

Abstract
Vanillin (VAN) and cinnamaldehyde (CIN) are dietary flavorings that exhibit antimutagenic activity against mutagen-induced and spontaneous mutations in bacteria. Although these compounds were antimutagenic against chromosomal mutations in mammalian cells, they have not been studied for antimutagenesis against spontaneous gene mutations in mammalian cells. Thus, we initiated studies with VAN and CIN in human mismatch repair-deficient (hMLH1−) HCT116 colon cancer cells, which exhibit high spontaneous mutation rates (mutations/cell/generation) at the HPRT locus, permitting analysis of antimutagenic effects of agents against spontaneous mutation. Long-term (1–3-week) treatments of HCT116 cells with VAN at minimally toxic concentrations (0.5–2.5 mM) reduced the spontaneous HPRT mutant fraction (MF, mutants/106 survivors) in a concentration-related manner by 19% to 73%. A similar treatment with CIN at 2.5–7.5 μM yielded a 13% to 56% reduction of the spontaneous MF. Short-term (4–h) treatments also reduced the spontaneous MF by 64% (VAN) and 31% (CIN). To investigate the mechanisms of antimutagenesis, we evaluated the ability of VAN and CIN to induce DNA damage (comet assay) and to alter global gene expression (Affymetrix GeneChip) after 4-h treatments. Both VAN and CIN induced DNA damage in both mismatch repair-proficient (HCT116 + chr3) and deficient (HCT116) cells at concentrations that were antimutagenic in HCT116 cells. There were 64 genes in common whose expression was changed similarly by both VAN and CIN; these included genes related to DNA damage, stress responses, oxidative damage, apoptosis, and cell growth. RT-PCR results paralleled the Affymetrix results for 4 selected genes (HMOX1, DDIT4, GCLM, and CLK4). Our results show for the first time that VAN and CIN are antimutagenic against spontaneous mutations in mammalian (human) cells. These and other data lead us to propose that VAN and CIN may induce DNA damage that elicits recombinational DNA repair and, consequently, reduces spontaneous mutation.
Keywords: Cinnamaldehyde, Vanillin, Spontaneous mutagenesis, Microarray, Comet assay, Human cells, HPRT
Introduction
Epidemiological studies have demonstrated that certain components of the diet can have beneficial effects to health, such as reducing risks for various types of cancers [1–4]. A wide array of dietary agents have been studied or are considered for chemoprevention of cancer [5], and many have antimutagenic activity in laboratory studies [6]. Vanillin (VAN) and cinnamaldehyde (CIN) are food flavorings that inhibit induced mutations in bacteria [7–10] and mammalian cells [11–14]. Although VAN and CIN inhibit spontaneous mutation in bacteria [15–19], this effect has not yet been reported in mammalian cells. VAN and CIN have been classified as bioantimutagens because they can modulate cellular processes such as DNA replication and repair [18]. Studies in bacteria led to the hypothesis that VAN and CIN inhibited mutagenesis by increasing repair of DNA damage through the recombinational repair pathway [10]. Later studies in Drosophila supported this view [20,21].
We showed previously that VAN and CIN reduced spontaneous mutations in Salmonella TA104 only at GC sites and not at AT sites and that this inhibition required the pKM101 plasmid [17]. Recently, we demonstrated that VAN and CIN required RecA recombinational repair, but not mismatch, SOS, or nucleotide excision repair, in order to reduce spontaneous mutation in Escherichia coli [19]. In addition, VAN, but not CIN, was actually mutagenic in a mismatch repair (MMR)-defective strain of E. coli [19]. Thus, we proposed that VAN and CIN produced DNA damage that elicited recombinational repair, but not other types of repair, resulting in a reduction of spontaneous mutation in bacteria [19].
To examine whether these compounds exhibited antimutagenic effects in mammalian (human) cells, we evaluated the ability of VAN and CIN to reduce spontaneous mutagenesis in mismatch repair-deficient (MMR−) human colon cancer HCT116 cells. These cells have an elevated spontaneous HPRT mutation rate of ~86 × 10−6 mutations/cell/generation, which is ~19 times higher than in the mismatch repair-corrected cell line HCT116 + chr3 (4.55 × 10−6) [22]. The spontaneous mutant fraction (MF) for the MMR− HCT116 cells is ~12.5 times higher (150 × 10−6) than in MMR+ HCT116 + chr3 cells (12 × 10−6) [23]. Cell with a high spontaneous mutation rate are needed to effectively detect treatment-induced reductions in spontaneous mutagenesis. HCT116 cells were used to demonstrate the antimutagenicity of dietary antioxidants such as ascorbate, α-tocopherol, (−) epigallocatechin gallate (EGCG), and lycopene against spontaneous mutation [22,24]. Lycopene was the most effective of these antimutagens, reducing the spontaneous mutation rate at HPRT by 70% [22].
Because VAN was mutagenic in mismatch repair-deficient E. coli [19] and both VAN and CIN required recombinational repair in order to be antimutagenic, we evaluated the reduction of the spontaneous MF at the HPRT locus in HCT116 cells as well as the induction of DNA damage (comet assay) by VAN and CIN in both HCT116 and HCT116 + chr3 cells. In addition, we examined the influence of VAN and CIN on global gene expression in these cells in order to identify genes and pathways that might play a role in the antimutagenic effects of these agents.
Materials and methods
Chemicals
VAN (>98% pure, Sigma, St. Louis, MO) and CIN (trans-cinnamaldehyde, >99% pure, Aldrich, Milwaukee, WI) were dissolved in dimethyl sulfoxide (DMSO, Burdick and Jackson, Muskegon, MI), and stored at 4°C as stock solutions at 5 M for VAN and 0.5 M for CIN. Fresh dilutions were made in medium immediately before each experiment. The final concentration of DMSO in the culture medium was 0.1%.
Cell culture
HCT116 cells are MMR-deficient (hMLH1−) human colon cancer cells derived from a hereditary non-polyposis colon cancer HNPCC patient (ATCC, Rockville, MD). The HCT + chr3 cell line used in the comet assay was kindly provided by Dr. Thomas Kunkel (National Institute of Environmental Health Sciences, RTP, NC) [25]. Cell lines were maintained in DF medium, which consisted of Dulbecco’s modified Eagle medium:Ham’s F12 (1:1) with 10% fetal bovine serum (FBS, Gemini Bio Products, Calabasas, CA). Cells were cultured at 37°C in an atmosphere of 5% CO2. To cleanse cultures of pre-existing HPRT mutants, cells were grown in HAT medium (100-μM hypoxanthine, 0.4-μM aminopterin, and 16-μM thymidine in DF medium) for 10–14 days before the start of each mutagenesis assay.
