

Abstract
Understanding how proteins are able to form stable complexes is of fundamental interest from the perspective of protein structure and function. Here we show that λ repressor fusions can be used to identify and characterize homotypic interaction domains encoded by the genome of Saccharomyces cerevisiae, using a selection for polypeptides that can drive the assembly of the DNA binding domain of bacteriophage λ repressor. Three high complexity libraries were constructed by cloning random fragments of S. cerevisiae DNA as λ repressor fusions. Repressor fusions encoding homotypic interactions were recovered, identifying oligomerization units in 35 yeast proteins. Seventeen of these interaction domains have not been previously reported, while the other 18 represent homotypic interactions that have been characterized at varying levels of detail. The novel interactions include several predicted coiled-coils as well as domains of unknown structure. With the availability of genomic sequences it should be possible to apply this approach, which provides information about protein-protein interactions that is complementary to that obtained from yeast two-hybrid screens, on a genome-wide scale in yeast or other organisms where large-scale protein-protein interaction data is not available.
Keywords: lambda repressor, chimeric proteins, Saccharomyces cerevisiae, protein oligomerization domains
Background
There is broad interest in the development of genome-wide methods for identifying protein-protein interactions. Recently, several large-scale yeast two-hybrid screens have been used to generate protein interaction maps for Saccharomyces cerevisiae (Fromont-Racine et al., 1997; Ito et al., 2000; Uetz et al., 2000; Ito et al., 2001; Schwikowski et al., 2000). The λ repressor fusion system is well suited for a complementary interaction hunt focused on identifying homotypic interaction domains. Bacteriophage λ repressor requires its C-terminal dimerization domain for proper biological activity. Removing the C-terminal domain inactivates the repressor. However, a heterologous oligomerization domain fused to the native DNA binding domain can reconstitute the activity of the repressor (Hu et al., 1990). Since its original description as a system to study the oligomerization properties of the Gcn4p leucine zipper (Hu et al., 1990), the bacteriophage λ repressor fusion system has been used to map and characterize oligomerization domains present in a large number of proteins from a variety of biological sources (reviewed in Mariño-Ramírez and Hu, 2001).
The widespread use of the repressor system suggested that it could also be used to identify new homotypic interactions on a genomic scale. However, initial efforts to use the repressor system to find homotypic oligomerization domains from S. cerevisiae (Zhang et al., 1999) and E. coli (Jappelli and Brenner, 1999) were discouraging, due to high backgrounds of self-assembling peptides that did not correspond to annotated open reading frames (ORFs).
Here, we describe the use of a modified version of the repressor system in a pilot screen to identify homotypic interaction domains encoded by the S. cerevisiae genome. The modified repressor fusion system uses a weak constitutive promoter to drive the expression of the fusions as well as an amber mutation at position 103 of λcI to allow rapid screening for insert dependence (Mariño-Ramírez and Hu, 2001). We show that our modified system recovers both known and previously unidentified interaction domains, and that the background of non-ORF encoded self-assembling peptides has been substantially reduced.
Results
Identification of homotypic ISTs using repressor fusions
The vectors used for these studies have three modifications compared to pJH391, the plasmid used in most published repressor fusion studies. First, we deleted a fragment containing the rop gene to increase the plasmid copy number and increase the yield of DNA for sequencing. Second, we replaced the lacUV5 promoter with the P7107 promoter (Zeng et al., 1997), a weak constitutive promoter, to decrease the expression of the fusion protein and eliminate the background of host mutations in the lacI gene. Third, we introduced an amber mutation at position 103 of λcI to facilitate testing the fusion constructs for insert dependence. Three plasmid vectors (pLM99-101) were constructed, allowing inframe fusion in all three forward reading frames (Figure 2).
Figure 2.
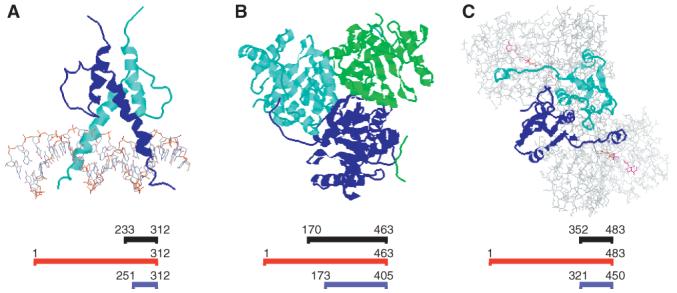
ISTs with structures or homologues in the PDB. (A) The basic helix-loop-helix domain from Pho4p bound to DNA (PDB Accession No. 1A0A). (B) The trimeric subdomain from E. coli α-ketogluatrate dehydrogenase E2 subunit (PDB Accession No. 1C4T). (C) Dimeric E. coli glutathione reductase (PDB Accession No. 1GER). In each panel, the residues encoded by the IST are shown as ribbon diagrams. Non-water atoms that are in the crystal structure but are not encoded in the IST are shown as wireframe. Alignments are shown below each structure. Residues in the IST are shown in black; the full-length yeast ORF is shown in red; the aligned protein from the PDB is shown in blue
Our general strategy is to start with a library of genomic DNA fragments cloned downstream of the repressor DNA-binding domain, select for those that confer immunity to phage infection, and then screen the survivors for those where the immune phenotype requires expression of the insert. The initial selection is done in a strain containing an amber suppressor (supF), while the screening for insert-dependence is done by comparing the ability of repressor fusions to repress a λPL-cat reporter in the presence and absence of the amber suppressor. In strains carrying this reporter, repressor activity turns off chloramphenicol resistance.
