

Abstract
Fifteen different taxoid conjugates were prepared by linking various anticancer compounds, including camptothecin (CPT), epipodophyllotoxin (EP), colchicine (COL), and glycyrrhetinic acid (GA), at the 2’- or 7-position on paclitaxel (TXL, 1) through an ester, imine, amine, or amide bond. Newly synthesized conjugates were evaluated for cytotoxic activity against replication of several human tumor cell lines. Among them, TXL-CPT conjugates, 8–10, were more potent than TXL itself against the human prostate carcinoma cell line PC-3 (ED50 = 14.8, 3.1, 19.4 nM compared with 55.5 nM), and conjugate 10 was also eightfold more active than TXL against the LN-CAP prostate cancer cell line. These compounds also possessed anti-angiogenesis ability as well as lower inhibitory effects against a normal cell line (MRC-5). Thus, conjugates 8–10 are possible antitumor drug candidates, particularly for prostate cancer.
Keywords: Paclitaxel, Conjugation, Cytotoxity, Prostate cancer
Paclitaxel (TXL, 1, Figure 1), a plant derived antimitotic agent,1–2 is currently in clinical use against ovarian and breast cancer. It promotes the irreversible assembly of tubulin into microtubules by binding to and stabilizing microtubules. This mechanism of action is unique among the established antitumor drugs, including the vinca alkaloids, vincristine and vinblastine, which prevent microtubule assembly by microtubule binding.3 Other natural products, including camptothecin (CPT),4 which is approved for clinical use in the United State, epipodophyllotoxin (EP),5 and colchicine (COL),6 also possess potent antitumor activities with different mechanisms of action3 from TXL. Some EP derivatives, such as etoposide (2d), topotecan, and irinotecan, are approved by the FDA for cancer treatment, although their therapeutic use is often limited by undesired side effects, such as myelosuppression, multidrug resistance and poor water solubility. In addition, glycyrrhetinic acid (GA),7 which is a well-known natural compound isolated from plants, shows antitumor-promoter activity, as well as anti-allergic, anti inflammatory, and anti-ulcer effects.
Figure 1.
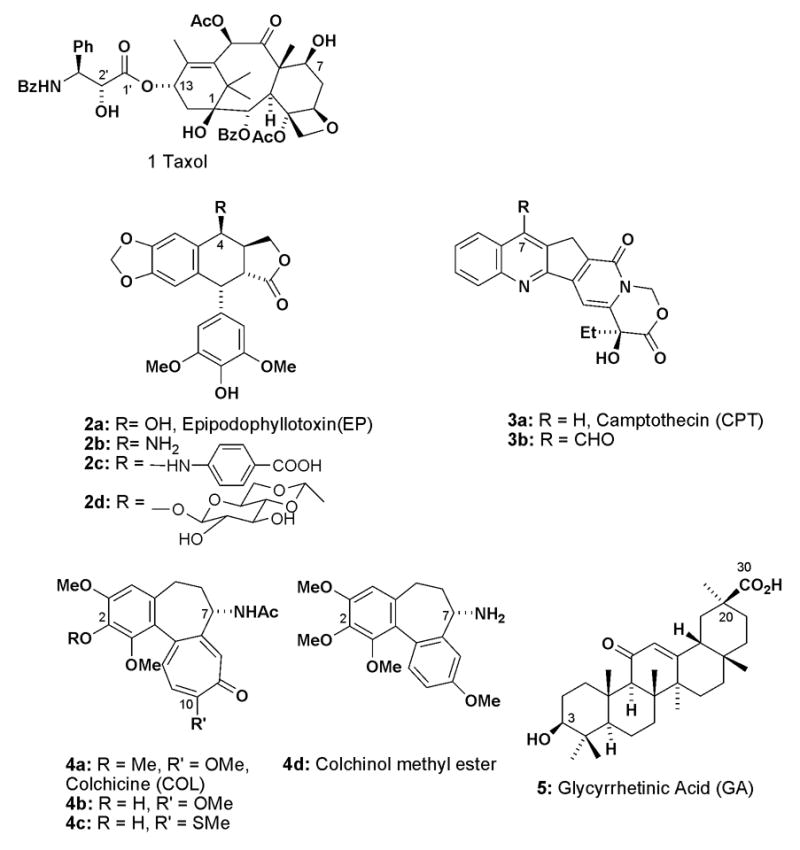
Structures of paclitaxel (1) and other antitumor agents used for conjugation.
