

Abstract
Prolonged exposure to organophosphate (OP) pesticides may produce cognitive deficits reflective of hippocampal injury in both humans and rodents. Recent work has indicated that microtubule trafficking is also adversely affected by exposure to the OP pesticide chlorpyrifos, suggesting a novel mode of OP-induced neurotoxicity. The present studies examined effects of prolonged exposure to chlorpyrifos-oxon (CPO) on acetylcholinesterase (AChE) activity, immunoreactivity (IR) of microtubule-associated proteins, neuronal injury, and tubulin polymerization using in vitro organotypic slice cultures of rat hippocampus and bovine tubulin. Cultures were exposed to CPO (0.1-10 μM) in cell culture medium for 1-7 days, a regimen producing progressive reductions in AChE activity of 15-60%. Cytotoxicity (somatic uptake of the non-vital marker propidium iodide, as well as, IR of α-tubulin and microtubule-associated protein-2 (a/b) [MAP-2] were assessed 1, 3, and 7 days after the start of CPO exposure. As early as 24- hr after the start of exposure, CPO-induced deficits in MAP-2 IR were evident and progressive in each region of slice cultures at concentrations as low as 0.1 μM. CPO exposure did not alter α-tubulin IR at any time point. Concentration-dependent injury in the CA1 pyramidal cell layer and to a lesser extent, CA3 and dentate cells, was evident 3 days after the start of CPO exposure (≥ 0.1 μM) and was greatest after 7 days. Tubulin polymerization assays indicated that CPO (≥ 0.1 μM) markedly inhibited the polymerization of purified tubulin and map-rich tubulin, though effects on MAP-rich tubulin were more pronounced. These data suggest that exposure to CPO produces a progressive decrease in neuronal viability that may be associated with impaired microtubule synthesis and/or function.
The broad spectrum organophosphorus (OP) insecticide chlorpyrifos (O,O-diethyl O-3,5,6-trichloro-2-pyridinyl phosphorothioate; CPF) is an inhibitor of cholinesterases, including acetylcholinesterase (AChE), and has until recently been widely used in residential settings in the United States. This compound remains one of the most widely used pesticides in agricultural settings. Emerging evidence, obtained largely through the use of rodents, suggests that acute or prolonged exposure to CPF and/or its metabolic product(s) may overtly injure the central nervous system or produce marked changes in neuronal function that persist after exposure has ceased, particularly during the early postnatal period (Olivier et al. 2001; Slotkin et al. 2001; Zheng et al. 2000). However, it must be noted that not all studies find evidence of persisting CNS abnormalities following the cessation of CPF exposure (Padilla et al. 2005).
While much is known about the lethality and neurotoxicity produced by acute CPF exposure, relatively little is known about the means by which chronic exposure to this compound, particularly low concentrations of CPF, may adversely affect neuronal function. Examining this issue is likely to be of significance in understanding the potential consequences of CPF exposure both in agricultural workers and in those exposed to CPF for prolonged periods of time in residential or educational settings. Prolonged exposure to CPF has recently been shown to produce delayed learning impairments in rodents (Bushnell et al. 1994; Terry et al. 2003) and neuropathies in humans (Kaplan et al. 1993) that are not correlated with the presence of overt toxicity. These effects are distinct from the OP-induced delayed neuropathies (OPIDN) produced by other OP agents largely because of the relatively greater potency of CPF in inhibiting AChE, as compared to neuropathy target esterase (Kropp and Richardson 2003), though OPIDN may be produced following administration of CPF doses in excess of its LD50 (Lotti et al. 1986; Richardson et al. 1993).
It has been widely postulated that CPF toxicity depends, in large part, on rapid CYP450-dependent metabolism of CPF to its oxygen analog CP-oxon (CPO), which is markedly more potent than CPF at inhibiting AChE (see Richardson 1995, for review). This may have significant public health implications given recent evidence of abiotic hypochlorous acid-dependent metabolism of CPF to CPO in chlorinated water (Wu and Laird 2003). However, in primary neuronal cell culture systems demonstrating neurotoxicity during CPF exposure, it is unclear to what extent CPO formation may occur, though applied CPO has demonstrated greater potency in this regard (Caughlan et al. 2004; Roy et al. 1998; Terry et al. 2003). One recent study has demonstrated that CPF does retain its ability to inhibit axonal outgrowth in the presence of the CYP-450 inhibitor SKF-525A (Howard et al 2005), suggesting that both the parent compound and CPO possess the ability to injure neurons. The hydrolytic product of CPF metabolism, 3,5,6-trichloro-2-pyridinol, is ineffective at producing neurotoxicity (Caughlan et al. 2004).
Prolonged inhibition of AChE by CPF or CPO may well contribute to the toxicity observed during CPF exposure, however, the extent of AChE inhibition does not readily correlate with behavioral abnormalities produced by CPF exposure (Howard et al. 2005; Terry et al. 2003). Additional modes of CPF-induced neurotoxicity have been proposed and include inhibition of numerous serine hydrolases (Casida and Quistad 2005); co-valent modification of M2 muscarinic receptors with resulting reductions in cellular cAMP content (Huff et al. 2001); inhibition of cannabinoid 1 receptors (Quistad et al. 2002) and impairment of microtubule function, reflected in inhibition of both fast antero- and retrograde axonal transport (Terry et al. 2003). These latter data are of particular interest in suggesting that microtubules, and possibly proteins such as α- and/or β-tubulin or microtubule-associated proteins, may be novel substrates for CPF and/or CPO action in the CNS. Alterations in tubulin polymerization, which may occur with changes in MAP-2 or α/β-tubulin activity or expression, induces the activity of multiple pro-apoptotic proteins (ie. Bax, Bid, Bim), mitochondrial release of cytochorome C and activation of effector caspases downstream of caspase 9 (Giacca 2005). This suggestion is consistent with recent findings that CPF and CPO exposure produces apoptosis in developing rodent brain (Caughlan et al. 2004; Roy et al. 1998).
The present studies examined the extent to which short-term and prolonged exposure to CPO altered neuronal viability, as well as, immunoreactivity of α-tubulin and microtubule-associated proteins (MAP) 2a and 2b in organotypic slice cultures of immature rat hippocampus. Further, these studies examined the extent to which CPO would alter polymerization of both purified and MAP-rich bovine tubulin in vitro.
EXPERIMENTAL PROCEDURES
Organotypic Hippocampal Slice Preparations
Complete brains from 8-day-old male and female Sprague-Dawley rat pups were aseptically extracted and transferred to dissection medium (4°C), consisting of Minimum Essential Medium (MEM) plus 25mM HEPES, 2mM L-glutamine, and 50μM streptomycin/penicillin. Bilateral hippocampi were dissected out and placed into chilled culture medium. Culture medium consisted of dissection medium with the addition of sterile H2O, 36mM glucose, 25% Hanks’ balanced salt solution (HBSS), and 25% heat-inactivated horse serum (HIHS). Unilateral hippocampi were coronally sectioned at 200μm using a McIllwain tissue chopper (Mickle Laboratory Engineering Co. Ltd., Gomshall, UK), yielding approximately 12 slices per unilateral hippocampus, and transferred to fresh chilled culture medium. Three slices of hippocampus were placed onto each Millicell-CM (0.4μm) biopore membrane insert with 1ml culture medium pre-warmed to 37°C and applied to the bottom of each well. Inserts were placed into 35mm 6- well culture plates and kept at 37°C in an incubator containing 5% C02, 21% oxygen, and 74% nitrogen with 95% humidity. After 5 days of slice attachment to inserts and stabilization in culture medium, experiments were conducted as described below. Treatment of all animals was carried out in accordance with the National Institute of Health Guide for the care and use of laboratory animals (NIH Publications No. 80-23). All HIHS was supplied by Sigma-Aldrich, Co. (St. Louis, MO, USA), and all other culture medium solutions were supplied by Gibco-BRL (Gaithersburg, MD, USA). All experiments were performed a minimum of three times, utilizing different litters of pups.