To test the feasibility of the approach we performed two reconstruction experiments. First, we constructed a library using genomic DNA from bacteriophage λ as the source of inserts. As expected, we were able to recover the C-terminal domain (amino acids 136-237) of λ repressor as well as inserts from several other λ genes, including the putative self-assembly domain from λ P (amino acids 39-233), which has previously been shown to form homodimers (Zylicz et al., 1984). Second, we tested the ability of well-characterized oligomeric proteins from yeast to drive sufficient self-assembly of repressor fusions to confer phage immunity. We cloned two known dimeric yeast proteins: full-length Tpi1p and Gcn4p as repressor fusions. Both reconstituted the activity of λ repressor in an insert-dependent manner.
We constructed libraries in each of our three vectors using quasi-random genomic DNA fragments of the S. cerevisiae strain BY4741, an S288C derivative (Brachmann et al., 1998). We estimate that each library contains ∼106 independent inserts; 95% of the clones contained a single genomic insert, with an average insert size of 1000±500 base pairs.
Each of the three repressor fusion libraries was then subjected to selection for phage immunity. Survivors were screened for insert-dependence and the positive clones were identified by DNA sequencing. Figure 1 shows a flow chart for the processing of the clones through each step in the screen. The positive clones identified fall into two categories: ORF-encoded and non-ORF-encoded. We identified 180 ORF-encoded interacting sequence tags (ISTs). These ISTs were clustered into families of overlapping fragments, identifying potential homotypic interactions in 35 yeast proteins (Table 1).
Figure 1.
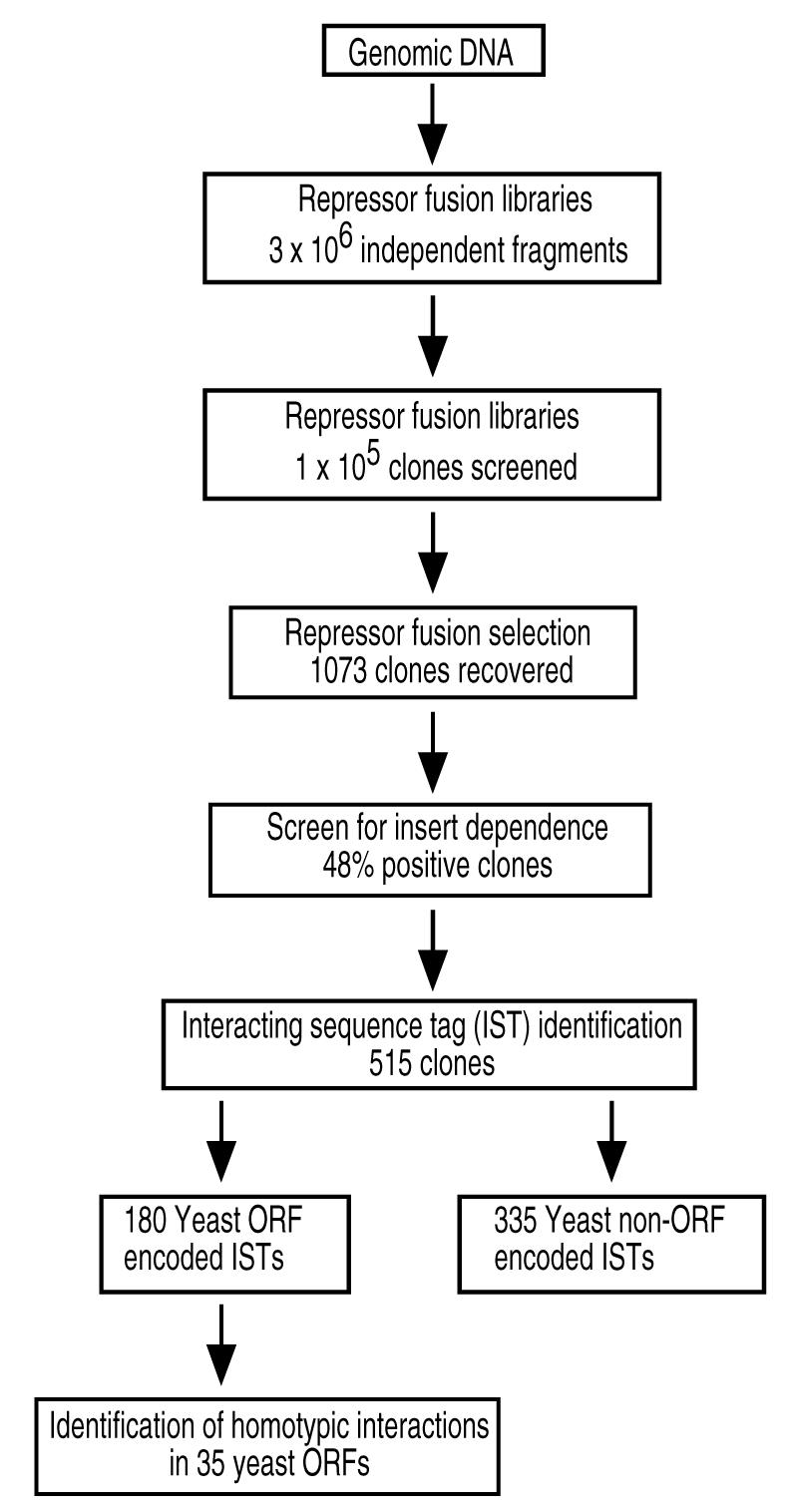
Outline of the strategy used for the identification and characterization of homotypic interactions using repressor fusions. Quasi-random fragments of yeast genomic DNA are cloned in three different repressor fusion vectors to generate libraries with a total number of 3×106 independent clones. Approximately 5.5×105 clones from each library have been subjected to a selection using λ phage allowing the recovery of 1073 clones. A screen for insert dependence ensures that the phenotype observed is due to the presence of the insert alone; 515 clones passed the insert dependence test. The positive clones that contained ORF encoded interacting sequence tags (ISTs) revealed homotypic interactions in 35 different yeast proteins
Table 1.