Conjugation of two antitumor agents with different mechanisms of action can possibly augment the potency of both compounds or reduce the side effects and drug-resistance development. We have already reported the unique antitumor activities of such conjugates, including TXL-CPT,8 TXL-EP,9 and CPT-EP.10 In our continuous effort to develop new classes of antitumor agents based on TXL, we investigated the syntheses and evaluation of TXL conjugates with the above mentioned antitumor or antitumor-promoter compounds through various linkages at the TXL C-2’ and C-7 positions as reported herein.
Newly synthesized conjugates are shown in Figure 2. In conjugates 6–15, EP, CPT, COL, and GA are connected to the TXL C-2’ position through various linkages, while conjugates 16–21 contain EP, COL, and GA connected to the C-7 position of TXL. As shown in Scheme 1, a linking group was introduced directly at the TXL C-2’ position using the appropriate anhydride or carboxylic acid to provide 22–25.11 For the C-7 linked conjugates, the TXL C-2’ hydroxyl group was first protected with a Cbz or TBS group, the linking carboxylic acid was added at C-7, and the protecting group was then removed to afford 26–28. Compounds 22–24, 26, and 27 were conjugated with EP derivative 2c,12 COL derivatives 4b–d,13 and GA (5), respectively, by using EDCI in the presence of DMAP to provide 6, 7, 11–13, 15–17, and 21. Heating intermediates 22 and 23 with 7-formyl-CPT (3b)14 in benzene produced the corresponding imines 8 and 10, respectively. In the same way, imines 14, 18, and 20 were prepared from 25 with 4d and 28 with 2b or 4d, respectively. Hydrogenation of 8 and 18 produced the corresponding amines 9 and 19.
Figure 2.
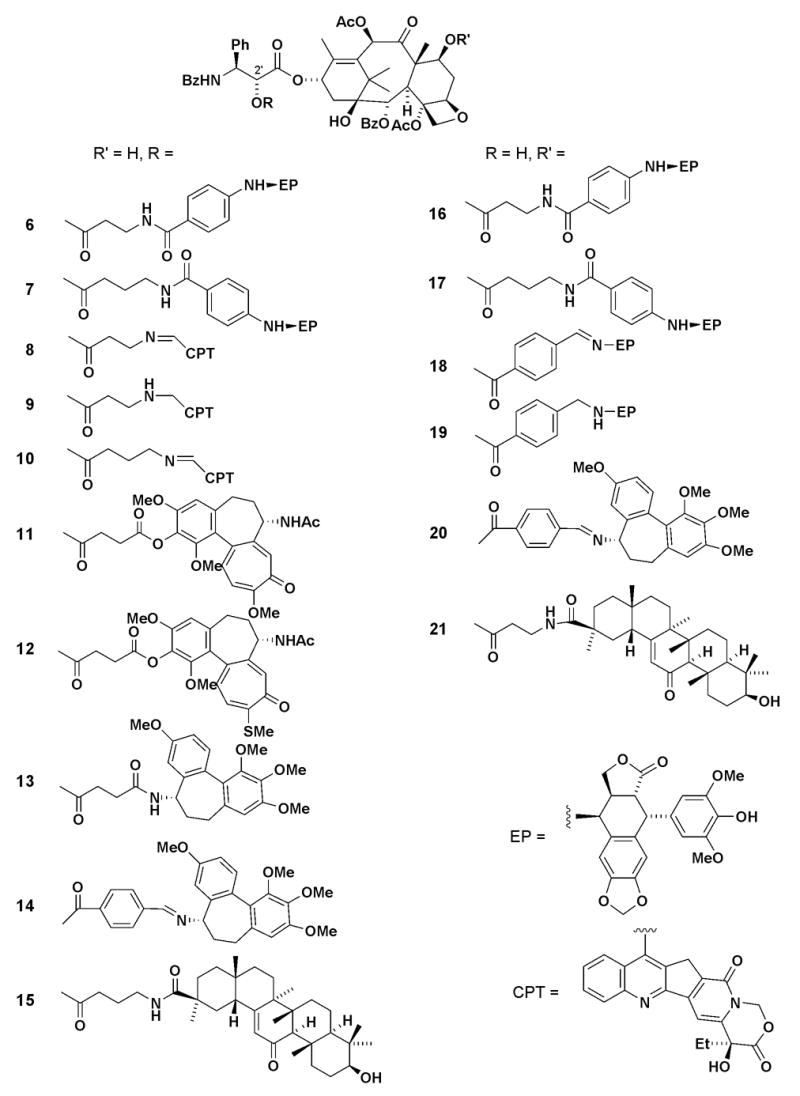
Structures of conjugates.
