

Abstract
Cardiac development proceeds via the activation of a complex network of regulatory factors which both directly and indirectly impact downstream cardiac structural genes. In Drosophila, the NK homeodomain transcription factor Tinman is critical to cardiac specification and development via the activation of a number of key regulatory genes which mediate heart development. In this manuscript we demonstrate that Tinman also functions in Drosophila to directly activate transcription of the ATP binding cassette gene Sulphonylurea receptor (Sur). Cardiac expression of Sur is regulated by Tinman via an intronic enhancer which first becomes active at stage 12 of embryogenesis and whose function is restricted to the Tin cardial cells by the end of embryogenesis. Cardiac Sur enhancer activity subsequently persists through larval and adult development, but interestingly becomes modulated in several unique subsets of Tin-expressing cardial cells. The cardiac enhancer contains four binding sites for Tinman protein; mutation of two of these sites significantly reduces enhancer activity at all stages of development, and activation of the wild-type enhancer by ectopic Tinman protein confirms Sur is a direct target of Tinman transcriptional activation. These findings delineate at the molecular level specific sub-types of Tin cardial cells, and define an important regulatory pathway between two Drosophila genes for which mutations in human homologs have been shown to result in cardiac disease.
Keywords: heart, Tinman, sulphonylurea receptor, Drosophila, NKX2.5, transcriptional networks
1. Introduction
Several cardiac-restricted transcription factors have been shown to be critical to the development of the circulatory system in animals, and recent research has defined a detailed cross-regulatory network for cardiogenesis involving these factors (reviewed in Brand, 2003; Cripps and Olson, 2002). Research in the model system Drosophila has placed the NK homeodomain transcription factor Tinman (Tin) at the centre of this complex regulatory network in flies, since tin null mutants fail to specify cardiac tissue (Bodmer, 1993; Azpiazu and Frasch, 1993). In support of a broad role for the tin gene in cardiac development, inactivation of the function of vertebrate tin orthologs, in particular nkx2.5, severely affects normal cardiogenesis (Lyons et al., 1995; Grow and Krieg, 1998). Furthermore, mutations in human nkx2.5 are associated with congenital cardiac defects (Schott et al., 1998). Clearly a detailed understanding of the regulation and function of tin will be critical to defining mechanisms of cardiac development and disease.
To date, most of the Drosophila targets of Tinman that have been identified have also been cardiac regulatory factors, including Myocyte enhancer factor-2 (Mef2; Gajewski et al., 1997, 1998; Cripps et al. 1999), the GATA factor gene pannier (Gajewski et al., 2001), the bHLH factor gene Hand (Han and Olson, 2005), the orphan nuclear receptor gene seven-up (Ryan et al., 2007), and tin itself (Xu et al., 1998). The observation that Tin directly impacts so many other cardiac transcription factors underlines its importance in the cardiac regulatory network.
Most of the cardiac enhancers for these Tin target genes contain at least two Tin binding sites within 300bp of each other, which are critical to normal cardiac function. Chen and Schwartz (1995) identified using site selection amplification binding assays a Tin consensus binding sequence of 5′-TNAAGTG, although most papers have quoted the site with a Y (C or T) at the second position based upon the highest affinity sites identified in that study. Knowledge of the target sequence for Tin has made a critical contribution to the identification of Tin target enhancers based upon the presence of clustered Tin sites (Gajewski et al., 1997; Akasaka et al., 2006; Ryan et al., 2007).
In addition to the Tin target genes that encode regulatory proteins, it is now appreciated that structural genes are also activated directly by Tin. In Drosophila, this is the case for the β-tubulin gene βTub60D (Kremser et al., 1999) and the transmembrane protein gene Toll (Wang et al., 2005). However since Tin activates the muscle differentiation factor gene Mef2 (Gajewski et al., 1997, 1998; Cripps et al., 1999), whether Tin is a general activator of cardiac structural genes remains to be determined.
One candidate structural gene for activation by Tin is Sur, encoding the sulphonylurea receptor (Nasonkin et al., 1999). In mammals, expression of SUR proteins is enriched in a number of tissues including heart, skeletal muscle and pancreatic cells (Chutkow et al., 1996; Hernandez Sanchez et al., 1997), where these ATP-binding molecules act to modulate the function of the inwardly-rectifying K+ channels with which they are associated (see for example Aguilar-Bryan et al., 1995; Inagaki et al., 1995). Importantly, mutations in human SUR2 (also called ABCC9) which have been characterized result in dilated cardiomyopathy and ventricular tachycardia in later life (Bienengraeber et al., 2004).