Chlorpyrifos Oxon (CPO) Exposure
At 5 DIV, slice cultures were transferred to new six-well plates containing 1 ml of regular cell culture medium on the bottom. Slices from a given rat were distributed throughout different treatment groups (N = 15-18/group for all studies). Cell cultures were exposed to medium (1 ml) alone, medium containing 2.5 μg/ml propidium iodide (3,8-diamino-5-(3-(diethylmethylamino) propyl)-6-phenyl phenanthridinium diiodide; PI) or medium containing PI and CPO (0.1-10 μM), which was slowly introduced to the top of insert membranes. Propidium iodide is a highly stable, polar fluorescent dye that only penetrates the membranes of damaged, and/or potentially dying cells, thereby binding nucleic acids and emitting a bright, intensified red fluorescence upon excitation, as described below (Zimmer et al. 2000). All cultures were then placed back in an incubator and were removed from the incubator 1, 3, or 7 days after the start of CPO exposure. PI fluorescence, as well as IR of microtubule-associated proteins was assessed as described below.
Measurement of Acetylcholinesterase Activity
Following 1, 3, or 7 days of exposure to CPO, 3 hippocampal slice cultures from a portion of wells were removed from membranes, placed into a microfuge tube with 120μl of PBS and sonicated for 14 sec using a Vibra Cell sonicating wand (Sonics and Materials, Inc., Newtown, CT) to homogenize tissue. Acetylcholinesterase (AChE) activity was assessed, after Ellman (1961) with modifications. Tissue homogenate (50 μl/sample) was added to wells of a 96-well plate containing 250 μl of the reaction mixture (4.8 μM acetylthiocholine iodide and 321 μM dithiobisnitrobenzoate in 0.1 M Na2HPO4 buffer, pH 8.0; Sigma, St. Louis, MO). Plates were then immediately loaded into a Beckman Coulter DTX 880 multimodal detector (Fullerton, CA) and absorbance at 412 nm was measured every 5 min for 60 min. The rate of AChE activity was then calculated for each time point of measurement using the formula (Δ absorbance/min)/(1.36 × 104).
Measurement of Immunoreactivity and Neurodegeneration
To measure IR of α-tubulin and MAP-2, cultures were washed twice in 0.9% phosphate-buffered saline and fixed for 30-min in 10% paraformaldehyde. Cultures were subsequently washed twice again in PBS and incubated for 45-min in PBS buffer containing 0.1% Triton-X and .005% bovine serum albumin to permeabilize membrane. Following two washes in PBS, cultures were incubated for 24-hr at 4°C in permeabilization buffer containing monoclonal primary antibodies (1/200 dilution) against mouse anti-bovine MAP-2 (2a + 2b) that reacts with rat MAP-2a and b or a mouse α-tubulin antibody (that strongly reacts with rat α-tubulin) raised against filaments of Strongylocentrotus purpuratus sperm axonemes (Sigma, St. Louis, MO). Following the 24-hr incubation with primary antibody, cultures were washed twice with PBS and placed in wells containing permeabilization buffer with the addition of a fluorescein isothiocyanate (FITC)- or tetramethylrhodamine isothiocyanate (TRITC)-conjugated secondary antibody (goat anti-mouse) for 24-hr at 4°C. After this incubation, slice cultures were washed twice in PBS and imaged, as described below.
Cell damage, as well as immunoreactivty of MAP-2 (2a + 2b) and α-tubulin, were subsequently detected by fluorescent microscopy. Fluorescence of PI, FITC-, or TRITC-conjugated antibodies was visualized with SPOT Advanced version 4.0.2 software for Windows (W. Nuhsbaum Inc., McHenry, IL) using a 5X or 20X objective on an inverted Leica DMIRB microscope (W. Nuhsbaum Inc.) fitted for fluorescence detection (mercury-arc lamp) and connected to a personal computer via a SPOT 7.2 color mosaic camera (W. Nuhsbaum Inc.; McHenry, IL). Propidium iodide fluorescence and secondary antibodies conjugated with TRITC were excited using a band-pass filter at 515-560 nm (615 nm emission) while those antibodies conjugated with FITC were excited using a band-pass filter at 495 nm (520 nm emission). Fluorescence of all fluorophors was measured only in the primary neuronal cell layers of the hippocampal complex: the molecular, granule and polymorphic layers of the dentate gyrus and the pyramidal cell layers of CA1 and CA3. These regions were outlined in whole and mean pixel intensity of PI fluorescence and were analyzed by densitometry using Image J 1.29x (National Institutes of Health, Bethesda, MD).
Tubulin Polymerization
Purified (< 3% MAP) and MAP-rich bovine α/β-tubulin dimer (> 99% pure) were obtained from Cytoskeleton, Inc. (Boulder, CO). Purified (MAP-deficient) tubulin (1.8 mg/ml) was diluted in tubulin polymerization buffer (pH 6.9) consisting of 80 mM piperazine-N,N’-bis[2-ethanesulfonic acid] sequisodium salt; 2.0 mM MgCl2; 0.5 mM ethylene glycol-bis(b-amino-ethyl ether) N,N,N’,N’-tetra-acetic acid (G-PEM). MAP-rich tubulin is diluted in G-PEM to produce a final protein concentration of 1.0 mg/ml. Paclitaxel, which promotes microtubule polymerization via interactions with β-tubulin, was employed as a positive control in all assays and was incubated for the 60 min kinetic assay (described below) with both purified and MAP-rich tubulin in G-PEM buffer at a concentration of 10 μM. To examine effects of CPO on polymerization of both purified and MAP-rich tubulin, both tubulins were diluted in G-PEM containing CPO (0.1-10.0 μM). Upon dilution to final concentrations, all samples were immediately aliquoted (100 μl) into a 96-well plate and inserted into a pre-warmed (37°) Beckman-Coulter DTX 880 Multimodal Detector (Beckman-Coulter, Fullerton, CA). Absorbance at 340 nm was measured every 5 min for 60 min, after Lee and Timasheff (1977), as well as, Shelanski et al. (1973). All assays were replicated in quadruplicate (N = 12-16).
Statistical Analysis
To examine effects of CPO on AChE activity, a two-way repeated measures ANOVA was used to compare the rate of AChE activity at each time point of measurements, as reflected in the in moles/liter/min after Ellman et al. (1961). For each slice stained with propidium iodide or antibody-conjugated fluorophores, a background optical intensity measurement was obtained and subtracted individually from measurements obtained for the primary cell body layer of each culture subregion (dentate gyrus, CA3, and CA1) prior to statistical analysis to account for potential daily variability in camera performance. Initially, three-way ANOVA (analysis of variance) analyses were conducted, using Sigma Stat software, to identify any significant effects across hippocampal region (dentate gyrus vs. CA3 vs. CA1), treatment, and gender groups. After no significant three-way interactions were observed, data for male and female cultures were collapsed for subsequent studies and analyzed using a two-way ANOVA in order to compare effects of different treatment groups within each hippocampal subregion (dentate gyrus vs. CA3 vs. CA1) at different times of observation. The Tukey test was used for all two-way ANOVAs as a post hoc analysis tool when appropriate with alpha < .05 used as significance criteria. Data were converted to percent control for the presentation of figures.