Homotypic ISTs found in the Saccharomyces cerevisiae genome
| Protein | Intact length (aa) | ISTs (aa) | No. of isolates | Min IST | Previously identified interaction |
|---|---|---|---|---|---|
| Ai2p | 854 | 151-191 | 1 | 41 | Novel interaction in this study |
| Bbp1p | 385 | 314-364 | 32 | 51 | Yeast 2-hybrid (Schramm et al., 2000) |
| Cbf2p | 956 | 671-956 | 1 | 286 | Gel filtration (Russell et al., 1999) |
| Cat8p | 1433 | 116-141 | 2 | 26 | Novel interaction in this study |
| Fap1p | 965 | 341-740 | 1 | 400 | Novel interaction in this study |
| Fin1p | 291 | 235-291 | 2 | 57 | Novel interaction in this study |
| Glr1p | 483 | 352-483 | 2 | 132 | (Jones and Williams, 1975) |
| Hsc82p | 705 | 417-688 | 1 | 272 | Novel interaction in this study |
| Hsp26p | 214 | 30-214 | 2 | - | Gel filtration (Haslbeck et al., 1999) |
| 33-214 | 3 | 182 | |||
| Hsp42p | 375 | 201-375 | 1 | 175 | Gel filtration (Wotton et al., 1996) |
| Hsp82p | 709 | 421-693 | 1 | 273 | Proteolysis; gel filtration (Nemoto et al., 1995) |
| Kgd2p | 463 | 170-463 | 4 | 293 | Sedimentation equilibrium (Repetto and Tzagoloff, 1991) |
| Mdj1p | 511 | 126-452; | 5 | - | Novel interaction in this study |
| 144-452; | 1 | - | |||
| 182-452 | 2 | 224 | |||
| Not5p | 560 | 67-108; | 6 | 42 | Novel interaction in this study |
| 67-137 | 1 | - | |||
| 67-146 | 4 | - | |||
| Pds1p | 373 | 30-140 | 1 | 108 | Novel interaction in this study |
| Prp19p | 503 | 61-144 | 2 | 80 | Gel filtration (Tarn et al., 1994) |
| Pho4p | 312 | 233-312 | 1 | 80 | Crystal Structure (Shimizu et al., 1997) homodimer |
| Pwp1p | 576 | 464-514 | 1 | 51 | Novel interaction in this study |
| Rec107p | 314 | 117-269 | 1 | 153 | Novel interaction in this study |
| Rep2 | 296 | 66-194 | 1 | 129 | Immunoprecipitation; Yeast 2-hybrid (Ahn et al., 1997; Sengupta et al., 2001) |
| Sec7p | 2009 | 1917-2009 | 5 | 93 | Novel interaction in this study |
| Skn7p | 622 | 204-331 | 3 | - | Yeast 2-hybrid; |
| 229-331 | 11 | 103 | Immunoprecipitation (Krems et al., 1996; Raitt et al., 2000) | ||
| 229-360 | 10 | - | |||
| Snz2p | 298 | 10-298 | 1 | 274 | Yeast 2-hybrid (Ito et al., 2001) |
| Snz3p | 298 | 10-298 | 1 | 274 | Yeast 2-hybrid (Ito et al., 2001) |
| Srl2p | 392 | 335-392 | 8 | - | Yeast 2-hybrid (Ito et al., 2001; Uetz et al., 2000) |
| 365-392 | 26 | 28 | |||
| Tup1p | 713 | 12-211 | 2 | - | Sedimentation equilibrium (Jabet et al., 2000; Varanasi et al., 1996) |
| 1-119 | 8 | 119 | Homotetramer | ||
| Ugp1p | 499 | 122-276 | 1 | 155 | Yeast 2-hybrid (Ito et al., 2001) |
| Yap5p | 245 | 40-132 | 7 | - | Novel interaction in this study |
| 61-132 | 8 | 72 | |||
| Ydr266cp | 639 | 94-198 | 1 | 105 | Novel interaction in this study |
| Ydr520cp | 722 | 146-197 | 1 | 52 | Novel interaction in this study |
| Yel015wp | 551 | 251-551 | 1 | 300 | Yeast 2-hybrid (Fromont-Racine et al., 2000; Ito et al., 2001) |
| Ygl068wp | 194 | 49-110 | 1 | 62 | Novel interaction in this study |
| Yhl010cp | 585 | 414-585 | 1 | 172 | Novel interaction in this study |
| Yil122wp | 351 | 21-171 | 3 | 151 | Novel interaction in this study |
| Ymr111cp | 462 | 93-191 | 2 | 99 | Peter Uetz (pers. comm.) |
We also identified 335 non-ORF ISTs, which cluster into 23 unique sequences. All of these contain runs of 5-31 contiguous cysteines, where the shorter oligocysteines are part of much longer Cysrich peptides. These non-ORF peptides are derived from the antisense strands of 20 different annotated ORFs containing poly Q, N or S sequences (see Table 2). Based on their simple sequences, these peptides could be easily identified and discarded.
Table 2.