Scheme 1.
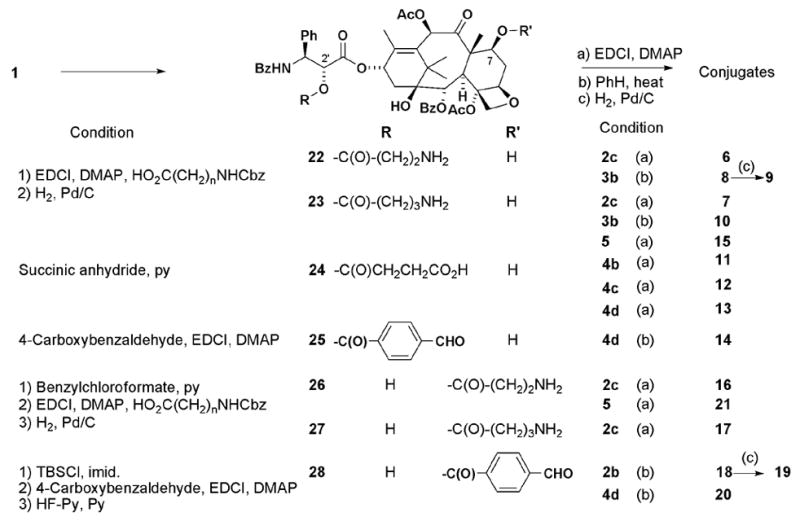
Synthesis of conjugates.
All synthesized compounds were evaluated for cytotoxic activity against replication of several human tumor cell lines, human ovarian carcinoma (1A9), human lung carcinoma (A549), breast cancer (MCF-7), human prostate carcinoma (LN-CAP, PC-3, DU-145), human epidermoid of the nasopharynx (KB), and multi-drug resistant expressing P-glycoprotein (KB-VIN) and against the normal cell line, human embryonic fibroblast (MRC-5). The results are shown in Table 1 with the values for TXL (1), EP derivatives 2b–c, etoposide (2d), CPT (3a), 7-formyl-CPT (3b), 2-demethyl-COL (4b), 2-demthylthio-COL (4c), and GA (5) for comparison. All conjugated compounds showed better activity than the partner compounds, although most of them were less potent than TXL itself. However, TXL-CPT conjugates 8–10 displayed different patterns of inhibition against the LN-CAP and PC-3 prostate cancer cell lines, with less effect on the normal cell line, MRC-5. Imine 10 had an ED50 value of 0.34 nM and was 7.7-fold more potent than TXL against LN-CAP cells, while compound 9 had an ED50 value of 3.1 nM and was 18-fold more potent than TXL against PC-3 cells.
Table 1.