RESULTS
Acetylcholinesterase (AChE) Activity
Following 1 day of exposure to the two highest concentrations of CPO (1.0 and 10.0 μM), AChE activity in the slice cultures was reduced by approximately 50% [F(3,206)= 107.86, P < 0.001], though no marked differences in AChE activity between these two groups were noted (post hoc= P <0.05 vs control and 0.1 μM). Exposure to the 0.1 μM concentration of CPO did not reduce AChE activity at this time point. In contrast, 3 days of exposure to CPO, at each concentration, markedly reduced AChE activity [F(3,187)= 10.68, p < 0.001], as compared to control values (post hoc = P < 0.05). Similar inhibition of AChE was observed following 7 days of exposure to each concentration of CPO [F(3,374)=54.11, P <0.001), though concentration dependence was observed in that AChE activity in tissue exposed to 10 μM CPO was significantly lower than that measured in tissue exposed to the lower concentrations of CPO, as well as controls (post hoc= P < 0.05). Effects of CPO exposure on AChE activity following 1, 3, or 7 days of exposure are shown in Figure 1.
Figure 1.
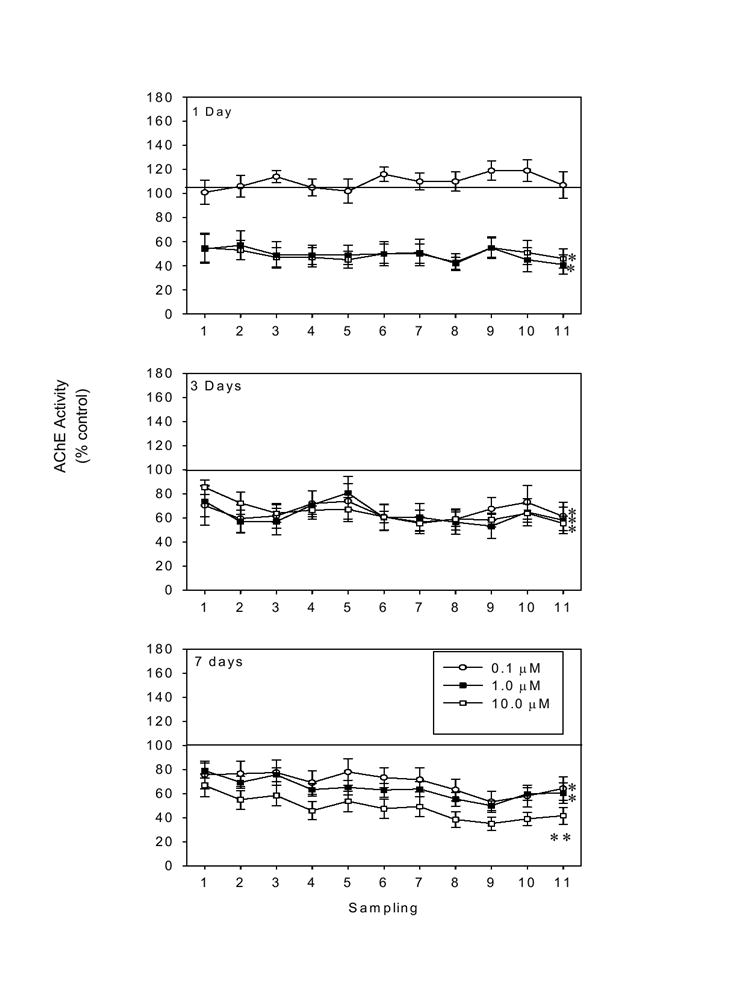
Effects of 1, 3, or 7 days of CPO exposure (0.1-10.0 μM) on acetylcholinesterase (AChE) activity (as % control) in homogenized hippocampal slice cultures. Significant inhibition of AChE activity was evident at 1 day (1.0-10.0 μM) and was sustained for 7 days, particularly with exposure to the highest concentration of CPO. * =P < 0.05 vs control; **=P < 0.05 vs all other groups.
α-tubulin/MAP-2 Immunoreactivity
Immunoreactivity (IR) of both microtubule-associated proteins was measured after 1, 3, and 7 days of continuous exposure to CPO (0.1-10 μM). α-tubulin IR was not altered by CPO exposure, at any time point of observation (data not shown). In contrast, CPO exposure produced marked decrements in immunoreactivity of MAP-2, evident as early as 1 day after the start of CPO exposure (Figure 2). This initial time point (1 day) is particularly interesting in this regard given that neurotoxicity was not evident. At this time, MAP-2 IR in the primary neuronal layers of the dentate gyrus [F(3,128)=2.86; P < 0.05], CA3 [F(3,124)=2.96; P < 0.05], and CA1, [F(3,128)=3.85, P < 0.001] regions was reduced by approximately, 18%, 18%, and 12%, respectively, following exposure to the highest concentration of CPO (post hoc = P < 0.05 versus control). However, three days of exposure to CPO (1-10 μM) produced significant reductions in MAP-2 immunoreactivity (to 75%-85% of control values) in each region of organotypic slice cultures (post hoc= P < 0.05 vs control).
Figure 2.
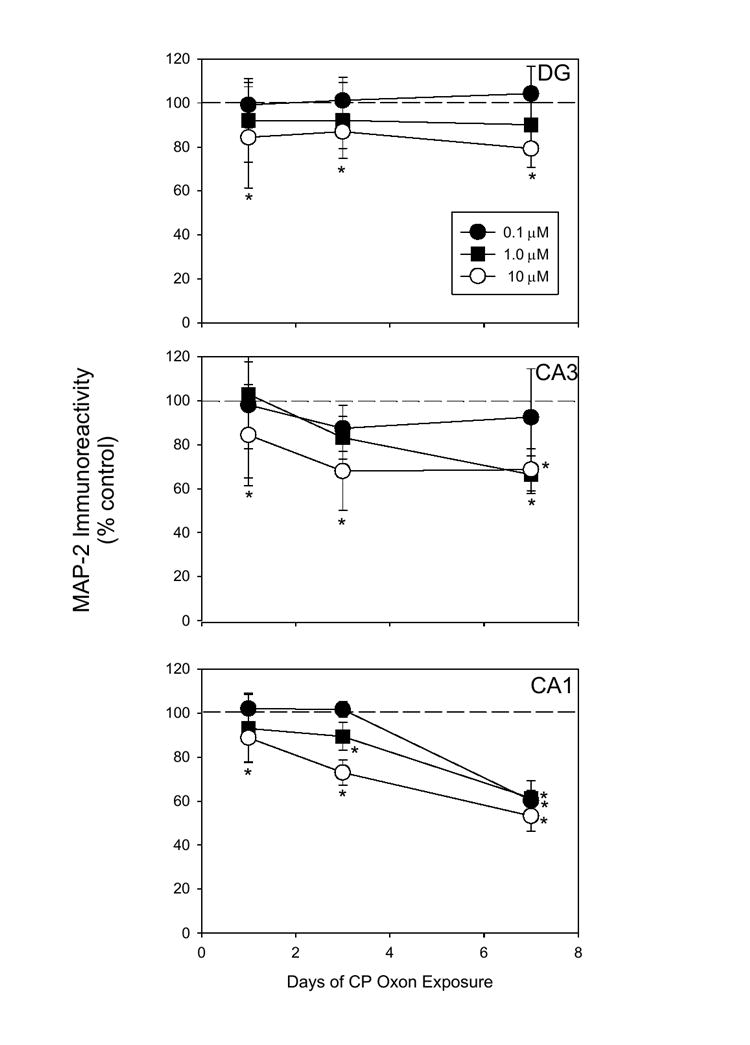
Effects of CPO exposure (0.1-10.0 μM) on immunoreactivity (IR) of MAP-2a/b (as % control) in hippocampal slice culture subregions 1, 3, and 7 days after the start of CPF exposure. Reductions in MAP-2a/b were evident in the primary neuronal layers of each region beginning on day 1 (≥1.0 μM) and progressively worsened in the pyramidal cell layers of CA1 and CA3 (≥0.1 μM). * = P < 0.05 vs control.