Non-ORF encoded cysteine-rich peptides found in the Saccharomyces cerevisiae genome
| Deduced amino acid sequencea | Number of isolates | Chromosomal location | Coding ORF |
|---|---|---|---|
| LWRHSSSPRFHRRCCCCCCCCYCCYCCYPCSY | 12 | I (113013-113329) | CCR4 (Poly N region) |
| PACCCCCCCCCCCG | 7 | I (113279-113320) | CCR4 (Poly Q region) |
| CCCCCCCCCCCPKPFDRG | 1 | II (455749-455697) | YBR108W (Poly Q region) |
| CNCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCACACACACACACACACACACACACACACACACACACACACACACA | 67 | II (463964-464236) | CYC8 (Poly Q region) |
| CCCCCCCCCCCCCCCCCCCCCWLFGETKSPFESTRG | 4 | V (83153-83256) | ANP1 (Poly Q region) |
| CCCCCCCCCCCCCCCCNCNCLCSSNSGCLTNR | 9 | VIII (169164-169258) | SLT2 (Poly Q region) |
| ESSPSLFLFCCCCCRSCCCKSCCCCCCCCCCCCCSTFCSSQLA | 3 | VIII (66497-66726) | OPT1 (Poly Q region) |
| LALCCCCCCCCCCCCCCCCCCCKTLA | 8 | IX (169442-169696) | YIL105C (Poly Q region) |
| LCCCCCCCCCCCCCCCC | 12 | IX (400057-399972) | DAL81 (Poly Q region) |
| NNMCCWCCCCCCCCWSG | 21 | XI (379746-379795) | IXR1 (Poly Q region) |
| CCCCCNCCCCCNCSCCCICCWW | 6 | XI (380574-380804) | IXR1 (Poly Q region) |
| YIQSHCCHCCHCCCCCCCCCCCLKIR R*NWTRKMKKKRRRRRRRRRKKKKKISLQSPSP | 5 | XI (613293-613514) | SRP40 (Poly S region) |
| CCCYCCCCCRRCYYCYCHCYCYCDCYYCCCCHCY | 10 | XI (627156-626933) | YKR096W (Poly N region) |
| LTPWSGVYPCCCCCCCCCCCCC | 1 | XII (556403-556211) | ENT2 (Poly Q region) |
| CDCCGCGIPCCGRGCCDICCCDICCCGCCCCCCCGCEFCV | 1 | XIII (354540-354307) | MCM1 (Poly Q region) |
| LGGTTSTLCCCSSCCCCCCCCCCC | 10 | XIII (44448-44281) | DAT1 (Poly Q region) |
| ILWVMNGLGGTTSTLCCCSSCCCCCCCCCCC | 7 | XIII (44469-44372) | DAT1 (Poly Q region) |
| KGRLSPTPCCCCCCCCCCCCCCCRTFSPGGKL | 17 | XIV (720244-720339) | POP2 (Poly Q region) |
| CCCCCCCCCCCCCCCIC | 12 | XV (1036126-1036386) | NDD1 (Poly Q region) |
| CWCDCCCCCCCCCRC | 49 | XV (534545-534451) | AZF1 (Poly Q region) |
| PTKNKGGLIGCCCCGCCNDDAVRFCCCCCCCCCCC | 1 | XVI (117290-116854) | YPL229W (Poly Q region) |
| LHLRVHLHYYYSNCYCYCYCYCRCRCCCCCCCCCCCYCCCCC*CC*CC | 1 | XVI (379312-379483) | RLM1 (Poly N region) |
| SESSCMKDCNGSLFKKFVEKRSCITD KLAPLEEDVNGSPNGFWGGNCCCCCCCCCCCCCC | 4 | XVI (606223-606404) | YPR022C (Poly Q region) |
The deduced sequence begins with the first amino acid ncoded by yeast DNA and continues until and ochre (UAA) or opal (UGA) stop codon is found.
In two cases there was an additional amber codon found, denoted with an asterisk.
In the JH787 background the amber (UAG) codon is replaced by a tyrosine.
Mapping assembly domains within ORFs
The ISTs identify not only those genes that encode proteins that could form homotypic oligomers, but also the regions within the genes that encode sequences that are sufficient to drive oligomerization. For seven of the 35 ORFs with ISTs, we found more than one fragment that encoded an IST. In each case the fragments were overlapping. The sizes of the shortest ISTs for each gene range from 26 amino acids (aa) for Cat8p to 400 aa for Fap1p.
Comparison with previously documented interactions
We performed literature and database searches (Cherry et al., 1998; Hodges et al., 1999) to determine which of the ISTs corresponded to interactions that had been observed previously by other methods. Among the 35 proteins identified here, homotypic interactions have been previously demonstrated for 17 of them by biochemical or genetic methods (Table 1). The evidence for interaction ranges from crystal structures of the self-assembling domain to positive results in yeast two-hybrid assays. In addition, in the cases where the oligomerization domain has been mapped (Hsp42p, Wotton et al., 1996); Hsp82p, Nemoto et al., 1995; Pho4p, Shimizu et al., 1997; Rep2, Sengupta et al., 2001; Tup1p, Varanasi et al., 1996; Yel015wp, Fromont-Racine et al., 2000) our ISTs contain the sequences shown to be needed for self-assembly.
Structures of ISTs
To determine whether the ISTs recovered represented known or novel structures, we performed BLAST searches comparing our ISTs to all of the polypeptide sequences of proteins whose 3-D structures have been deposited in the Protein Data Bank (PDB). Figure 2 shows the structures of three ISTs where structures are known or can be inferred from homology. In only one case, Pho4p, we found a structure and an IST for the same protein from S. cerevisiae. The Pho4p IST encodes a homodimeric basic helix-loop-helix region required for Pho4p activity.