Survival assays
To determine the optimal non-toxic dose range for VAN and CIN for the antimutagenesis studies, 7 day “in situ” clonal survival assays were conducted as per Mure and Rossman [22]. Briefly, 300 cells were seeded per 60-mm dish, in triplicate, with VAN (0.5–5.0 mM) or CIN (5–50 μM). After 7 days growth in the presence of the compound, the surviving colonies were stained and counted. Concentrations of VAN and CIN that yielded greater than 70% survival were identified.
For determination of long-term (1–3 wk) survival, HCT116 cells were seeded at a density of 2.5 × 106 cells per 100-mm dish and grown in the presence of the indicated concentration of VAN or CIN for 1–3 weeks. Fresh VAN or CIN was added to the cell cultures at every cell population expansion (once per week), resulting in discontinuous but repeated exposures to the compounds that can be likened to occasional, non-daily human dietary exposures. At the end of 1, 2, or 3 weeks, the cells were trypsinized and then reseeded at a density of 300 cells per 60-mm dish without VAN or CIN. After 7 days of growth, colonies were fixed in methanol, stained with 0.5% crystal violet in 50% methanol, and counted. Two independent assays were performed.
In the short-term (4 h) studies, cell survival was determined by reseeding 300 cells per 60-mm dishes, in triplicate, immediately following the end of the 4-h treatment with 2.5-mM VAN or 7.5-μM CIN. Surviving clones were stained and counted after 7 days growth in DF medium without VAN or CIN.
Long-term mutant frequency assays
Accumulating spontaneous mutations were measured over several weeks in cultured mammalian cells as described previously [26]. Briefly, 5 × 105 cells from HAT-treated cultures were seeded in nonselective DF medium with or without VAN or CIN present during population growth expansion [22]. Mutants were then allowed to accumulate while the cultures grew for 15–18 generations over 3 weeks. At 1, 2, and 3 weeks, 106 cells were sampled and reseeded into medium containing 6.7 μg/ml of 6-thioguanine (6TG, Sigma, St. Louis, MO) at a density of 105 cells per 100-mm dish for mutant selection. The plating efficiency in nonselective medium and the total cell count were determined each week at the time of sampling. 6TG medium was replenished after 7 days, and mutant colonies were stained after 12–14 days with 0.5% crystal violet in 50% methanol and then scored. Mutant fraction (MF) was calculated by dividing the number of mutant colonies by the number of cells seeded, corrected for plating efficiency.
For VAN, we carried out three independent assays, each including treatments of 0 (control), 0.5, 1.0, and 2.5-mM VAN. For CIN, we originally carried out three independent assays, each including treatments of 0 (control), 2.5, 5.0, 7.5-μM CIN. Because the highest treatment concentration in the third assay was invalidated by contamination, we added a fourth assay that included only the control and the highest concentration.
Short-term mutant frequency assay
Two days before treatment with VAN or CIN, HAT-treated HCT116 cells were seeded in two T-175 flasks at 2 × 106 cells per flask in DF medium supplemented with 10% FBS. At 60% confluence (~3.5 × 106 cells per flask), cells were treated with 7.5-μM CIN or 2.5-mM VAN for 4 h at 37°C in DF and then harvested. Cells were seeded for survival as noted above. Concurrently, two T-75 flasks were each seeded with 5 × 105 untreated cells or VAN- or CIN-treated cells and incubated for 1 week for mutant expression to deplete HPRT enzyme. The cells were then reseeded in medium containing 6.7 μg/ml of 6TG at 105 cells per 100-mm dish for mutant selection. 6TG medium was replenished after 7 days, and mutant colonies were stained after 12–14 days with 0.5% crystal violet in 50% methanol and scored. The plating efficiency in nonselective medium was also determined at the time of plating into 6TG medium for mutant fraction calculations, with mutant fraction expressed as mutants/106 surviving cells and corrected for plating efficiency. Three independent assays were performed.
Comet assay
HCT116 and HCT116 + chr3 cells were exposed to VAN (0, 0.5, 1.0, and 2.5 mM) and CIN (0, 2.5, 5.0, and 7.5 μM) in DF medium without FBS for 4 h at 37°C to evaluate DNA damage via a modified version of the comet assay described by Singh et al. [27]. Briefly, ~50,000 cells were used to prepare each slide. Slides were placed in in a horizontal electrophoresis tank containing 0.3-M NaOH and 1-mM NA2EDTA, pH 13 to unwind the DNA for 20 min before electrophoresis at a constant voltage of 25V for 40 min. After electrophoresis, the slides were neutralized, dried, and stained with 20 μg/ml of ethidium bromide (dissolved in water), and 50 comets/slide were scored using Komet 5.0 (Kinetic Imaging Ltd., Liverpool, UK). Results were expressed as Olive Tail Moment (OTM). All experiments were done in low light to reduce background UV induction of DNA damage. Two independent experiments were conducted with VAN and CIN in both the HCT116 and HCT + chr3 cell lines, and 2 slides were prepared for each concentration of VAN and CIN. Thus, for each concentration, 200 cells from 4 slides were scored to evaluate the effects of VAN and CIN in each cell line.
Microarray analysis
Gene expression analysis was conducted using Affymetrix Human U133A Genechip® arrays (Affymetrix, Santa Clara, CA), which assays approximately 14,500 human genes with 22,215 probesets. Total RNA from untreated HCT116 cells and cells exposed for 4 h to VAN (2.5 mM) and CIN (7.5 μM) from three independent MF assays described previously was isolated using an RNeasy® Minikit (QIAGEN Inc., Valencia, CA). Total RNA from each of these experiments was amplified using the Affymetrix 1-Cycle cDNA Synthesis protocol. Starting with 1 μg of total RNA, cRNA was produced according to the manufacturer’s protocol. For each array, 15 μg of amplified cRNAs were fragmented and hybridized to the array for 16 h in a rotating hybridization oven using the Affymetrix Eukaryotic Target Hybridization Controls and protocol. Slides were stained and washed as indicated in the Antibody Amplification Stain for Eukaryotic Targets protocol using the Affymetrix Fluidics Station FS450. Arrays were then scanned with an Affymetrix Scanner 3000. Probe-level intensities were obtained using the Genechip® Operating Software (Version 1.2.0.037).
Validation of Affymetrix array results using real-time PCR amplification (RT-PCR) was conducted on four genes (HMOX1, GCLM, CLK4, and DDIT4), each of which had 1.5-fold or greater changes in gene expression for at least one of the two compounds. Briefly, cDNA was generated from 450 ng of RNA from each sample using a High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. RT-PCR reactions consisted of 3 μl of cDNA (a total of 27 ng), 1 X TaqMan® Universal Master Mix (Applied Biosystems), and 1 X primer/probe mix for the four genes: HMOX1 (hCG40033), GCLM (hCG3194), CLK4 (hCG20100), and DDIT4 (hCG2024489, Applied Biosystems) in a final volume of 10 μl. Signals were recorded during PCR using an ABI PRISM® instrument (Applied Biosystems). All gene expression results were normalized to 18S rRNA expression (Hs99999901 Applied Biosystems) and were calculated using the delta Ct method (Applied Biosystems).