Dramatic reductions in MAP-2 immunoreactivity were observed following 7 days of exposure to CPO, primarily in the CA1 and CA3 subregions of slice cultures. In these regions, exposure to each concentration of CPO, including 0.1 μM, produced reductions in MAP-2 immunoreactivity of 35%-45%. In the dentate gyrus, MAP-2 immunoreactivity was not reduced at the 7 day time point, as compared to the 3 day time point. As with the 3 day time point, exposure to either of the two higher concentrations of CPO produced significant reductions in MAP-2 immunoreactivity.
Propidium Iodide (PI)
In the CA1 and CA3 regions of cultures, CPO exposure produced both time- and concentration-dependent increases in neuron injury, as reflected in increased PI fluorescence with each additional measurement. No regional injury was observed 24 hrs after the start of CPO exposure, with any concentration of CPO, though increases in PI uptake of approximately 8% were observed in tissue exposed to the two highest concentrations of CPO in the CA1 region. After three days of exposure to CPO, significant increases in PI uptake [significant effect for treatment; F(3,100) =7.64, P < 0.001; post hoc = P < 0.05] were evident in the CA1 region (to approximately 122% of control values); CA3 region [F(3,97)=4.45; P < 0.05] (to approximately 135% of control values); and dentate gyrus [F(3,97)=3.50; P <0.05] (to approximately 121% of control values). PI uptake caused by exposure to the two highest concentrations of CPO in the CA1 and CA3 regions was significantly greater than that in controls and those exposed to the lowest concentration of CPO. When measured 7 days after the start of CPO exposure, PI fluorescence had increased markedly above that observed at earlier time points in the pyramidal cell layer of the CA3 (to approximately 145-165% of control levels) and CA1 (to approximately 135-163% of control values) regions following exposure to each concentration of CPO (Figure 3). No further elevations in PI uptake were observed in dentate gyrus after the 3 day observation time. A significant effect of time was also observed, post hoc analysis indicated that PI fluorescence was significantly greater at 7 days, than at all other time points of observation, in all groups. Representative images of PI fluorescence and MAP-2 immunoreactivity are illustrated in Figures 4 (20-40X magnification) and 5 (5X magnification), respectively.
Figure 3.

Effects of CPO exposure (0.1-10.0 μM) propidium iodide (PI) fluorescence (as % control) in hippocampal slice culture subregions 1, 3, and 7 days after the start of CPF exposure. CPO produced increases in PI fluorescence, evident by day 3 of exposure (≥10.0 μM) that progressively worsened in the pyramidal cell layers of CA1 and CA3 (≥1.0 μM). * = P <0.05 vs control. ** = P < 0.05 vs all other groups.
Figure 4.
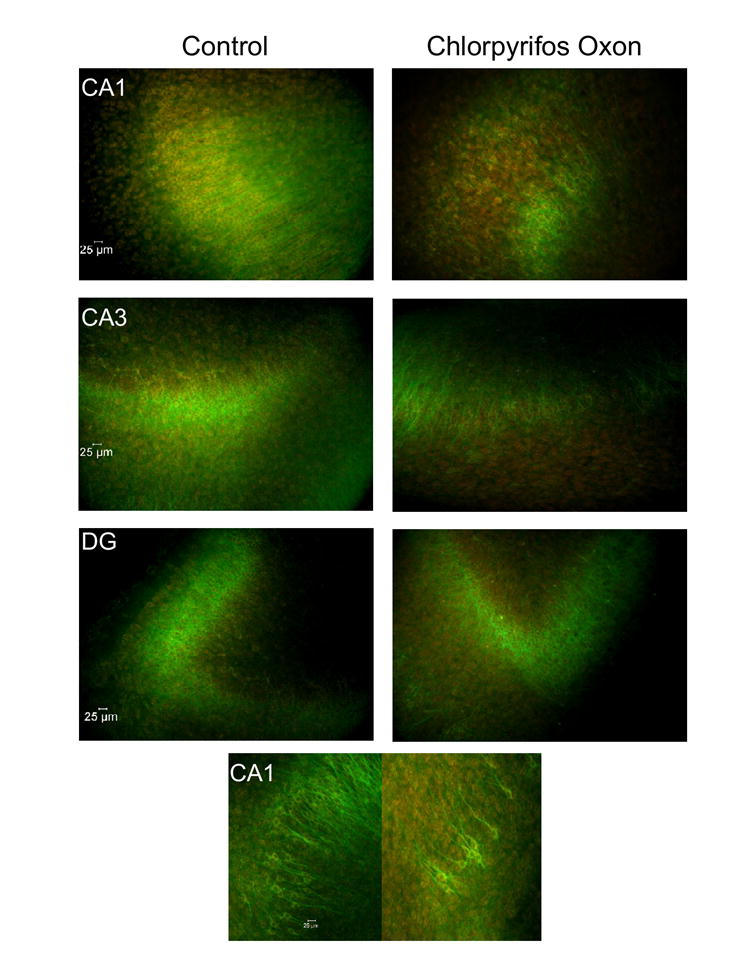
Representative images of MAP-2 immunoreactivity (FITC) in hippocampal cultures exposed to CPO at 20X (top, in all regions) and 40X (bottom, in CA1). MAP-2 images are merged with images of propidium iodide fluorescence (red), a marker of somatic injury.
Tubulin Polymerization
Purified and MAP-rich bovine tubulin were used to examine the effects of acute CPO exposure on tubulin polymerization. It was hypothesized, based on evidence of rapid losses in MAP-2 IR during CPO exposure, that CPO exposure would preferentially disrupt MAP-dependent, rather than MAP–independent, tubulin polymerization. Figure 6 illustrates the effects of MAP and the microtubule stabilizing agent paclictaxel on tubulin polymerization. Initial studies monitored polymerization for 60 min, however, no further polymerization was observed after 40 min in any group. A two-way analysis of variance revealed a significant effect of treatment [F(2,7) = 25.54, P < 0.001] while post hoc analysis indicated that both map-deficient and map-rich tubulin produced significant increases in polymerization that were further potentiated by the addition of paclitaxel. Paclitaxel co-exposure with purified tubulin produced more than a 100% increase in tubulin polymerization at each time point where polymerization of MAP-rich tubulin was increased by approximately 175-200%. Further, polymerization of MAP-rich tubulin was significantly greater (approximately 200%) than that of MAP-deficient tubulin, at each time point.
Figure 6.