In two other cases, clear homology was found to one or more proteins in the PDB. KGD2 encodes dihydrolipoamide succinyltransferase, the E2 component of the yeast α-ketoglutarate dehydrogenase complex. As an essential component of the TCA cycle, ketoglutarate dehydrogenase subunits are found throughout evolution, and structures of subdomains of the E2 complex from bacterial orthologues are found in the PDB. The Kgd2p IST is 57% identical to the corresponding sequence within its E. coli orthologue (PDB Accession No. 1C4T), and 37% identical to the E2 component of pyruvate dehydrogenase in Bacillus stearothermophilus (PDB Accession No. 1B5S). As in the related pyruvate dehydrogenase complex, the E2 component of ketoglutarate dehydrogenase forms trimers. In the E. coli E2, these trimers form the vertices of a cube with 24 subunits (Knapp et al., 1998) while in B. stearothermophilus the trimers assemble into a dodecahedron with 60 subunits (Izard et al., 1999). Thus, although Kgd2p is known to form homo-oligomers in vivo (Repetto and Tzagoloff, 1991), it is unclear what oligomeric form we are detecting with the λ repressor fusions.
Glr1p encodes the yeast thioredoxin-dependent glutathione reductase. Glr1p is known to function as a homodimer. The IST from Glr1p, which encompasses the C-terminal 132 aa of the protein has 56% identity with residues 321-450 at the C-terminal end of the E. coli orthologue (PDB Accession No. 1GER) and 58% identity with residues 349-478 of the human orthologue (PDB Accession No. 3GRS). This segment of E. coli and human glutathione reductase forms a homodimeric core with a mixed α/β structure and is located at the dimer interface (Mittl and Schulz, 1994).
We also examined ISTs for their propensity to form coiled-coils, which are commonly found in protein-protein interaction interfaces. The COILS algorithm originally developed by Lupas et al. (1991) predicts coiled-coils with probabilities >80% in nine of the proteins containing ISTs identified in this study (Figure 3). In each case the IST covers part or the entire predicted coiled-coil region. The predicted coiled-coil in Tup1p has been demonstrated experimentally to be helical and sufficient to direct assembly of homotetramers (Jabet et al., 2000).
Figure 3.
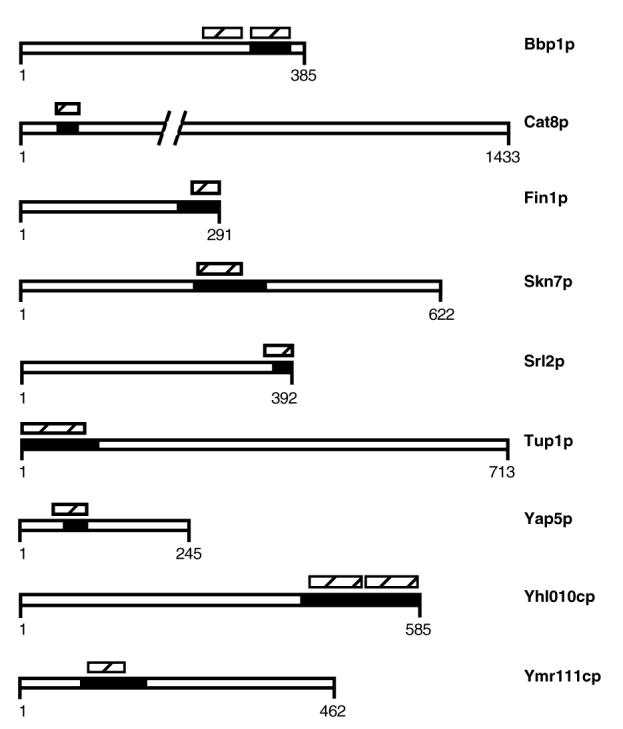
Predicted coiled-coils in proteins containing ISTs. The location of the minimal IST is represented by the black boxes. The slashed boxes represent regions predicted to contain a coiled coil region by COILS (http://tofu.tamu.edu/COILS2/). The predicted regions have coiled-coil probabilities >0.8 in COILS
Discussion
Using λ repressor fusions we were able to identify potential homotypic interactions in 35 proteins encoded by the yeast genome, including one protein from the 2μ plasmid present in the strain used to make the libraries. About half of the ISTs represent previously identified interactions, while the rest have not been described before. The ISTs we identify also represent a combination of proteins of known structure, those for which structures can be predicted with reasonably high confidence, and proteins of unknown structure.
In principle, all of the identities of yeast proteins capable of self-assembly should show up in an all vs. all interaction screen, such as the large-scale two-hybrid studies being undertaken by several laboratories. What, then, is the benefit of using the repressor fusion approach? Figure 4 shows a Venn diagram representation of the homotypic interactions found in this study and in three different two-hybrid studies: two using full-length ORFs from Ito et al. (2001) and Uetz et al. (2000) and one using predicted coiled-coil domains from Newman et al. (2000). There is minimal overlap between our results and the three large-scale yeast two-hybrid studies. The overlap among the yeast two-hybrid datasets is similarly small. Thus, most of the homotypic interactions we have found were not found in the earlier studies.
Figure 4.
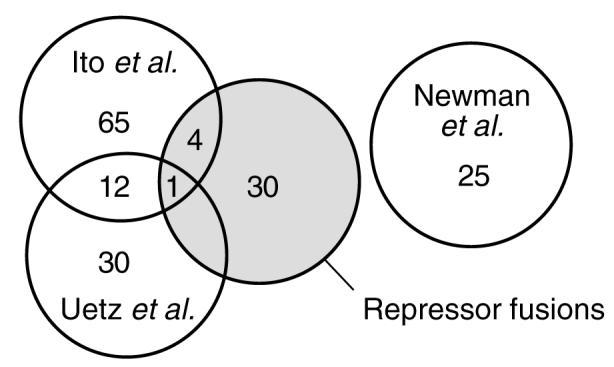
Venn diagram showing homotypic interactions found in three large-scale two-hybrid projects and our repressor fusion libraries. The homotypic interaction subset from Ito et al. (2001) was extracted from the full dataset (http://genome.c.kanazawa-u.ac.jp/Y2H/). The homotypic interaction subset from Uetz et al. (2000) was extracted from (http://depts.wasngton.edu/sfields/yplm/data/)
As noted by Hazbun and Fields (2001), despite the efforts to make each of the studies comprehensive, many interactions known from biochemical data have not been found by any of the large-scale interaction screens. In the study reported here we are clearly far from saturation. Interactions known to be detectable in reconstruction experiments, most notably the Gcn4p leucine zipper, have not yet been found. Although it is likely that additional screening of existing libraries will yield new ISTs, our libraries are also likely to be biased by the non-random cleavage of CviTI sites in our partial digests. New libraries based on other ways to fragment the target DNA may be a richer source of new ISTs.