Data analysis
We analyzed MF as an outcome in the long-term assays assuming that the observed MF had an underlying Poisson distribution with possible extra-Poisson variation. Our analysis used generalized linear mixed models with Poisson error distribution and its canonical link function (logarithm); in essence, our analysis was a mixed-effects analysis of variance for Poisson-distributed data. We conducted separate analyses for each compound. Our analysis estimated a separate geometric mean value of mutant fraction for each week and concentration of VAN and CIN and accounted for relevant sources of variation, including the multiple independent assays and possibly differing week- or concentration-response patterns among those assays.
We also used a mixed-function-effects analysis-of-variance approach similar to the previous one to analyze MF as an outcome in the short-term assays. Here, our analysis estimated separate geometric mean values for control, 7.5-μM CIN, or 2.5-mM VAN and accounted for assay-to-assay variation.
We analyzed the log-transformed median OTM of 50 cells per slide as an outcome in the comet assay. Based on examination of the residual plots, and from our experience with comet data from previous studies (unpublished data), we concluded that these transformed values had an approximately normal distribution and carried out a mixed-model analysis of variance separately for each compound. Our analysis fit a separate mean value for each cell line and each concentration level and accounted for relevant sources of variation, including independent assays, differing concentration- or cell-line response patterns between assays, and slide-to-slide variation.
All of the preceding statistical analyses were carried out using SAS software (version 9.0, SAS Institute, Cary, NC). In particular, we fitted the generalized linear mixed models with the GLIMMIX macro <http://support.sas.com/ctx/samples/index.jsp?sid=536>.
Expression levels for each gene in each sample were obtained from probe-level intensities using Robust Multichip Average analysis [28], and the resulting data were uploaded into GeneSpring® (Version 7.2). The variance for each gene was estimated using the Cross-Gene Error Model (Rocke-Lorenzo; GeneSpring® version 7.2; Redwood City, CA). Fold change was calculated as the ratio of the mean of three experimental expression measures to the mean of three control expression measures. Functional annotation for selected genes was obtained from GeneCards (http://www.genecards.org), an integrated database of human genes that includes automatically mined genomic, proteomic, and transcriptomic information.
Results
Survival and antimutagenesis for VAN and CIN
Before antimutagenicity testing, the cytotoxicities of VAN and CIN were evaluated in 7 day “in situ” clonal survival assays and in 1–3-week exposure assays. In the 7-day clonal survival assays, survival curves were generated for HCT116 cells with 5–50-μM CIN and 0.5–5.0-mM VAN (data not shown). At the highest concentrations tested for both CIN and VAN, cell survival was 10% or less. We chose concentrations that produced ~70% survival, 7.5 uM CIN and 2.5-mM, as the maximum concentration of each compound for further studies. The 1–3-week survival data (Fig. 1A and 1B) confirmed that the minimally toxic VAN concentrations of 0.5 mM, 1 mM, and 2.5 mM and CIN concentrations of 2.5 μM, 5 μM, and 7.5 μM were suitable for both the long- and short-term assays.
Fig. 1.
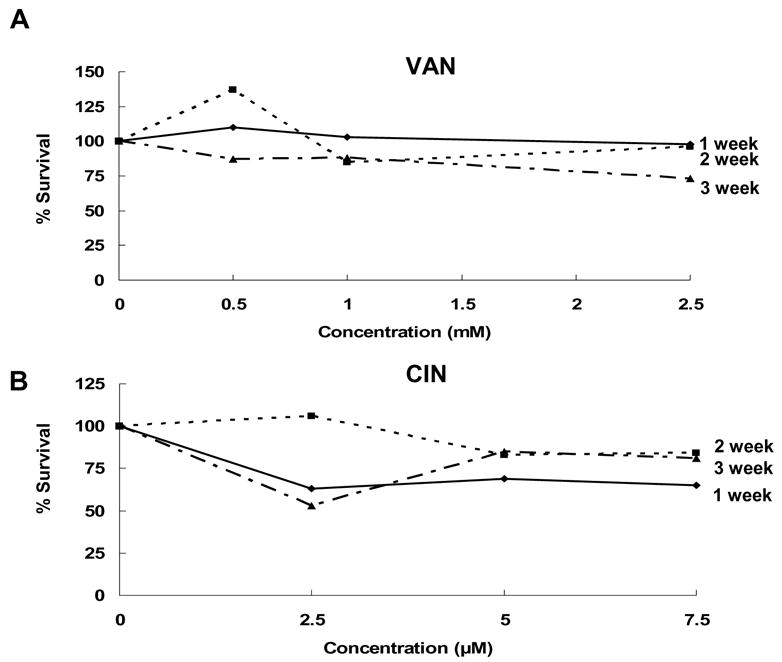
Clonal survival of HCT116 cells exposed to VAN (A) or CIN (B) for 1–3 weeks. Data represent the average of two independent experiments.
Typical long-term mammalian mutagenesis studies show exponential accumulations of mutants in continuous culture, and repeated dosing by effective antimutagens reduces this accumulation [22]. The MF concentration-response data for VAN and CIN (Fig. 2) represent the average of three independent experiments. VAN over a concentration range of 0.5–2.5 mM for 1–3 weeks suppressed the accumulation of HPRT mutants in HCT116 cells compared to untreated control cells in a concentration-related manner (overall trend test across all weeks, p < 0.001). For example, 2.5-mM VAN reduced the MF by 53% (95% CI; 32, 67), 72% (58, 82), and 54% (36, 66) in weeks 1, 2, and 3, respectively (Fig. 2). CIN also suppressed spontaneous MF over a concentration range of 2.5–7.5 μM, in a concentration-related manner (overall trend test across all weeks, p < 0.001). For example, at 7.5 μM CIN reduced the spontaneous HPRT MF by 56% (33, 71), 31% (5, 49), and 45% (25, 60) at 1, 2, and 3 weeks, respectively (Fig. 2).
Fig. 2.