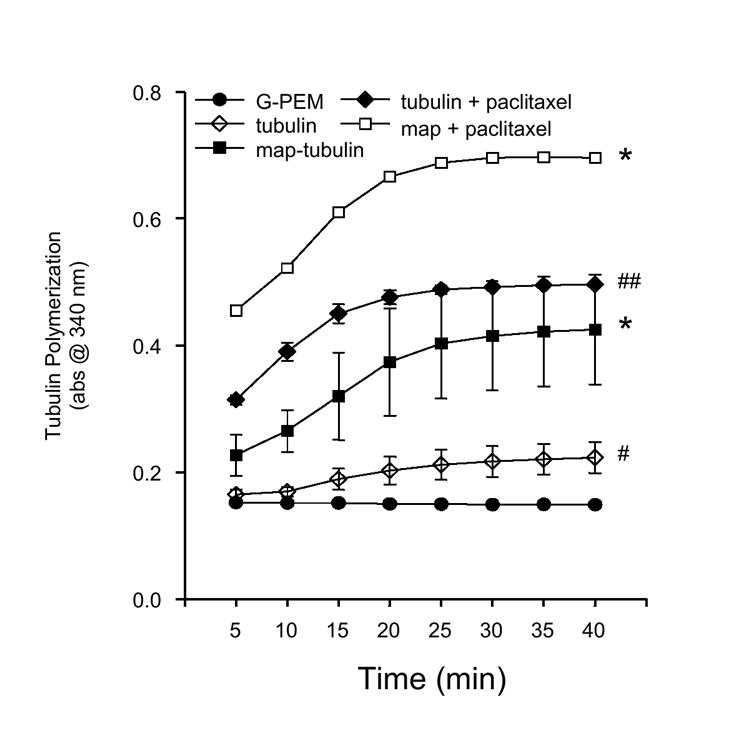
Comparison of the polymerization profiles of purified and MAP-rich tubulin. Polymerization of MAP-rich tubulin was markedly greater than that of purified tubulin. Exposure to the microtubule stabilizing agent paclitaxel increased total polymerization of MAP-deficient and MAP-rich tubulin . ** = P < 0.05 vs all groups; ## = P < 0.05 vs tubulin, G-PEM, MAP-tubulin; * = P < 0.05 vs tubulin, G-PEM; # = P < 0.05 vs control.
Analysis of the effects of CPO exposure of purified tubulin (MAP-deficient) polymerization produced a significant two-way interaction [F(28,80)=2.79, P < 0.001]. Post hoc analysis indicated that exposure to each concentration of CPO significantly reduced tubulin polymerization (~60-70%) at each time point with the exception of the first. Curiously, concentration-dependence of this effect was not observed , in that exposure to each concentration of CPO (0.1-10 μM) suppressed polymerization to a very similar extent (to 60-70% of control levels). CPO produced a 2-fold greater inhibition of polymerization of map-rich tubulin relative to map-deficient tubulin. In contrast to MAP-deficient tubulin, polymerization of MAP-rich tubulin was reduced in a concentration-dependent manner by CPO [F(3,400) = 7.02; P < 0.001; Figure 7). The lowest concentration of CPO (0.1 μM) reduced polymerization by 20-60% from the second to the last measurement, while the highest concentration (10.0 μM) reduced polymerization by 30-80% over the course of these measurements. The 1.0 μM concentration produced inhibition of polymerization that was nearly equal to that produced by the highest concentration. A significant effects of time was also observed [F(19,400)=6.67, P < 0.001]. Post hoc analysis indicated that polymerization at all time points after the 25 minute observation point was significantly greater than that observed during each of the first measurement periods.
Figure 7.
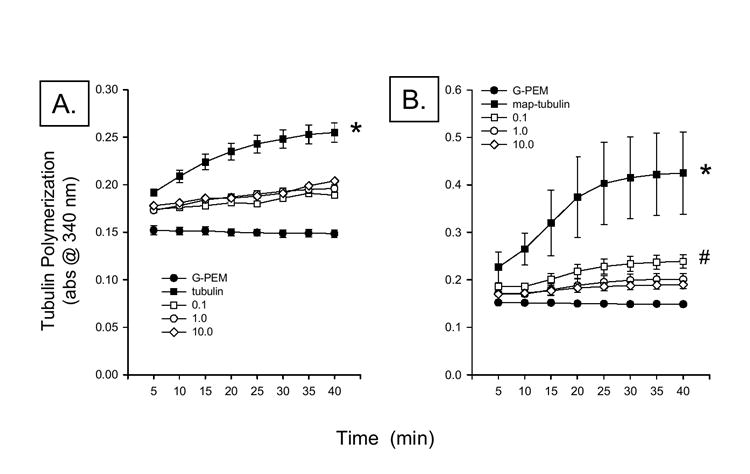
Effects of CPO (0.1-10 μM) on polymerization of purified (A.) and MAP-rich (B.) tubulin. CPO, at all concentrations, reduced polymerization by approximately 25%. In contrast, CPO reduced polymerization of MAP-rich tubulin by approximately 45-60%. * = P < 0.05 vs control (G-PEM) and CPO. # = P < 0.05 vs 10.0 μM CPO.
DISCUSSION
The present studies were designed to examine the effects of prolonged exposure to CPO on AChE activity, as well as, the bioavailability of microtubule-related proteins α-tubulin and MAP-2a/b in a neonatal rat slice culture preparation. Further, theses studies examined effects of CPO exposure on these proteins in parallel with the time course of cytotoxicity. Lastly, effects of CPO exposure on polymerization of purified and MAP-rich bovine tubulin were assessed to examine whether CPO affected microtubule formation in the absence of AChE. Findings of these studies suggest that CPO exposure produces concentration- and time-dependent reductions in hippocampal AChE activity and selective reductions in the availability and functional effects of MAP-2 proteins that precede neuronal injury. However, it must be noted that the relatively modest levels of AChE inhibition produced by exposure to the higher concentrations of CPO (~50-60%) were unexpected given evidence that the IC50 value of CPO for cholinesterases in rat brain is known to be in the low nM range (Mortensen et al. 1998). It is possible that the compound possess affinity for constituents of the heat-inactivated horse serum used in the medium, potentially affecting availability of free CPO. As such, it would be of use to examine these parameters using cell culture medium without the addition serum.
Deficits in MAP-2 immunoreactivity were evident in each slice culture region (dentate gyrus, CA3, and CA1) only 24 hours after the start of exposure to the highest concentration of CPO. Increased cytotoxicity, determined using propidium iodide uptake, was not evident until 72 hours. MAP-2 deficits in the CA1 were concentration-dependent. In the CA3 region, further reductions in MAP-2 were evident at 72 hours, including those produced by exposure to 1 μM CPO, but no further decline was observed at the later time point. In the dentate gyrus, a maximal reduction in MAP-2 availability occurred within 24 hours of starting CPO exposure. This heightened sensitivity of the CA1 region to injury may reflect a greater density of neurons since CA1 pyramidal cells display the greatest density of thionin staining using this organotypic preparation (Prendergast et al. 2001) and the most immunoreactivity for MAP-2 in CA1 projection layers. However, the predominant localization of cholinergic receptors in the hippocampus, at least nicotinic acetylcholine receptors, is in the hilus region (Prendergast et al 2001) and cholinergic afferents synapse almost exclusively in the molecular layer of the dentate gyrus (Tsai et al. 2002). Thus, it is unclear why pyramidal cells of the CA1 layer demonstrate the greatest sensitivity to the toxic effects of CPO.
It is critical to note that the concentration-response curves for severity (PI uptake) and time-course of cytotoxicity observed in each subregion were closely associated with those for MAP-2. More specifically, those concentrations of CPO that produced MAP-2 deficits in a time-dependent manner also produced neurotoxicity in a very similar, time-dependent manner, suggesting an association between MAP-2 deficits and subsequent increases in cytotoxicity. In that MAP-2 is present in only dendrites and soma membranes, these reductions may reflect dendritic pruning or somatic loss. However, evidence that PI increases only occur after reductions in MAP-2 content are observed suggests that the MAP-2 deficits do not merely reflect reduced neuronal content. Interestingly, MAP-2 proteins have multiple serine residues which could be covalently modified by CPO. Notably, the linear amino acid sequences surrounding serines889, 892, 894, 1512, 1515 in rat MAP2 (NCBI locus NP_037198) are similar to the sequence around the active-site serine residue in AChE, which is covalently modified by CPO, thereby inactivating AChE. Covalent modification of MAP2 by CPO could mask epitopes recognized by MAP antibodies (decrease MAP2 immunoreactivity) and/or adversely affect MAP2 function. Phosphorylation of MAPs decreases MAP-microtubule interactions, which promotes disassembly of microtubules (Raffaelli et al. 1992; Yamamoto et al. 1983), perhaps CPO-mediated phosphorylation of MAPs and subsequent destabilization of MAPs plays a role in CPF and CPO toxicity. It is importance to note, however, that PI uptake was monitored only during a relatively brief period of CPO exposure (7 days) and no analysis of tissue occurred following removal of CPO. To fully validate this model of subchronic CPO exposure in future studies, it will be of importance to examine the persistence of this apparent cytotoxicity.
