sensitivity was already lower than that of mzef. Again mzef had a better performance, especially at the exon level, and it is more specific (fewer false positives).
Table 4.
app gene summary
| Method | ex | lt | rt | ol | fp | tl | sn | sp | SN | SP | CC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| grail2 | 7 | 8 | 10 | 12 | 13 | 17 | 0.41 | 0.35 | 0.64 | 0.62 | 0.63 |
| hexon | 9 | 9 | 10 | 10 | 38 | 17 | 0.53 | 0.19 | 0.54 | 0.29 | 0.39 |
| mzef | 11 | 11 | 12 | 12 | 10 | 17 | 0.65 | 0.52 | 0.64 | 0.69 | 0.66 |
mzef is available at the anonymous ftp site phage.cshl.org in the directory pub/science/mzef (the author may be contacted at mzhang@cshl.org). It is also available through the World Wide Web at the URL of http://www.cshl.org/genefinder. Users can set a prior probability parameter p0, (for example, set p0 = 0.04 for higher specificity in gene-poor loci, or set p0 = 0.08 for higher sensitivity in gene-rich loci, or set p0 = 0.02 for a cosmid). Users can also choose the overlapping parameter ovlp other than 0 (for example, set ovlp = 2 to get up to two more overlapping exons ranked by posterior probabilities). This is important because sometimes the true exon may lie in the top few of the overlapping exon list, or these overlapping exons may represent alternatively spliced isoforms. As the program also outputs separate frame scores, 3′ss score, exon score, and 5′ss score, it can help users make their own selections if desired. In our experience, users should use at least three different programs. Agreement among different predictions usually ensures a higher confidence for experimental pursuit. As the sequence data base grows very rapidly, a first scan with a similarity-based program is also absolutely essential.
Acknowledgments
I thank T. G. Marr for continuous encouragement and support, R. Guigo for sending the preprint and ALLSEQ data before publication, and Prof. K. Murakami for sending the app sequence and some testing results before publication. I also thank my colleagues: A. Krainer, B. Stillman, M. Wigler, and W. McCombie for reading the manuscript and J. Lugert, P. Monardo, and C. Tanck for system/network/web page assistance. This work is supported by National Institutes of Health Grant KO1 HG00010-04.
Footnotes
Abbreviations: QDA, quadratic discriminant analysis; ss, splice site.
Murakami, K. & Tsukuni, S., Workshop on Gene-Finding and Gene Structure Prediction, Oct. 13–14, 1995, University of Pennsylvania, Philadelphia.
References
- 1.Anonymous Nat Genet. 1995;13:1–5. [Google Scholar]
- 2.Wooster R, Bignell G, Lancaster J, Swift S, Seal S, et al. Nature (London) 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 3.Tartaglia L A, Dembski M, Weng X, Deng N, Culpepper J, et al. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 4.Collins F, Galas D. Science. 1993;267:43–46. doi: 10.1126/science.8211127. [DOI] [PubMed] [Google Scholar]
- 5.Gardiner K, Mural R J. Trends Genet. 1995;11:77–79. doi: 10.1016/S0168-9525(00)89003-4. [DOI] [PubMed] [Google Scholar]
- 6.Morgan J G, Dolganov G M, Robbins S E, Hinton L M, Lovett N. Nucleic Acids Res. 1992;20:5173–5179. doi: 10.1093/nar/20.19.5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan W F, Wei X, Shukla H, Parimoo S, Xu H, et al. Genomics. 1993;17:575–581. doi: 10.1006/geno.1993.1374. [DOI] [PubMed] [Google Scholar]
- 8.Rommens R, Lin B, Hutchinson G B, Andrew S E, Goldberg Y P, et al. Hum Mol Genet. 1993;2:901–907. doi: 10.1093/hmg/2.7.901. [DOI] [PubMed] [Google Scholar]
- 9.Buckler A J, Chang D D, Graw S L, Brook J D, Haber D A. Proc Natl Acad Sci USA. 1991;88:4005–4009. doi: 10.1073/pnas.88.9.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krizman D B, Berget S M. Nucleic Acids Res. 1993;21:5198–5202. doi: 10.1093/nar/21.22.5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Legouis R, Hardelin J P, Levilliers J, Claverie J-M, Compain S, et al. Cell. 1991;67:423–435. doi: 10.1016/0092-8674(91)90193-3. [DOI] [PubMed] [Google Scholar]
- 12.Claverie J-M. Genomics. 1994;23:575–581. doi: 10.1006/geno.1994.1545. [DOI] [PubMed] [Google Scholar]
- 13.Khan A S, Wilcox A S, Polymeropoulos M H, Hopkins J A, Stevens T J, et al. Nat Genet. 1992;2:180–185. doi: 10.1038/ng1192-180. [DOI] [PubMed] [Google Scholar]
- 14.Adams M D, Dubnick M, Kerlavage A R, Moreno R, Kelley J M, et al. Nature (London) 1992;355:632–634. doi: 10.1038/355632a0. [DOI] [PubMed] [Google Scholar]
- 15.Gish W, States D J. Nat Genet. 1993;3:266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- 16.Gelfand M S. J Comp Biol. 1995;1:87–115. doi: 10.1089/cmb.1995.2.87. [DOI] [PubMed] [Google Scholar]
- 17.Burset M, Guigo R. Genomics. 1996;34:353–367. doi: 10.1006/geno.1996.0298. [DOI] [PubMed] [Google Scholar]
- 18.McLachlan G J. Discriminant Analysis and Statistical Pattern Recognition. New York: Wiley; 1992. [Google Scholar]
- 19.Fisher R A. Ann Eugen. 1936;7:179–188. [Google Scholar]
- 20.Solovyev V V, Salamov A A, Lawrence C B. Nucleic Acids Res. 1994;22:5156–5163. doi: 10.1093/nar/22.24.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krzanowski W J. Principles of Multivariate Analysis. Oxford: Clarendon; 1993. p. 347. [Google Scholar]
- 22.Zhang, M. Q. & Marr. T. G. (1997) Genome Res., in press.
- 23.Cinkosky M J, Fickett J W, Gilna P, Burks C. Science. 1991;252:1273–1277. doi: 10.1126/science.1925538. [DOI] [PubMed] [Google Scholar]
- 24.Stormo G D. In: Nucleic Acid and Protein Sequence Analysis. Bishop M J, Rawlings C J, editors. Oxford: IRL; 1987. pp. 231–258. [Google Scholar]
- 25.Fickett J W, Tung C S. Nucleic Acids Res. 1992;20:6441–6450. doi: 10.1093/nar/20.24.6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snyder E E, Stormo G D. J Mol Biol. 1995;248:1–18. doi: 10.1006/jmbi.1995.0198. [DOI] [PubMed] [Google Scholar]
- 27.Uberbacher E C, Mural R J. Proc Natl Acad Sci USA. 1991;88:11261–11265. doi: 10.1073/pnas.88.24.11261. [DOI] [PMC free article] [PubMed] [Google Scholar]