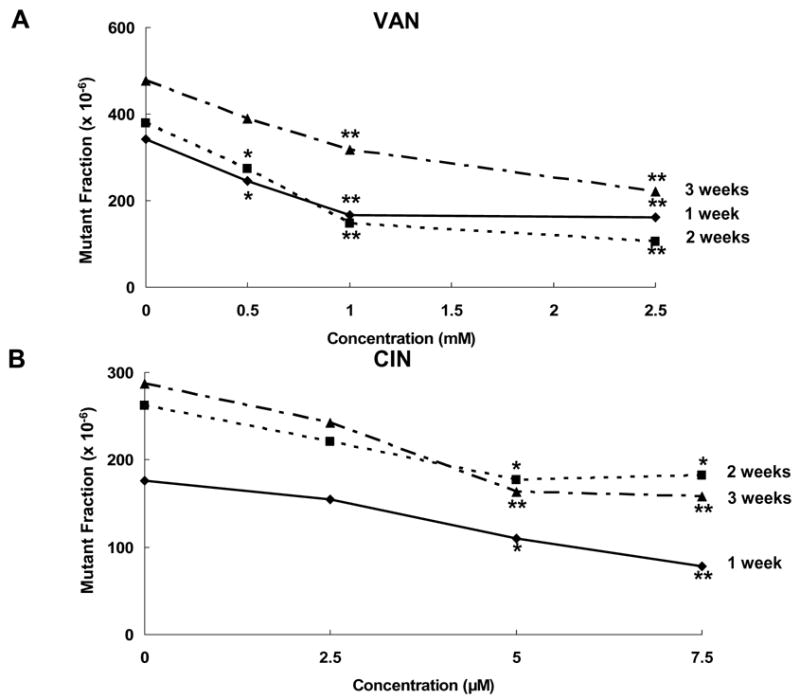
Reduction of spontaneous MF in HCT116 cells by VAN and CIN. The concentration-response curves for VAN (A) represent the data from three independent experiments. The concentration-response curves for CIN (B) represent data from 4 independent experiments. * indicates a significant decrease in MF relative to untreated cells at p < 0.05. ** indicates a significant change at p < 0.01.
In addition to the long-term exposures, HCT116 cells were exposed for 4 h to 2.5-mM VAN and 7.5-μM CIN in three replicate experiments. Cell survival under these exposure conditions was 95% for CIN and 82% for VAN. At 2.5 mM, VAN significantly reduced the mutant fraction by 64% (p = 0.006); at 7.5 μM, CIN reduced the mutant fraction by 31%; however, this was not statistically significant (p = 0.06).
Comet assay
VAN and CIN were evaluated in the comet assay for DNA damaging effects in both the HCT116 and HCT116 + chr3 cell lines after 4-h exposures. At concentrations that were antimutagenic against spontaneous HPRT mutations, VAN induced significant, concentration-related increases in DNA damage (trend test, p < 0.01) in both HCT116 and HCT116 + chr3 cells (Fig. 3A). At 2.5 mM VAN, DNA damage was slightly higher in the MMR+ cell line HCT116 + chr3 than in the MMR− cell line HCT116, and DNA damage levels were significantly different between the two cell lines (p = 0.004). CIN also induced significant, concentration-related increases in DNA damage (trend test, p < 0.01) in both cell lines (Fig. 3B). However, in contrast to VAN, the levels of DNA damage induced in the two cell lines by CIN were not significantly different (p = 0.80). Thus, both VAN and CIN induced concentration-related increases in DNA damage in both the MMR+ and MMR- cell lines.
Fig. 3.
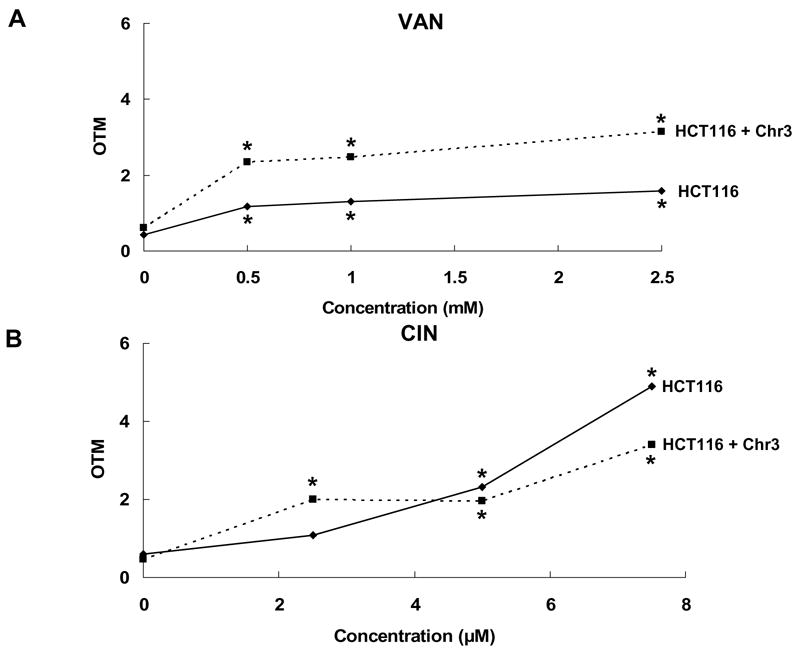
Effects of VAN (A) and CIN (B) on DNA damage in HCT116 and HCT116 + Chr3 cell lines in the comet assay. * indicates a significant difference relative to untreated cells at p < 0.01.
Gene expression
Transcriptional changes after 4-h exposures to 2.5-mM VAN or 7.5-μM CIN were evaluated from three independent experiments. Most statistically significant changes in gene transcription were modest, ranging from 1.20 to 5.05 fold (Table 1). Genes with probe-level intensities below 60 were considered noise, thus removing 13,875 probesets from the analysis. Differential expression was assessed at p < 0.01 and p < 0.05 by one-way analysis of variance for each treatment compared to control. For p < 0.05 VAN had 459 significant probesets and CIN had 291 significant probesets. The gene lists were compared, identifying 67 genes in common. Of these 67 genes, 64 genes changed from 14% to 52% in the same direction for both VAN and CIN. Table 1 shows a list of 23 selected genes that may have potential roles in DNA damage, oxidative and stress responses. For example, HMOX1and HSPA1B were up-regulated by both VAN and CIN. Expression of the damage-inducible transcript DDIT4 was increased only after VAN exposure. In addition, genes in the glutathione synthesis pathway, GCLM and GCLC, were up-regulated by both VAN and CIN. A number of genes involved in growth and cell proliferation, including MAP2K2, FGFR2, TGFB1I1, JUND, INSIG1, and CTGF, were down-regulated to a similar extent by both VAN and CIN. For validation of the microarray results, RT-PCR was conducted for 4 genes (HMOX1, DDIT4, GCLM, and CLK4) with 1.5-fold or greater expression differences for at least one of the two compounds. Fold changes for these 4 genes were similar for both RT-PCR and microarray (Fig. 4).
Table 1.