Cytotoxic activity data of taxol conjugates
| Cmpd | ED50 (nM)a |
ED50 (μM)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 1A9 | A549 | MCF-7 | LN-CAP | PC-3 | DU-145 | KB | KB-VIN | MRC-5 | |
| 1 | 1.0 | 2.3 | 1.1 | 2.6 | 55.5 | 1.3 | 1.8 | 311 | NT a |
| 2b | 739 | 1074 | 686 | 680 | 1234 | 3107 | 111.2 | 2050 | NT |
| 2d | 284.9 | 1609 | 11659 | 530.1 | 9452 | 1600 | 1365 | 14447 | >30 |
| 3a | 3.2 | 6.9 | 3.2 | 2.0 | 16.7 | 10.9 | 20.9 | 15 | NT |
| 3b | 4.3 | 12.8 | 4.3 | 4.3 | 18.4 | 7.7 | 80.6 | 25 | NT |
| 4b | 183.7 | 950.6 | 361.0 | 324.7 | 475.3 | 611.9 | 146.1 | 28977 | NT |
| 4c | 43.6 | 99.8 | 59.9 | 59.9 | 112.2 | 113.5 | 47.9 | 7426 | NT |
| 5 | >20 b | >20 b | >20 b | >20 b | >20 b | >20 b | >20 b | >20 b | NT |
| 6 | 2.9 | 10.1 | 6.4 | 4.2 | 12.1 | 8.6 | 8.4 | 9964 | >10 c |
| 7 | 8.1 | 23.7 | 22.1 | 10.2 | 42.8 | 32.5 | 13.9 | 6231 | 6174 |
| 8 | 1.6 | 2.6 | 26.7 | 3.0 | 14.8 | 3.7 | 1.3 | 62.4 | 177.7 |
| 9 | 1.5 | 2.6 | 2.7 | 1.2 | 3.1 | 2.3 | 1.9 | 2050 | 6133 |
| 10 | 1.0 | 1.9 | 34.1 | 0.34 | 19.4 | 1.9 | 1.5 | 56.3 | 243.6 |
| 11 | 10.6 | 27.3 | 26.5 | 12.9 | 50.7 | 31.9 | 19.0 | >20 b | NT |
| 12 | 10.3 | 23.2 | 24.7 | 19.5 | 71.8 | 29.8 | 17.0 | >20 b | NT |
| 13 | 14.0 | 28.5 | 29.2 | 23.7 | 70.4 | 34.4 | 21.57 | >20 b | NT |
| 14 | 2.3 | 3.8 | NT | NT | NT | NT | 5.4 | 308 | NT |
| 15 | 45.3 | 77.4 | 38.7 | 46.2 | 103.6 | 68.3 | 53.6 | >20 b | NT |
| 16 | 20.7 | 45.6 | 43.9 | 22.1 | 86.3 | 83.4 | 31.6 | >20 b | 3279 |
| 17 | 104 | 1319 | 125.0 | NT | NT | NT | 347.2 | NA | NT |
| 18 | 60.1 | 111.0 | 48.6 | 90.1 | 145.3 | 139.1 | 53.8 | 6220 | NT |
| 19 | 75.7 | 123.5 | 68.2 | 128.5 | 191.5 | 167.5 | 87.4 | 5200 | NT |
| 20 | 2.3 | 3.8 | NT | NT | NT | NT | 2.3 | 308 | NT |
| 21 | 154.3 | 224.8 | 129.8 | 192.7 | 652.1 | 311.0 | 147.3 | >20 b | NT |
Human ovarian carcinoma (1A9), human lung carcinoma (A549), breast cancer (MCF-7), human prostate carcinoma (LN-CAP, PC-3, DU-145), human umbilical vein endothelial cell (HUVEC), human epidermoid carcinoma of the nasopharynx (KB), multi-drug resistant expressing P-glycoprotein (KB-VIN) and human embryonic fibroblast (MRC-5).
Cytotoxicity as ED50 values for each cell line, the concentration of compound that caused 50% reduction in absorbance at 562 nm relative to untreated cells using the sulforhodamine B assay.
Test compound (20 μg/mL) did not reach 50% inhibition.
Cytotoxic activity was somewhat dependant on the conjugated position on TXL as well as the type of linkage. From comparison of the TXL-EP conjugates 6, 7, 16, and 17, the conjugates linked at the C-2’ position (6, 7) showed better activity than conjugates linked at the C-7 position (16, 17) against all cell lines. Moreover, the linkage with two methylenes (6, 16) gave better results than the one with three methylenes (7, 17). However, TXL-COL conjugates 14 (2’-linkage) and 20 (7-linkage) showed similar potency against most cell lines. A phenyl imino linkage was better than the linear amido linkage, as TXL-COL conjugate 14 was four- to nine-fold more potent than 13 against 1A9, A549, and KB cells.
The results of selected compounds in an anti-angiogenesis assay are shown in Table 2. Compared with the other conjugates, imines 8 and 10 possessed significant activity with ED50 values of 0.73 and 0.98 μg/mL, respectively.
Table 2.