A role for AChE inhibition in producing the time-dependent neurotoxicity observed can not be excluded, however. Bigbee et al. (1999) and others have demonstrated that AChE activity is critical for axonal outgrowth during early CNS development, likely via extra-synaptic effects. Thus, AChE inhibition may interfere with neurite development and contribute to initiation of pro-apoptotic cascades sensitive to impairment of cytoskeletal integrity (Bhalla 2003; Ting-Ting et al. 2005). However, it is evident that a close temporal association between AChE inhibition by CPO and increased PI uptake was not observed. For example, 24 hr of exposure to CPO, at 1.0 μM markedly reduced AChE activity, though cytotoxicity was not observed in tissue exposed to this concentration of CPO until PI uptake was assessed after 7 days of exposure. Though one can not fully discount a role for AChE inhibition in this form of cytotoxicity without further investigation, it is possible if not likely that the CPO-induced injury observed was more closely associated with deficits in MAP-2 bioavailability.
Functional studies examining the polymerization of purified and MAP-rich bovine tubulin support the contention that microtubules and MAP-2 may represent substrates in CPO toxicity. Co-exposure to CPO markedly attenuated the polymerization of both purified tubulin (that was comprised of less than 3% map) and MAP-rich tubulin. However, marked differences in the extent of inhibition of polymerization were observed. For example, CPO reduced polymerization of purified tubulin by approximately 25% during each phase of microtubule formation. Clearly then, CPO inhibits microtubule formation in vitro independently of MAP-2, through an uncharacterized mechanism not involving a loss of α-tubulin content. Though MAP-2 is only observed in post-mitotic cells (Izant and McIntosh 1980), evidence of MAP-independent effects on tubulin polymerization suggest that CPO may adversely effect neurogenesis by impairing mitotic spindle formation and/or function. This suggestion is strongly supported by previous findings that CPF exposure impairs both anterograde and retrograde axonal transport (Terry et al. 2003), effects that almost certainly involve alterations in function of the cytoskeletal motor proteins dynein and kinesin. Both kinesin and dynein are intimately involved in mitotic spindle formation and function (Cassimeris 2004; Mazumdar and Misteli 2005) and impaired function would be expected to produce mitotic arrest. Indeed CPF and CPO have been shown to inhibit mitosis and produce apoptosis in embryonic and neonatal brain, effects not closely correlated with relative AChE inhibition (Roy et al. 1998; Song et al. 1998). It will be of interest to further investigate the effects of CPO exposure on α/β-tubulin dimer formation and Υ-tubulin-mediated nucleation, for example, to identify map-independent effects of CPO that attenuate microtubule formation. However, it was evident that the ability of CPO to inhibit microtubule formation was markedly greater when MAP-rich tubulin was used. CPO reduced polymerization of map-rich tubulin to a much greater extent than purified tubulin, by approximately 45-80%.
MAP-2 proteins promote microtubule formation in post-mitotic cells by facilitating polymerization of nascent α/β-tubulin dimmers with existing protofilaments and providing structural support to formed protofilaments (Chen et al. 1992). Thus, impairing MAP-2 function likely leads to microtubule depolymerization and instability. The functional consequences of this CPO-induced microtubule attenuation may be relevant to understanding the neurotoxic effects of exposure to CPO. Disruption of normal microtubule polymerization and depolymerization cycles produces a significant pro-apoptotic signal. Agents, particularly antineoplastic compounds, that either promote tubulin polymerization (ie. paclitaxel) or promote depolymerization (ie. nocodazole) promote the expression of multiple pro-apoptotic proteins such as the Bcl-2 proteins BAX and BAK and the more distantly-related BH3-only protein BIM (Bhalla 2003; Ting-Ting et al. 2005). Some of these agents may also reduce expression of antiapoptotic Bcl-2 proteins (Beswick et al. 2006).
The toxic and adverse behavioral effects of exposure to CPF or its primary toxic metabolite CPO likely involve a broad array of effects on cytoskeletal components, function of multiple neurotransmitter receptors, and intracellular signaling pathways. For example, the present studies and others indicate that CPF and CPO exposure may produce alterations in axonal and dendritic development (Bigbee et al. 1999; Howard et al. 2005), as well as, impair axonal transport (Terry et al. 2003) and change expression or function of several cholinergic, dopaminergic, and serotonergic (5-HT) markers (Liu et al. 1999; Padilla et al. 2005; Slotkin and Seidler 2005; Slotkin et al. 2001; Zheng et al. 2000). Intracellular signaling may be altered during or following CPF exposure given evidence that CPF and CPO exposure alters function of multiple ATPases (Barber et al. 2001) and impairs signaling in the adenylate cyclase-cAMP pathway (Olivier et al. 2001).
The extent to which many of these neuronal changes associated with CPF or CPO exposure are directly related to AChE inhibition is unclear. Clearly, some adverse effects of CPF or CPO exposure, particulary during CNS development, may involve consequences of marked AChE inhibition. For example, AChE clearly has an extrasynaptic morphogenic role in neurite development and its inhibition attenuates neurite growth as well as recovery from axotomy (Dupree and Bigbee 1996; Dupree et al. 1995; Robertson 1987). In contrast, others have shown that CPF exposure produces abnormalities in axonal and dendritic growth in developing sympathetic neurons at concentrations far below those that inhibit AChE (Howard et al. 2005). It is clear that, despite deafferentation, post synaptic expression of AChE persists in the isolated organotypic slices used in the present studies (Sáez-Valero et al. 2003) and the concentrations of CPO examined are high enough to inhibit AChE. Further, the cultured tissue employed was derived from neonatal rats and it is possible that continuing axonal and dendrite pathfinding, for example, persists at the in vitro ages encompassed by the present experiments. However, in vitro studies of bovine tubulin polymerization clearly demonstrate the AChE-independence of CPO effects on microtubule formation. Clearly then, CPF and CPO effects on neuronal viability are highly complex involving both AChE-dependent and independent mechanisms.
A further potentially critical factor mediating CPF and CPO toxicity is the exposure regimen required to produce behavioral abnormalities and/or neuronal injury. That many of the changes to cytoskeleton, transmitter markers and/or intracellular signaling may persist following cessation of pesticide exposure is somewhat unclear. Padilla et al. (2005) have recently reported that CPF effects on muscarinic receptor and dopamine transporter density recovered to control levels following three months of withdrawal from CPF exposure. In contrast, Slotkin and Seidler (2005) reported that changes in 5-HT1A and 5-HT2 receptors, as well as, the 5-HT transporter persisted for at least 5 months after a four-day CPF exposure regimen. Further, brief gestational exposure to CPF was shown to produce changes in expression of cholinergic markers (ie. ChAT and the choline transporter) and neuronal development that were first observed several weeks after gestational exposure. It is likely, however, that persistence of these changes is not necessary to produce neuronal injury, at least in the developing brain. Several studies have reported that even a single exposure to CPF or CPO produces marked apoptosis in neuronal cultures of young brain (Caughlan et al. 2004). Subchronic exposure (8 days) has also been reported to produce marked neuronal injury in cell culture of neonatal rodent brain (Terry et al. 2003). Clearly, neuronal development is markedly altered by both acute and chronic CPF or CPO exposure, possibly suggesting that the developing brain may be more sensitive to toxic effects of CPF or CPO exposure (Rice and Barone 2000). Age-dependent CPF/CPO toxicity may be related to a greater sensitivity of the rodent neonatal brain AChE, as compared to that of the adult brain, to inhibition by CPF (ie. Liu et al. 1999). However, it must be noted that ample evidence exists that CPF or CPO exposure does indeed produce marked behavioral toxicity in adult rodents following prolonged exposure to doses of CPF that produce no overt toxicity (Terry et al. 2003), though recovery of behavior occurred with prolonged abstinence from CPF exposure. Further, while neonatal rats may be more sensitive to the acutely toxic effects of exposure to high doses of CPF, some investigators have reported that no age-dependent effects on cholinergic markers were evident following repeated exposure to low doses of CPF (Zheng et al. 2000). It will be of importance to further clarify which effects of CPF or CPO exposure do persist and those which do not as this may be relevant to the persistence of pesticide-related neurological abnormalities.