Effect of VAN and CIN on expression of selected genes after a 4-h exposure
| Fold change
|
|||
|---|---|---|---|
| Gene symbol | Gene name | VAN | CIN |
| DDIT4 | Damage-inducible transcript 4 | 1.6a | −1.3a |
| HMOX1 | Heme oxygenase (decycling) | 1.5a | 5.1a |
| CLK2 | CDC-like kinase 2 | 1.4a | −1.0a |
| SLC7A11 | solute carrier family 7 | 1.3a | 2.0a |
| TRIM16 | Tripartite motif-containing 16 | 1.2a | 2.2a |
| HSPA1B | heat shock 70kDa protein 1B | 1.2a | 1.4a |
| GCLM | glutamate-cysteine ligase, modifier subunit | 1.2a | 1.6a |
| GCLC | glutamate-cysteine ligase, catalytic subunit | 1.1a | 1.6a |
| TP53 | tumor protein p53 (Li-Fraumeni syndrome) | −1.1a | −1.4a |
| UNG2 | uracil-DNA glycosylase 2 | −1.1a | −1.3a |
| MAP2K2 | mitogen-activated protein kinase kinase 2 | −1.2a | −1.3a |
| SLC35D2 | solute carrier family 35, member D2 | −1.2a | −1.2a |
| FGFR2 | fibroblast growth factor receptor | −1.2a | −1.3a |
| TGFB1I1 | transforming growth factor beta 1 induced transcript 1 | −1.3a | −1.2a |
| DUSP6 | dual specificity phosphatase 6 | −1.3a | −1.1a |
| MARK4 | MAP/microtubule affinity-regulating kinase 4 | −1.3a | −1.3a |
| JUND | jun D proto-oncogene | −1.3a | −1.3a |
| INSIG1 | insulin induced gene 1 | −1.3a | −1.1a |
| CTGF | connective tissue growth factor | −1.4a | −1.1a |
| FZD2 | frizzled homolog 2 (Drosophila) | −1.5a | 1.0a |
| PPP1R10 | protein phosphatase 1, regulatory subunit 10 | −1.6a | −1.2a |
| UGP2 | UDP-glucose pyrophosphorylase 2 | 1.2b | 1.2b |
| DNMT3B | DNA (cytosine-5-)-methyltransferase 3 beta | −1.2b | −1.2b |
Fold changes relative to untreated cells. Genes were differentially transcribed at p < 0.01.
Genes were differentially transcribed at p < 0.05.
Fig. 4.
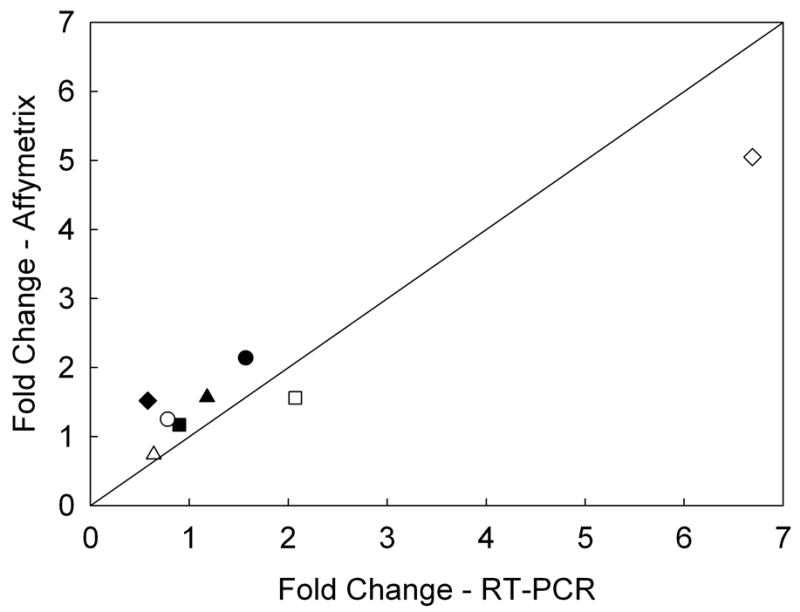
Comparison of changes in gene expression after cells were exposed to VAN or CIN for 4 h among selected genes as measured by microarray versus RT-PCR. Filled symbols indicate fold changes with VAN treatment, and open symbols indicate fold changes with CIN treatment (r2 = 0.97, p = 0.0001), ◆ = HMOX1, ⎵ = DDIT4, ● = CLK4, ■ = GCLM. The solid line indicates identical fold changes from both microarray and RT-PCR results.
Discussion
Antimutagenic effects of VAN and CIN against a variety of chemical and physical mutagens have been reported in numerous studies in bacteria [6–9], mammalian cells [11–14,29,30], and in vivo systems [11,31–33]. Only a few studies have reported on the antimutagenic effects of VAN and CIN against endogenous mutations in bacteria [15–17] and for VAN in Drosophila [34]. In the present study, we found that VAN and CIN inhibited spontaneous mutagenesis in the human colon cancer cell line HCT116 in a concentration-dependent manner in long-term exposures (Fig. 2) and were also effective in short-term exposures.
Both VAN and CIN reduced MF relative to that of the untreated cells in all replicate experiments at nontoxic concentrations yielding 70–90% cell survival. In the long-term assays, VAN reduced MF at all time points (1, 2, and 3 weeks) in a concentration-related manner in all replicate experiments. For example, 2.5-mM VAN reduced the spontaneous MF by ~50% in weeks 1 and 3, and by 72% at 2 weeks (Fig. 2). CIN also consistently reduced spontaneous MF at weeks 1, 2, and 3 in a concentration-related manner. For example, 7.5-μM CIN reduced MF by ~50% at 1 and 3 weeks, and by 31% at 2 weeks (Fig. 2). In the short-term assays (4-h exposures), VAN and CIN had consistent, inhibitory effects on the spontaneous MF. After short-term exposures, the MF was reduced by 31% by CIN (7.5 μM) and by 69% by VAN (2.5 mM).
The underlying mechanisms for the antimutagenic effects of VAN or CIN are still not completely understood. Results from previous studies in bacterial strains, have suggested that VAN and CIN reduce spontaneous and induced mutagensis by similar mechanisms, principally through enhancement of recombinational repair. However, results from the present study, along with recently published studies in mammalian cells [30,35–37], suggest that VAN and CIN may enhance recombinational repair through different mechanisms, possibly by inducing different types of DNA damage. Based on observations of the requirement for recA gene function in E. coli for the antimutagenic and survival-enhancing effects of VAN and CIN [7,8,10], Ohta et al. [10] hypothesized that these antimutagens may inhibit mutations induced by chemical mutagens (e.g., 4-nitroquinoline) or physical mutagens (e.g., UV-light) by enhancing recombinational repair. In Drosophila, VAN inhibited mitomycin C-induced mutations and dramatically increased recombination [20]. VAN also significantly increased recombination in Drosophila exposed to bleomycin [21], indicating that VAN promotes homologous recombination in mutagen-treated organisms. We have shown that the recA recombination function in E. coli lacI strains is required for inhibition of spontaneous mutations [19]. VAN and CIN reduced spontaneous mutations in the wild-type strain NR9102 as well as in the nucleotide excision repair-defective strain NR11634 and the SOS repair-defective strain NR11475. However, in the recA53 strain NR11317, which is deficient in both SOS repair and recombination, both VAN and CIN were mutagenic, supporting a role for recombination in antimutagenesis by VAN and CIN.