Anti-angiogenesis assay data of selected compounds in HUVEC cells
| Compound | ED50 (nM) | Compound | ED50 (nM) |
|---|---|---|---|
| 6 | 4.93 | 13 | 15.8 |
| 8 | 0.73 | 14 | 43.0 |
| 9 | 2.09 | 18 | 230 |
| 10 | 0.98 | 20 | 293 |
| 11 | 13.84 | 21 | 150 |
| 12 | 9.33 |
In conclusion, we have synthesized fifteen different conjugates between paclitaxel (TXL) and other antitumor agents, including camptothecin (CPT), epipodophyllotoxin (EP), colchicine (COL), and glycyrrhetinic acid (GA). The two components were joined by an ester, imine, amine or amide linkage at the 2’- or 7-position of TXL. Among them, TXL-CPT conjugates, 8–10, showed different in vitro cytotoxic activity profiles against human prostate carcinoma, LN-CAP and PC-3, with less effect against normal cells. These compounds also possessed anti-angiogenesis ability; therefore, conjugates 8–10 are possible antitumor drug candidates, particularly for prostate cancer.
Acknowledgments
This study was supported by grant CA-17625 from the National Cancer Institute, NIH, awarded to K. H. L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT, Sim GA. J Am Chem Soc. 1971;93:2325. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 2.Itokawa H, Lee KH. Taxus. Taylor & Francis; London and New York: 2003. [Google Scholar]
- 3.Islam MN, Iskander MN. Mini-Rev in Med Chem. 2004;4:1077. doi: 10.2174/1389557043402946. [DOI] [PubMed] [Google Scholar]
- 4.Recent review, Li QY, Zu YG, Shi RZ, Yao LP. Curr Med Chem. 2006;13:2021. doi: 10.2174/092986706777585004.
- 5.Lee KH, Zhiyan X. Anticancer Agents from Natural Products. 2005:71. [Google Scholar]
- 6.Capraro HG, Brossi A. In: The Alkaloids. Brossi A, editor. Academic Press; New York: 1984. [Google Scholar]
- 7.Recent review, Baltina LA. Curr Med Chem. 2003;10:155. doi: 10.2174/0929867033368538.
- 8.Ohtsu H, Nakanishi Y, Bastow KF, Lee FY, Lee KH. Bioorg Med Chem. 2003;11:1851. doi: 10.1016/s0968-0896(03)00040-3. [DOI] [PubMed] [Google Scholar]
- 9.Shi Q, Wang HK, Bastow KF, Tachibana Y, Chen K, Lee FY, Lee KH. Bioorg Med Chem. 2001;9:2999. doi: 10.1016/s0968-0896(01)00206-1. [DOI] [PubMed] [Google Scholar]
- 10.Bastow KF, Wang HK, Cheng YC, Lee KH. Bioorg Med Chem. 1997;5:1481. doi: 10.1016/s0968-0896(97)00102-8. [DOI] [PubMed] [Google Scholar]
- 11.Deutsh HM, Glinski JA, Hernandez M, Haugwits RD, Narayanan VL, Suffness M, Zalkow LH. J Med Chem. 1989;32:788. doi: 10.1021/jm00124a011. [DOI] [PubMed] [Google Scholar]
- 12.Xiao Z, Bastow KF, Vance JR, Sidwell RS, Wang HK, Chen MS, Shi Q, Lee KH. J Med Chem. 2004;47:5140. doi: 10.1021/jm030609l. [DOI] [PubMed] [Google Scholar]
- 13.a) Nakagawa-Goto K, Chen CX, Hamel E, Wu CC, Bastow KF, Brossi A, Lee KH. Bioorg Med Chem Lett. 2005;15:235. doi: 10.1016/j.bmcl.2004.07.098. [DOI] [PubMed] [Google Scholar]; b) Nakagawa-Goto K, Jung MK, Hamel E, Wu CC, Bastow KF, Brossi A, Ohta S, Lee KH. Heterocycles. 2005;65:541. [Google Scholar]
- 14.a) Wang HK, Liu SY, Hwang KM, Taylor G, Lee KH. Bioorg Med Chem. 1994;2:1397. doi: 10.1016/s0968-0896(00)82091-x. [DOI] [PubMed] [Google Scholar]; b) Miyasaka T, Sawada S, Nokata K. Heterocycles. 1981;16:1719. [Google Scholar]