In conclusion, the present findings are consistent with previous work and extend our understanding of the means by which CPF and CPO produce neuronal injury that is independent of AChE inhibition and may involve degradation of microtubules, possibly mediated by interactions with MAP-2 proteins. Thus, it is possible that prolonged CPF exposure in vivo may significantly impact not only early CNS development but neurogenesis in the adult brain.
Figure 5.
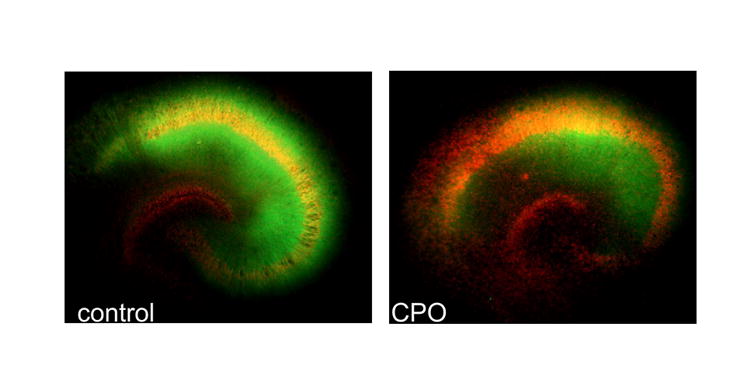
Representative images (5X magnification) of MAP-2 immunoreactivity (FITC, green) and propidium iodide (PI) fluorescence in hippocampal cultures exposed to CPO (1.0 μM) for 7 days. Marked attenuation of MAP-2a/b(FITC, green) immunoreactivity and increased PI signal (red) was observed following 7 days of exposure to CPO (1.0 μM).
Acknowledgments
This work was supported by NIEHS (ES012241)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barber D, Hunt J, Ehrich M. Inhibition of calcium-stimulated ATPase in the hen brain P2 synaptosomal fraction by organophosphorus esters: relevance to delayed neuropathy. J Toxicol Environ Health A. 2001;63:101–113. doi: 10.1080/15287390151126423. [DOI] [PubMed] [Google Scholar]
- Beswick RW, Ambrose HE, Wagner SD. Nocodazole, a microtubule depolymerising agent, induces apoptosis of chronic lymphocytic leukaemia cells associated with changes in Bcl-2 phosphorylation and expression. Leuk Res. 2006;30:427–436. doi: 10.1016/j.leukres.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Bhalla KN. Microtubule-targeted anticancer agents and apoptosis. Oncogene. 2003;22:9075–9086. doi: 10.1038/sj.onc.1207233. [DOI] [PubMed] [Google Scholar]
- Bigbee JW, Sharma KV, Gupta JJ, Dupree JL. Morphogenic role for acetylcholinesterase in axonal outgrowth during neural development. Environ Health Per. 1999;107:81–87. doi: 10.1289/ehp.99107s181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell PJ, Kelly KC, Ward TR. Repeated inhibition of cholinesterase by chlorpyrifos in rats: behavioral, neurochemical and pharmacological indices of tolerance. J Pharmacol Exp Ther. 1994;270:15–25. [PubMed] [Google Scholar]
- Casida JE, Quistad GB. Serine hydrolase targets of organophosphorus toxicants. Chem Biol Interact. 2005:157–158. 277–283. doi: 10.1016/j.cbi.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Cassimeris L. Cell division: eg’ing on microtubule flux. Curr Biol. 2004;14:R1000–1002. doi: 10.1016/j.cub.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Caughlan A, Newhouse K, Namgung U, Xia Z. Chlorpyrifos induces apoptosis in rat cortical neurons that is regulated by a balance between p38 and ERK/JNK MAP kinases. Toxicol Sci. 2004;78:125–134. doi: 10.1093/toxsci/kfh038. [DOI] [PubMed] [Google Scholar]
- Chen J, Kanai Y, Cowan NJ, Hirokawa N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature. 1992;360:674–677. doi: 10.1038/360674a0. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Bigbee JW. Retardation of neuritic outgrowth and cytoskeletal changes accompany acetylcholinesterase inhibitor treatment in cultured rat dorsal root ganglion neurons. J Neurosci Res. 1994;39:567–575. doi: 10.1002/jnr.490390508. [DOI] [PubMed] [Google Scholar]
- Dupress JL, Maynor EN, Bigbee JW. Inverse correlation of acetylcholinesterase (AChR) activity with the presence of neurofiliments inclusions in dorsal root ganglion neurons cultured in the presence of a reversible inhibitor of AChE. Neurosci Lett. 1995;197:37–40. doi: 10.1016/0304-3940(95)11895-4. [DOI] [PubMed] [Google Scholar]
- Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colormetric determination of acetylcholinsterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Giacca M. HIV-1, Tat, apoptosis and the mitochondria: a tubulin link ? Retrovirology. 2005;2:7. doi: 10.1186/1742-4690-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard AS, Bucelli R, Jett DA, Bruun D, Yang D, Lein PJ. Chlorpyrifos exerts opposing effects on axonal and dendritic growth in primary neuronal cultures. Toxicol Appl Pharmacol. 2005;207:112–124. doi: 10.1016/j.taap.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Huff RA, Abu-Ware AW, Abou-Donia MB. Effects of sub-chronic in vivo chlorpyrifos exposure on muscarinic receptors and adenylate cyclase of rat striatum. Arch Toxicol. 2001;75:480–486. doi: 10.1007/s002040100269. [DOI] [PubMed] [Google Scholar]
- Izant JG, McIntosh JR. Microtubule-associated proteins: a monoclonal antibody to MAP2 binds to differentiated neurons. Proc Natl Acad Sci USA. 1980;77:L4741–4745. doi: 10.1073/pnas.77.8.4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JG, Kessler J, Rosenberg N, Pack D. Sensory disturbances associated with Dursban (chlorpyrifos) exposure. Neurology. 1993;43:2193–2196. doi: 10.1212/wnl.43.11.2193. [DOI] [PubMed] [Google Scholar]
- Kropp T, Richardson R. Relative inhibitory potencies of chlorpyrifos oxon, chlorpyrifos methyl oxon, and mipafox for acetylcholinesterase versus neuropathy target esterase. J Toxicol Environ Health A. 2003;66:1145–1157. doi: 10.1080/15287390306360. [DOI] [PubMed] [Google Scholar]
- Lee JC, Timasheff SN. In vitro reconstitution of calf brain microtubules: effects of solution variables. Biochemistry. 1977;16:1754–1764. doi: 10.1021/bi00627a037. [DOI] [PubMed] [Google Scholar]
- Lotti M, Moretto A, Zoppellari R, Dainese R, Rizzuto N, Barusco G. Inhibition of lymphocytic neuropathy target esterase predicts the development of organophosphate-induced delayed polyneuropathy. Arch Toxicol. 1986;59:176–179. doi: 10.1007/BF00316329. [DOI] [PubMed] [Google Scholar]
- Liu J, Olivier K, Pope CN. Comparative neurochemical effects of repeated methyl parathion or chlorpyrifos exposures in neonatal and adult rats. Toxicol and Appl Pharmacol. 1999;158:186–196. doi: 10.1006/taap.1999.8693. [DOI] [PubMed] [Google Scholar]