Our observations that VAN and CIN were mutagenic in the recombination-defective E. coli strain NR11317 and that VAN was also slightly mutagenic in the mutl strain NR9319 [19], led us to examine the effects of VAN and CIN on DNA damage and on transcriptional changes in mammalian cells after 4-h exposures to minimally toxic concentrations of VAN and CIN in the present study. Although VAN and CIN clearly reduced the spontaneous MF in HCT116 cells after 4-h exposures, they also induced significant levels of DNA damage, detected as increased OTM in the comet assay, in both HCT116 and HCT116 + chr 3 cell lines (Fig. 3). The levels of DNA damage induced by VAN were greater in the MMR+ cell line compared to the MMR− cell line, whereas the levels of DNA damage induced by CIN were not significantly different between the two cell lines. This observation suggests that the ability of VAN, but not CIN, to induce DNA damage is influenced by the MMR status of the cells.
In order to evaluate transcriptional changes after 4-h VAN or CIN treatment, we isolated RNA from each of three replicate antimutagenesis experiments for use in microarray experiments. Except for a 5-fold increase in heme oxygenase 1 (HMOX1), most statistically significant transcriptional changes were modest, with 2-fold or lower increases or decreases for all other genes analyzed, 23 of which are shown on Table 1. Among the genes up-regulated by VAN and CIN were HMOX1, a gene up-regulated in response to oxidative stress [38,39], and the heat shock 70 kDa protein 1B (HSPA1B). In addition, two genes in the glutathione synthesis pathway, GCLC and GCLM, were up-regulated after CIN exposure.
Several growth factor- and cell proliferation-related genes were down-regulated in response to VAN and CIN treatment, including MAP2K2, FGFR2, TGFB1L1, and CTGF. Down-regulation of growth factors and cell proliferation genes may also be consistent with a response to DNA damaging agents [40–43]. Gene expression changes in HCT116, a colon cancer cell line that was chosen for these antimutagenesis experiments because of its high spontaneous background, must be interpreted with caution because other cellular functions in these cells may also be dysregulated. However, the clear evidence of increased DNA strand breakage in the comet assay shows that VAN and CIN do induce DNA damage in mammalian cells.
Although the exact type of DNA damage induced by CIN is not known, results from several studies suggest that CIN may increase oxidative damage indirectly. At concentrations of CIN higher than used in our study, other researchers found that CIN depleted intracellular glutathione and protein thiol levels and increased reactive oxygen species (ROS) levels in mammalian cells [36,37]. Increased levels of ROS in turn induced apoptosis via decreased mitochondrial transmembrane potential and release of cytochrome c [37]. In the present study, increased transcription of the glutathione synthesis genes GCLM and GCLC is suggestive of glutathione depletion after CIN treatment.
In contrast to oxidant damage induced by CIN, VAN has been shown to have antioxidant and free-radical scavenging effects. VAN inhibited both singlet oxygen-induced single-strand breaks in the plasmid pBR322 and singlet oxygen-induced protein oxidation [44], scavenged free radicals [45,46], and inhibited lipid peroxidation [47,48]. Although VAN has exhibited antimutagenic effects against numerous genotoxic agents, co-treatment with VAN increased the HPRT mutant frequency in ethyl methanesulfonate (EMS)-treated V79 cells [30], and VAN increased recombination in Drosophila exposed to bleomycin without decreasing mutations [21]. These results suggest that the antimutagenic effect of VAN depends on the type of DNA lesion induced and the specific DNA repair pathway that corrects the DNA damage. In this regard, Durant et al. [35] reported that VAN inhibited non-homologous DNA end-joining (NHEJ) in human cell extracts by specifically inhibiting DNA PK enzyme activity without affecting the level of RAD50/RPA foci formed during homologous recombination (HR) repair of DNA double-strand breaks. Thus, VAN may alter the balance between NHEJ, a potentially error-prone pathway, and HR, a relatively error-free pathway.
In summary, we have demonstrated for the first time that VAN and CIN reduced the spontaneous MF at minimally toxic concentrations in mammalian cells. Increased DNA damage,resulting from VAN and CIN treatment in the HCT116 and HCT116 + chr3 cell lines as detected in the comet assay, along with limited evidence of gene transcriptional responses to DNA damage, supports our hypothesis that VAN and CIN induce DNA damage, although the specific types of damage may differ between these compounds As suggested by our bacterial studies [19], this damage may then elicit recombinational repair, correcting not only the DNA damage induced by VAN and CIN, but also removing endogenous DNA damage and contributing to the overall antimutagenic effect of these compounds.