In practice, comprehensive identification of protein-protein interactions will involve complementary information from a variety of genetic and biochemical approaches. Among the genetic approaches, repressor fusions are well suited to identify homotypic interactions. Newman et al. (2000) has argued that homotypic interactions, especially those involving homodimers are likely to be underrepresented in yeast two-hybrid screens due to preferential interaction of baits within a dimeric DNA binding protein over preys coming from solution. A wide variety of technical limitations will affect the recovery of ISTs from yeast two-hybrid pairs, repressor fusions or both, e.g. post-translational modifications required for folding in assembly in yeast are unlikely to be recapitulated in E. coli. Nevertheless, both two-hybrid methods and repressor fusions can clearly provide identities of many proteins involved in homotypic interactions.
A genome-wide survey of protein-protein interactions should provide two kinds of information: not only what proteins can interact, but also what parts of the proteins are involved in the interactions. One of the most useful kinds of information provided by repressor fusions is the localization of oligomerization domains on a genome-wide scale. Because repressor fusions require only single libraries of hybrid proteins to identify homotypic ISTs, the number of subdomains that can be tested scales linearly with the number of clones that can be subjected to selection for repressor activity. By contrast, detecting a homotypic interaction in a two-hybrid system requires that both the bait and prey be present in the same cell. This means that the number of protein fragments that can be tested scales only as the square root of the library size.
These considerations, along with the higher transformation efficiency of E. coli, allowed us to use random fragments of genomic DNA instead of the full-length ORFs favored by the large-scale yeast two-hybrid approaches. Thus, our ISTs provide mapping information about the location of the oligomerization domains within proteins as well as the identities of the proteins involved in self-assembly. In general, the ISTs we find are much smaller than the proteins that contain them. Where different, overlapping ISTs are recovered from the same protein (as for Hsp26p, Mdj1p, Not5p, Skn7p, Srl2p, Tup1p and Yap5p) the endpoints of the ISTs can be used to delimit the minimal region required for oligomerization. In the case of Tup1p, amino acids 1-72 have been shown to be sufficient for oligomerization (Tzamarias and Struhl, 1994; Varanasi et al., 1996). The shortest IST we found covered amino acids 1-119, while the overlap between two ISTs suggested that residues 12-119 might be sufficient to form an oligomer.
Self-assembling domains derived from IST analysis will expand our understanding of the many ways nature builds protein complexes. The domains may provide more tractable targets for structure determination than the intact proteins from which they come. Additionally, isolated interaction domains may provide useful tools for functional genomics; expression of the domains in yeast could yield dominant negative phenotypes. While this may not provide much new information in a genetically well-characterized system like S. cerevisiae, a similar approach may be useful for a variety of genetically less tractable organisms. In cases where assembly domains prove to be involved in an important cellular function, the repressor fusions themselves can provide screens for drug discovery.
Finally, detailed study of homotypic interaction domains is likely to identify new structural motifs that will be found in other proteins. Although the interactions we identify are homotypic, it is likely that in many cases, evolutionarily related structures are also used for heterotypic interactions. Examples of structures used in both homotypic and heterotypic interactions include the HLH (Robinson and Lopes, 2000) and leucine zipper (Hurst, 1994) motifs. Other interaction domains that function in both homo- and hetero-oligomers may provide additional mechanisms of regulation by combinatorial assembly of different subunits.
Materials and methods
Strains and plasmids
All the strains used in this study are derivatives of AG1688 [F’128 lacIq lacZ::Tn5/araD139, Δ(ara-leu)- 7697, Δ(lac)X74, galE15, galK16, rpsL(StrR), hsdR2, mcrA, mcrB1] (Hu et al., 1993). The repressor fusion libraries were transformed into JH787 [AG1688 (ϕ80 Su-3)]. The screening for insert dependence was done on LM58 [JH787 (λLM58)] and LM59 [AG1688 (λLM58)]. λLM58 carries a PL-cat reporter. The repressor fusion vectors (Figure 5) used to generate the libraries were pLM99 (GenBank Accssion No. AF308739), pLM100 (GenBank Accession No. AF308740) and pLM101 (GenBank Accession No. AF308741). These vectors contain an amber mutation at position 103 in the repressor, between the DNA binding domain and the DNA insert (Mariño-Ramírez and Hu, 2001).
Figure 5.

Schematic representation of repressor fusion vectors used in this study. A. Sequence of the multiple cloning site region in pLM99. B. Sequence of the multiple cloning site region in pLM100. C. Sequence of the multiple cloning site region in pLM101
Repressor fusion library construction and characterization
We prepared yeast nuclei and extracted genomic DNA from S. cerevisiae BY4741 as described (Shimizu et al., 1991). The DNA was partially digested with CviTI (Megabase Research) to generate blunt ends. The DNA was cloned into the SmaI site of pLM99, pLM100 and pLM101 to generate three libraries in different reading frames to increase genome coverage. Inserts from 60 randomly chosen clones were examined by PCR amplification to establish the percentage of recombinants and average fragment size. Amplification reactions were done by PCR using Taq DNA polymerase (Promega) and two flanking primers: the cI primer (5′-AGGGATGTTCTCACCTAAGCT-3′) and T-phi primer (5′-CTCAGCGGTGGCAGCAGCCAA-3′).