- Mazumdar M, Misteli T. Chromokinesins: multitalented players in mitosis. Trends Cell Biol. 2005;15:349–355. doi: 10.1016/j.tcb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Mortensen SR, Hooper MJ, Padilla S. Rat brain acetylcholinesterase activity: developmental profile and maturation sensitivity to carbamate and organophosphorus inhibitors. Toxicology. 1998;125:13–19. doi: 10.1016/s0300-483x(97)00157-1. [DOI] [PubMed] [Google Scholar]
- Olivier K, Jr, Liu J, Pope C. Inhibition of forskolin-stimulated cAMP formation in vitro by paraoxon and chlorpyrifos oxon in cortical slices from neonatal, juvenile and adult rats. J Biochem Mol Toxicol. 2001;15:263–269. doi: 10.1002/jbt.10002. [DOI] [PubMed] [Google Scholar]
- Padilla S, Marshall RS, Hunter DL, Oxendine S, Moser VC, Southerland SB, Mailman RB. Neurochemical effects of chronic dietary and repeated high-level acute exposure to chlorpyrifos in rats. Toxicol Sci. 2005;88:161–171. doi: 10.1093/toxsci/kfi274. [DOI] [PubMed] [Google Scholar]
- Prendergast MA, Harris BR, Mayer S, Holley RC, Pauly JR, Littleton JM. Nicotine exposure reduces N-methyl-D-asparate toxicity in the hippocampus: relation to distribution of the alpha7 nicotinic acetylcholine receptor subunit. Med Sci Monit. 2001;7:1153–1160. [PubMed] [Google Scholar]
- Quistad GB, Nomura DK, Sparks SE, Segall Y, Casida JE. Cannabinoid CB1 receptor asa a target for chlorpyrifos oxon and other organophosphorus pesticides. Toxicol Lett. 2002;135:89–93. doi: 10.1016/s0378-4274(02)00251-5. [DOI] [PubMed] [Google Scholar]
- Raffaelli N, Yamauchi PS, Purich DL. Microtubule-associated protein autophosphorylation alters in vitro microtubule dynamic instability. FEBS Lett. 1992;296:21–24. doi: 10.1016/0014-5793(92)80394-v. [DOI] [PubMed] [Google Scholar]
- Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108(Suppl 3):511–533. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ. Assessment of the neurotoxic potential of chlorpyrifos relative to other organophosphorus compounds: a critical review of the literature. J Toxicol Environ Health. 1995;44:135–165. doi: 10.1080/15287399509531952. [DOI] [PubMed] [Google Scholar]
- Richardson RJ, Moore TB, Kayyali US, Fowke JH, Randall JC. Inhibition of hen brain aceytlcholinesterase and neurotoxic esterase by chlorpyrifos in vivo and kinetics of inhibition by chlorpyrifos oxon in vitro: application to assessment of neuropathic risk. Fundam Appl Toxicol. 1993;20:273–279. doi: 10.1006/faat.1993.1036. [DOI] [PubMed] [Google Scholar]
- Robertson RT. A morphological role for transiently expressed acetylcholinesterase activity in developing thalamocrotical systems ? Neurosci Lett. 1987;75:259–264. doi: 10.1016/0304-3940(87)90531-3. [DOI] [PubMed] [Google Scholar]
- Roy TS, Andrews JE, SEidler FJ, Slotkin TA. Chlorpyrifos elicits mitotic abnormalities and apoptosis in neuroepithelium of cultured rat embryos. Teratoloty. 1998;59:323–324. doi: 10.1002/(SICI)1096-9926(199808)58:2<62::AID-TERA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Sáez-Valero J, Gonzalez-Garcia C, Cena V. Acetylcholinesterase activation in organotypic rat hippocampal slice cultures deprived of oxygen and glucose. Neurosci Lett. 2003;348:123–125. doi: 10.1016/s0304-3940(03)00790-0. [DOI] [PubMed] [Google Scholar]
- Shelanski ML, Gaskin F, Cantor CR. Microtubule assembly in the absence of added nucleotides. Proc Natl Acad Sci USA. 1973;70:765–768. doi: 10.1073/pnas.70.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin TA, Cousins MM, Tate CA, Seidler FJ. Persistent cholinergic presynaptic deficits after neonatal chlorpyrifos exposure. Brain Res. 2001;902:229–243. doi: 10.1016/s0006-8993(01)02387-3. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Seidler FJ. The alterations in CNS serotonergic mechanisms caused by neonatal chlorpyrifos exposure are permanent. Dev Brain Res. 2005;158:115–119. doi: 10.1016/j.devbrainres.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Song X, Violin JD, Seidler FJ, Slotkin TA. Modeling the developmental neurotoxicity of chlorpyrifos in vitro: macromolecule synthesis in PC112 cells. Toxicol Appl Pharmacol. 1998;151:182–191. doi: 10.1006/taap.1998.8424. [DOI] [PubMed] [Google Scholar]
- Tan TT, Degenhardt K, Nelson DA, Beaudion B, Nieves-Neira W, Bouillet P, Villunger A, Adams JM, White E. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell. 2005;7:227–238. doi: 10.1016/j.ccr.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Terry AV, Jr, Stone JD, Buccafusco JJ, Sickles DW, Sood A, Prendergast MA. Repeated exposures to subthreshold doses of chlorpyrifos in rats: hippocampal damage, impaired axonal transport and deficits in spatial learning. J Pharm Exp Ther. 2003;305:375–384. doi: 10.1124/jpet.102.041897. [DOI] [PubMed] [Google Scholar]
- Tsai ES, Haraldson SJ, Baratta J, Lander AD, Yu J, Robertston RT. Basal forebrain cholinergic cell attachement and neurite outgrowth on organotypic slice cultures of hippocampal formation. Neuroscience. 2002;115:815–827. doi: 10.1016/s0306-4522(02)00460-8. [DOI] [PubMed] [Google Scholar]
- Wu J, Laird DA. Abiotic transformation of chlorpyrifos to chlorpyrifos oxon in chlorinated water. Environ Tox Chem. 2003;22:261–264. [PubMed] [Google Scholar]
- Yamamoto H, Fukunaga K, Tanaka E, Miyamoto E. Ca2+- and calmodulin-depdendent phosphorylation of micorotubule-associated protein 2 and tau factors, and inhibition of microtubule assembly. J Neurochem. 1983;41:1119–1125. doi: 10.1111/j.1471-4159.1983.tb09060.x. [DOI] [PubMed] [Google Scholar]
- Zheng Q, Olivier K, Won YK, Pope CN. Comparative cholinergic neurotoxicity of oral chlorpyrifos exposures in preweanling and adult rats. Toxicol Sci. 2000;55:124–132. doi: 10.1093/toxsci/55.1.124. [DOI] [PubMed] [Google Scholar]
- Zimmer J, Kristensen BW, Jakobsen B, Noraberg J. Excitatory amino acide neurotoxicity and modulation of glutamate receptor expression in organotypic brain slice cultures. 2000 doi: 10.1007/s007260070029. [DOI] [PubMed] [Google Scholar]