Acknowledgments
This research was supported in part by NIH R03 CA89732 (to C.B.K.), by the NYU/NIEHS Center ES00260, by the NYU Cancer Center CA16087, and by an NIEHS T32 ES007324 training grant (A.K.). This research was also supported in part by the Intramural Research Program of the National Institute of Environmental Health Sciences, NIH, DHHS. D.T.S. acknowledges support from an NIEHS Intramural Research Training Award. We thank the NIEHS Microarray Center for assistance with the gene expression results presented in this work. We also thank K. Witt (NIEHS) and S. Warren, R. Owen, and J. Ross (US EPA) for their helpful comments on this manuscript. This manuscript was reviewed by the National Health and Environmental Effects Research Laboratory, US Environmental Protection Agency, and approved for publication. Approval does not signify that the contents necessarily reflect the views and policies of the agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Key TJ, Schatzkin A, Willett WC, Allen NE, Spencer EA, Travis RC. Diet, nutrition and the prevention of cancer. Public Health Nutr. 2004;7:187–200. doi: 10.1079/phn2003588. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy ET. Evidence for nutritional benefits in prolonging wellness. Am J Clin Nutr. 2006;83:410S–414S. doi: 10.1093/ajcn/83.2.410S. [DOI] [PubMed] [Google Scholar]
- 3.Serra-Majem L, Roman B, Estruch R. Scientific evidence of interventions using the Mediterranean diet: a systematic review. Nutr Rev. 2006;64:S27–47. doi: 10.1111/j.1753-4887.2006.tb00232.x. [DOI] [PubMed] [Google Scholar]
- 4.DeMarini DM. Dietary interventions of human carcinogenesis. Mutat Res. 1998;400:457–465. doi: 10.1016/s0027-5107(98)00052-9. [DOI] [PubMed] [Google Scholar]
- 5.Brenner DE, Gescher AJ. Cancer chemoprevention: lessons learned and future directions. Br J Cancer. 2005;93:735–739. doi: 10.1038/sj.bjc.6602765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohta T. Modification of genotoxicity by naturally occurring flavorings and their derivatives. Crit Rev Toxicol. 1993;23:127–146. doi: 10.3109/10408449309117114. [DOI] [PubMed] [Google Scholar]
- 7.Ohta T, Watanabe K, Moriya M, Shirasu Y, Kada T. Analysis of the antimutagenic effect of cinnamaldehyde on chemically induced mutagenesis in Escherichia coli. Mol Gen Genet. 1983b;192:309–315. doi: 10.1007/BF00392167. [DOI] [PubMed] [Google Scholar]
- 8.Ohta T, Watanabe K, Moriya M, Shirasu Y, Kada T. Antimutagenic effects of cinnamaldehyde on chemical mutagenesis in Escherichia coli. Mutat Res. 1983;107:219–227. doi: 10.1016/0027-5107(83)90164-1. [DOI] [PubMed] [Google Scholar]
- 9.Ohta T, Watanabe M, Watanabe K, Shirasu Y, Kada T. Inhibitory effects of flavourings on mutagenesis induced by chemicals in bacteria. Food Chem Toxicol. 1986;24:51–54. doi: 10.1016/0278-6915(86)90264-4. [DOI] [PubMed] [Google Scholar]
- 10.Ohta T, Watanabe M, Shirasu Y, Inoue T. Post-replication repair and recombination in uvrA umuC strains of Escherichia coli are enhanced by vanillin, an antimutagenic compound. Mutat Res. 1988;201:107–112. doi: 10.1016/0027-5107(88)90116-9. [DOI] [PubMed] [Google Scholar]
- 11.Imanishi H, Sasaki YF, Matsumoto K, Watanabe M, Ohta T, Shirasu Y, Tutikawa K. Suppression of 6-TG-resistant mutations in V79 cells and recessive spot formations in mice by vanillin. Mutat Res. 1990;243:151–158. doi: 10.1016/0165-7992(90)90038-l. [DOI] [PubMed] [Google Scholar]
- 12.Fiorio R, Bronzetti G. Effects of cinnamaldehyde on survival and formation of HGPRT- mutants in V79 cells treated with methyl methanesulfonate, N-nitroso-N-methylurea, ethyl methanesulfonate and UV light. Mutat Res. 1994;324:51–57. doi: 10.1016/0165-7992(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 13.Sanyal R, Darroudi F, Parzefall W, Nagao M, Knasmuller S. Inhibition of the genotoxic effects of heterocyclic amines in human derived hepatoma cells by dietary bioantimutagens. Mutagenesis. 1997;12:297–303. doi: 10.1093/mutage/12.4.297. [DOI] [PubMed] [Google Scholar]
- 14.Sasaki YF, Imanishi H, Watanabe M, Ohta T, Shirasu Y. Suppressing effect of antimutagenic flavorings on chromosome aberrations induced by UV-light or X-rays in cultured Chinese hamster cells. Mutat Res. 1990;229:1–10. doi: 10.1016/0027-5107(90)90002-l. [DOI] [PubMed] [Google Scholar]
- 15.de Silva HV, Shankel DM. Effects of the antimutagen cinnamaldehyde on reversion and survival of selected Salmonella tester strains. Mutat Res. 1987;187:11–19. doi: 10.1016/0165-1218(87)90071-1. [DOI] [PubMed] [Google Scholar]
- 16.De Flora S, Bennicelli C, Rovida A, Scatolini L, Camoirano A. Inhibition of the ‘spontaneous’ mutagenicity in Salmonella typhimurium TA102 and TA104. Mutat Res. 1994;307:157–167. doi: 10.1016/0027-5107(94)90288-7. [DOI] [PubMed] [Google Scholar]
- 17.Shaughnessy DT, Setzer RW, DeMarini DM. The antimutagenic effect of vanillin and cinnamaldehyde on spontaneous mutation in Salmonella TA104 is due to a reduction in mutations at GC but not AT sites. Mutat Res. 2001;480–481:55–69. doi: 10.1016/s0027-5107(01)00169-5. [DOI] [PubMed] [Google Scholar]
- 18.Kada T, Inoue T, Ohta T, Shirasu Y. Antimutagens and their modes of action. Basic Life Sci. 1986;39:181–196. doi: 10.1007/978-1-4684-5182-5_15. [DOI] [PubMed] [Google Scholar]
- 19.Shaughnessy DT, Schaaper RM, Umbach DM, DeMarini DM. Inhibition of spontaneous mutagenesis by vanillin and cinnamaldehyde in Escherichia coli: dependence on recombinational repair. Mutat Res. 2006 doi: 10.1016/j.mrfmmm.2006.08.006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santos JH, Graf U, Reguly ML, Rodrigues de Andrade HH. The synergistic effects of vanillin on recombination predominate over its antimutagenic action in relation to MMC-induced lesions in somatic cells of Drosophila melanogaster. Mutat Res. 1999;444:355–365. doi: 10.1016/s1383-5718(99)00101-1. [DOI] [PubMed] [Google Scholar]
- 21.Sinigaglia M, Reguly ML, de Andrade HH. Effect of vanillin on toxicant-induced mutation and mitotic recombination in proliferating somatic cells of Drosophila melanogaster. Environ Mol Mutagen. 2004;44:394–400. doi: 10.1002/em.20067. [DOI] [PubMed] [Google Scholar]