Selection and screening procedure
Detailed procedures for selection and screening have been described (Mariño-Ramírez and Hu, 2001). Briefly, selection of immune clones was done by plating ∼107 JH787 cells containing amplified fusion libraries on LB-ampicillin-kanamycin plates seeded with 108 pfu/plate λKH54 and λKH54h80. The amber suppressor in JH787 allows the expression of full-length fusions. M13 transducing stocks from the surviving colonies were prepared and used to individually transduce the repressor fusions to suppressor (supF; LM58) and non-suppressor (sup0; LM59) strains. In clones where the active repressor phenotype is dependent on self-assembly of the insert-encoded domain, phage immunity is dependent on suppression of the amber mutation. Clones identified as insert-dependent by differential repression of PL-cat in supF and sup0 strains were picked for further study; any clone where immunity was not insert-dependent was discarded.
Identification of interacting fragments
Plasmid DNA was extracted from the positive clones and the inserts were identified by automated dye-terminator DNA sequencing from the cI and T-phi primers. DNA sequencing reactions were done using the ABI Big Dye terminator kit (Applied Biosystems) and sequences were obtained at the Gene Technologies Laboratory in the Department of Biology at Texas A&M University. The sequences were identified by BLAST (Altschul et al., 1997) searches to the yeast protein database (NCBI) to identify the open reading frame (ORF) containing a homotypic interaction. The sequences of the interacting sequence tags (ISTs) encoding self-interaction domains were inferred from the reference yeast genome sequence.
Acknowledgements
The authors thank Mike Cherry for making the homotypic interaction data available in the Saccharomyces Genome Database (SGD). We thank Svenja Simon-Marshall and W. Brian Hatten for invaluable help with library screening. We thank Peter Uetz and Stan Fields for sharing data prior to publication. We also like to thank the members of the Hu laboratory, Debby Siegele, Wei Wang, Tom Kodadek and Stan Fields, for critical comments on the manuscript. This work was supported by funding from the National Science Foundation (MCB-9808474), the Robert A. Welch Foundation (A-1354) and the Advanced Research Program of the Texas Higher Education Coordinating Board (Award 999902-116). L.M. was supported by a fellowship from Fulbright/Colciencias/IIE.
References
- Ahn YT, Wu XL, Biswal S, et al. The 2μ plasmid-encoded Rep1 and Rep2 proteins interact with each other and colocalize to the Saccharomyces cerevisiae nucleus. J Bacteriol. 1997;179:7497–7506. doi: 10.1128/jb.179.23.7497-7506.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Cherry JM, Adler C, Ball C, et al. SGD: Saccharomyces genome database. Nucleic Acids Res. 1998;26:73–79. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromont-Racine M, Mayes AE, Brunet-Simon A, et al. Genome-wide protein interaction screens reveal functional networks involving Sm-like proteins. Yeast. 2000;17:95–110. doi: 10.1002/1097-0061(20000630)17:2<95::AID-YEA16>3.0.CO;2-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromont-Racine M, Rain JC, Legrain P. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nature Genet. 1997;16:277–282. doi: 10.1038/ng0797-277. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Walke S, Stromer T, et al. Hsp26: a temperature-regulated chaperone. EMBO J. 1999;18:6744–6751. doi: 10.1093/emboj/18.23.6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazbun TR, Fields S. Networking proteins in yeast. Proc Natl Acad Sci U S A. 2001;98:4277–4278. doi: 10.1073/pnas.091096398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges PE, McKee AH, Davis BP, Payne WE, Garrels JI. The Yeast Proteome Database (YPD): a model for the organization and presentation of genome-wide functional data. Nucleic Acids Res. 1999;27:69–73. doi: 10.1093/nar/27.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Newell N, Tidor B, Sauer R. Probing the roles of residues at the e and g positions of the GCN4 leucine zipper by combinatorial mutagenesis. Protein Sci. 1993;2:1072–1084. doi: 10.1002/pro.5560020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JC, O’Shea EK, Kim PS, Sauer RT. Sequence requirements for coiled-coils: analysis with lambda repressor-GCN4 leucine zipper fusions. Science. 1990;250:1400–1403. doi: 10.1126/science.2147779. [DOI] [PubMed] [Google Scholar]
- Hurst HC. Transcription factors 1: bZip proteins. Protein Profile. 1994;2:101–168. [PubMed] [Google Scholar]
- Ito T, Tashiro K, et al. Toward a protein-protein interaction map of the budding yeast: A comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc Natl Acad Sci USA. 2000;97:1143–1147. doi: 10.1073/pnas.97.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Chiba T, Ozawa R, et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci U S A. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izard T, Aevarsson A, Allen MD, et al. Principles of quasi-equivalence and Euclidean geometry govern the assembly of cubic and dodecahedral cores of pyruvate dehydrogenase complexes. Proc Natl Acad Sci U S A. 1999;96:1240–1245. doi: 10.1073/pnas.96.4.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabet C, Sprague ER, VanDemark AP, Wolberger C. Characterization of the N-terminal domain of the yeast transcriptional repressor Tup1. Proposal for an association model of the repressor complex Tup1×Ssn6. J Biol Chem. 2000;275:9011–9018. doi: 10.1074/jbc.275.12.9011. [DOI] [PubMed] [Google Scholar]