- 22.Mure K, Rossman TG. Reduction of spontaneous mutagenesis in mismatch repair-deficient and proficient cells by dietary antioxidants. Mutat Res. 2001;480–481:85–95. doi: 10.1016/s0027-5107(01)00172-5. [DOI] [PubMed] [Google Scholar]
- 23.Glaab WE, Skopek TR. Cytotoxic and mutagenic response of mismatch repair-defective human cancer cells exposed to a food-associated heterocyclic amine. Carcinogenesis. 1999;20:391–394. doi: 10.1093/carcin/20.3.391. [DOI] [PubMed] [Google Scholar]
- 24.Glaab WE, Hill RB, Skopek TR. Suppression of spontaneous and hydrogen peroxide-induced mutagenesis by the antioxidant ascorbate in mismatch repair-deficient human colon cancer cells. Carcinogenesis. 2001;22:1709–1713. doi: 10.1093/carcin/22.10.1709. [DOI] [PubMed] [Google Scholar]
- 25.Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;54:4308–4312. [PubMed] [Google Scholar]
- 26.Rossman TG, Goncharova EI, Nadas A. Modeling and measurement of the spontaneous mutation rate in mammalian cells. Mutat Res. 1995;328:21–30. doi: 10.1016/0027-5107(94)00190-g. [DOI] [PubMed] [Google Scholar]
- 27.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 28.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 29.Sasaki YF, Imanishi H, Ohta T, Shirasu Y. Effects of vanillin on sister-chromatid exchanges and chromosome aberrations induced by mitomycin C in cultured Chinese hamster ovary cells. Mutat Res. 1987;191:193–200. doi: 10.1016/0165-7992(87)90153-9. [DOI] [PubMed] [Google Scholar]
- 30.Tamai K, Tezuka H, Kuroda Y. Different modifications by vanillin in cytotoxicity and genetic changes induced by EMS and H2O2 in cultured Chinese hamster cells. Mutat Res. 1992;268:231–237. doi: 10.1016/0027-5107(92)90229-u. [DOI] [PubMed] [Google Scholar]
- 31.Inouye T, Sasaki YF, Imanishi H, Watanebe M, Ohta T, Shirasu Y. Suppression of mitomycin C-induced micronuclei in mouse bone marrow cells by post-treatment with vanillin. Mutat Res. 1988;202:93–95. doi: 10.1016/0027-5107(88)90168-6. [DOI] [PubMed] [Google Scholar]
- 32.Sasaki YF, Ohta T, Imanishi H, Watanabe M, Matsumoto K, Kato T, Shirasu Y. Suppressing effects of vanillin, cinnamaldehyde, and anisaldehyde on chromosome aberrations induced by X-rays in mice. Mutat Res. 1990;243:299–302. doi: 10.1016/0165-7992(90)90146-b. [DOI] [PubMed] [Google Scholar]
- 33.Imai T, Yasuhara K, Tamura T, Takizawa T, Ueda M, Hirose M, Mitsumori K. Inhibitory effects of cinnamaldehyde on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung carcinogenesis in rasH2 mice. Cancer Lett. 2002;175:9–16. doi: 10.1016/s0304-3835(01)00706-6. [DOI] [PubMed] [Google Scholar]
- 34.de Andrade HH, Santos JH, Gimmler-Luz MC, Correa MJ, Lehmann M, Reguly ML. Suppressing effect of vanillin on chromosome aberrations that occur spontaneously or are induced by mitomycin C in the germ cell line of Drosophila melanogaster. Mutat Res. 1992;279:281–287. doi: 10.1016/0165-1218(92)90245-u. [DOI] [PubMed] [Google Scholar]
- 35.Durant S, Karran P. Vanillins--a novel family of DNA-PK inhibitors. Nucleic Acids Res. 2003;31:5501–5512. doi: 10.1093/nar/gkg753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janzowski C, Glaab V, Mueller C, Straesser U, Kamp HG, Eisenbrand G. Alpha,beta-unsaturated carbonyl compounds: induction of oxidative DNA damage in mammalian cells. Mutagenesis. 2003;18:465–470. doi: 10.1093/mutage/geg018. [DOI] [PubMed] [Google Scholar]
- 37.Ka H, Park HJ, Jung HJ, Choi JW, Cho KS, Ha J, Lee KT. Cinnamaldehyde induces apoptosis by ROS-mediated mitochondrial permeability transition in human promyelocytic leukemia HL-60 cells. Cancer Lett. 2003;196:143–152. doi: 10.1016/s0304-3835(03)00238-6. [DOI] [PubMed] [Google Scholar]
- 38.Foucaud L, Bennasroune A, Klestadt D, Laval-Gilly P, Falla J. Oxidative stress induction by short time exposure to ozone on THP-1 cells. Toxicol In Vitro. 2006;20:101–108. doi: 10.1016/j.tiv.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 39.Ryter SW, Choi AM. Heme oxygenase-1: molecular mechanisms of gene expression in oxygen-related stress. Antioxid Redox Signal. 2002;4:625–632. doi: 10.1089/15230860260220120. [DOI] [PubMed] [Google Scholar]
- 40.Jelinsky SA, Samson LD. Global response of Saccharomyces cerevisiae to an alkylating agent. Proc Natl Acad Sci USA. 1999;96:1486–1491. doi: 10.1073/pnas.96.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heinloth AN, Shackelford RE, Innes CL, Bennett L, Li L, Amin RP, Sieber SO, Flores KG, Bushel PR, Paules RS. ATM-dependent and -independent gene expression changes in response to oxidative stress, gamma irradiation, and UV irradiation. Radiat Res. 2003;160:273–290. doi: 10.1667/rr3047. [DOI] [PubMed] [Google Scholar]
- 42.Maude SL, Enders GH. Cdk inhibition in human cells compromises chk1 function and activates a DNA damage response. Cancer Res. 2005;65:780–786. [PubMed] [Google Scholar]
- 43.Workman CT, Mak HC, McCuine S, Tagne JB, Agarwal M, Ozier O, Begley TJ, Samson LD, Ideker T. A systems approach to mapping DNA damage response pathways. Science. 2006;312:1054–1059. doi: 10.1126/science.1122088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar SS, Ghosh A, Devasagayam TP, Chauhan PS. Effect of vanillin on methylene blue plus light-induced single-strand breaks in plasmid pBR322 DNA. Mutat Res. 2000;469:207–214. doi: 10.1016/s1383-5718(00)00074-7. [DOI] [PubMed] [Google Scholar]
- 45.Santosh Kumar S, Priyadarsini KI, Sainis KB. Free radical scavenging activity of vanillin and o-vanillin using 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical. Redox Rep. 2002;7:35–40. doi: 10.1179/135100002125000163. [DOI] [PubMed] [Google Scholar]
- 46.Zhou YC, Zheng RL. Phenolic compounds and an analog as superoxide anion scavengers and antioxidants. Biochem Pharmacol. 1991;42:1177–1179. doi: 10.1016/0006-2952(91)90251-y. [DOI] [PubMed] [Google Scholar]
- 47.Murcia MA, Martinez-Tome M. Antioxidant activity of resveratrol compared with common food additives. J Food Prot. 2001;64:379–384. doi: 10.4315/0362-028x-64.3.379. [DOI] [PubMed] [Google Scholar]
- 48.Kamat JP, Ghosh A, Devasagayam TP. Vanillin as an antioxidant in rat liver mitochondria: inhibition of protein oxidation and lipid peroxidation induced by photosensitization. Mol Cell Biochem. 2000;209:47–53. doi: 10.1023/a:1007048313556. [DOI] [PubMed] [Google Scholar]