- Jappelli R, Brenner S. A genetic screen to identify sequences that mediate protein oligomerization in Escherichia coli. Biochem Biophys Res Commun. 1999;266:243–247. doi: 10.1006/bbrc.1999.1806. [DOI] [PubMed] [Google Scholar]
- Jones ET, Williams CH., Jr The sequence of amino acid residues around the oxidation-reduction active disulfide in yeast glutathione reductase. J Biol Chem. 1975;250:3779–3784. [PubMed] [Google Scholar]
- Knapp JE, Mitchell DT, Yazdi MA, et al. Crystal structure of the truncated cubic core component of the Escherichia coli 2-oxoglutarate dehydrogenase multienzyme complex. J Mol Biol. 1998;280:655–668. doi: 10.1006/jmbi.1998.1924. [DOI] [PubMed] [Google Scholar]
- Krems B, Charizanis C, Entian KD. The response regulator-like protein Pos9/Skn7 of Saccharomyces cerevisiae is involved in oxidative stress resistance. Curr Genet. 1996;29:327–334. doi: 10.1007/BF02208613. [DOI] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Mariño-Ramírez L, Hu JC. Using λ repressor fusions to isolate and characterize self-assembling domains. In: Golemis E, Serebriiskii I, editors. Protein-Protein Interactions: A Laboratory Manual. Cold Spring Harbor Laboratory Press; New York: 2001. [Google Scholar]
- Mittl PR, Schulz GE. Structure of glutathione reductase from Escherichia coli at 1.86 Å resolution: comparison with the enzyme from human erythrocytes. Protein Sci. 1994;3:799–809. doi: 10.1002/pro.5560030509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto T, Ohara-Nemoto Y, Ota M, Takagi T, Yokoyama K. Mechanism of dimer formation of the 90 kDa heat-shock protein. Eur J Biochem. 1995;233:1–8. doi: 10.1111/j.1432-1033.1995.001_1.x. [DOI] [PubMed] [Google Scholar]
- Newman JR, Wolf E, Kim PS. A computationally directed screen identifying interacting coiled coils from Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2000;97:13203–13208. doi: 10.1073/pnas.97.24.13203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raitt DC, Johnson AL, Erkine AM, et al. The Skn7 response regulator of Saccharomyces cerevisiae interacts with Hsf1 in vivo and is required for the induction of heathishock genes by oxidative stress. Mol Biol Cell. 2000;11:2335–2347. doi: 10.1091/mbc.11.7.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repetto B, Tzagoloff A. In vivo assembly of yeast mitochondrial α-ketoglutarate dehydrogenase complex. Mol Cell Biol. 1991;11:3931–3939. doi: 10.1128/mcb.11.8.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KA, Lopes JM. Survey and summary: Saccharomyces cerevisiae basic helix-loop-helix proteins regulate diverse biological processes. Nucleic Acids Res. 2000;28:1499–505. doi: 10.1093/nar/28.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell ID, Grancell AS, Sorger PK. The unstable F-box protein p58-Ctf13 forms the structural core of the CBF3 kinetochore complex. J Cell Biol. 1999;145:933–950. doi: 10.1083/jcb.145.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm C, Elliotthis S, Shevchenko A, Schiebel E. The Bbp1p-Mps2p complex connects the SPB to the nuclear envelope and is essential for SPB duplication. EMBO J. 2000;19:421–433. doi: 10.1093/emboj/19.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwikowski B, Uetz P, Fields S. A network of protein-protein interactions in yeast. Nature Biotechnol. 2000;18:1257–1261. doi: 10.1038/82360. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Blomqvist K, Pickett AJ, et al. Functional domains of yeast plasmid-encoded Rep proteins. J Bacteriol. 2001;183:2306–2315. doi: 10.1128/JB.183.7.2306-2315.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M, Roth SY, Szent-Gyorgyi C, Simpson RT. Nucleosomes are positioned with base pair precision adjacent to the α2 operator in Saccharomyces cerevisiae. EMBO J. 1991;10:3033–3041. doi: 10.1002/j.1460-2075.1991.tb07854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Toumoto A, Ihara K, et al. Crystal structure of PHO4 bHLH domain-DNA complex: flanking base recognition. EMBO J. 1997;16:4689–4697. doi: 10.1093/emboj/16.15.4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarn WY, Hsu CH, Huang KT, et al. Functional association of essential splicing factor(s) with PRP19 in a protein complex. EMBO J. 1994;13:2421–2431. doi: 10.1002/j.1460-2075.1994.tb06527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzamarias D, Struhl K. Functional dissection of the yeast Cyc8-Tup1 transcriptional co-repressor complex. Nature. 1994;369:758–761. doi: 10.1038/369758a0. [DOI] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Varanasi US, Klis M, Mikesell PB, Trumbly RJ. The Cyc8 (Ssn6)-Tup1 co-repressor complex is composed of one Cyc8 and four Tup1 subunits. Mol Cell Biol. 1996;16:6707–6714. doi: 10.1128/mcb.16.12.6707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotton D, Freeman K, Shore D. Multimerization of Hsp42p, a novel heat shock protein of Saccharomyces cerevisiae, is dependent on a conserved carboxyl-terminal sequence. J Biol Chem. 1996;271:2717–2723. doi: 10.1074/jbc.271.5.2717. [DOI] [PubMed] [Google Scholar]
- Zeng X, Herndon AM, Hu JC. Buried asparagines determine the dimerization specificities of leucine zipper mutants. Proc Natl Acad Sci U S A. 1997;94:3673–3678. doi: 10.1073/pnas.94.8.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Murphy A, Hu JC, Kodadek T. Genetic selection of short peptides that support protein oligomerization in vivo. Curr Biol. 1999;9:417–420. doi: 10.1016/s0960-9822(99)80188-2. [DOI] [PubMed] [Google Scholar]
- Zylicz M, Gorska I, Taylor K, Georgopoulos C. Bacteriophage λ replication proteins: formation of a mixed oligomer and binding to the origin of λ DNA. Mol Gen Genet. 1984;196:401–406. doi: 10.1007/BF00436186. [DOI] [PubMed] [Google Scholar]