Sequence and functional analysis of the single Drosophila Sur gene revealed that it comprises the ATP-binding function of the mammalian protein complex (Nasonkin et al., 1999), and that this gene is also expressed at high levels in the Tin-expressing cardiac cells of the Drosophila embryo (Nasonkin et al., 1999; Lo and Frasch, 2001). Given the co-incidence of tin and Sur expression in cardiac cells in this system, we hypothesized that Tin functioned as a direct activator of Sur during development.
In this paper we identify and characterize the cardiac enhancer sequences of the Drosophila Sur gene. We find that an enhancer located in the first intron of Sur confers cardiac-specific expression upon a lacZ reporter gene, and that the Tin transcription factor acts directly upon this enhancer to control its expression. Moreover, we demonstrate that the Sur enhancer functions in Tin cells throughout the life of the animal, yet its expression is modulated in functionally distinct subsets of Tin cells. These findings define an important transcriptional pathway in the development of the functional cardiac system, and provide an important regulatory link between two genes whose human homologs are each associated with cardiac disease.
2. Results
2.1 Identification of Tin binding sites in the Sur gene
To determine if expression of the Sur gene depended directly upon transcriptional activation by Tinman, we first sought to identify potential Tin binding sites in the vicinity of Sur. Analysis of ∼19kb of genomic DNA encompassing the Sur transcribed region identified 12 sequences matching the consensus 5′-TYAAGTG (Figure 1A). Of these, two sites in the first intron were relatively closely apposed, being separated by 271 nucleotides.
Figure 1. Identification of Tin binding sites in the Sur genomic region.

A: GBrowse image from FlyBase (Grumbling et al., 2006; url: http://flybase.bio.indiana.edu/) depicting the genomic region of the Sur gene. Sur is located on 2L and is transcribed towards the telomere (from right to left on the image). Sur-RA describes the exon structure of Sur. Directly below this, black vertical bars indicate the presence within the sequence of consensus Tin binding sites. Note that two sites are close together in the first intron. The horizontal black bar indicates the region tested for enhancer activity (see text). B: Tin protein binds specifically to the two Tin sites within the first intron. Shown is an autoradiograph of an electrophoretic mobility shift assays in which TinA (left four lanes) or TinB (right four lanes) were tested. A Tin-DNA complex (Bound) was formed in the presence of Tin protein and target site, but was not formed in the lane containing DNA plus unprogrammed lysate (Un.); the formation of this radioactive complex was competed by addition of 100-fold excess cold wild-type sequence (TA or TB), but not by addition of cold mutant sequence (mTA or mTB). NS indicates a non-specific complex formed in the absence or presence of Tin protein. Free indicates unbound probe DNA. TinA is the centromere-proximal site.
To determine if these two sites were capable of specifically binding Tin protein in vitro, we carried out an electrophoretic mobility shift assay using radioactively labeled dsDNA corresponding to each site and Tin protein synthesized in vitro. We found that Tin protein was able to bind to each labeled consensus site, as determined by the presence of a complex of reduced mobility compared to free labeled probe DNA (Figure 1B). The formation of each complex was effectively competed by the addition of non-radioactive competitor dsDNA corresponding to the wild-type sequences, but was not competed by addition of non-radioactive mutant competitor dsDNA. These results indicated that a strong Tin-binding region existed within the first intron of Sur, and identified this region as a possible location for the cardiac enhancer.
2.2 The Sur enhancer shows cardiac-specific expression and is responsive to Tin protein
To determine if the Sur first intron contained cardiac enhancer activity, we generated by PCR a fragment of DNA encompassing the two Tin sites (see Figure 1A). This DNA was fused to a minimal promoter-lacZ reporter gene and the activation of lacZ expression by the enhancer was analyzed in vivo in transgenic animals. In stage 16 transgenic embryos, the activity of the Sur enhancer closely mirrored the expression of the endogenous Sur gene (Figure 2A-C). β-galactosidase accumulation occurred specifically in the dorsal vessel, and overlapped significantly with the accumulation of Tin protein in the muscular cardial cells. Closer examination of the stain in Figure 2C revealed that not all Tin-positive cells of the dorsal vessel accumulated β-galactosidase protein (arrowheads in Figure 2B, C). The Tin-positive/β-galactosidase-negative cells are probably Tin pericardial cells, many of which lie slightly ventral to the dorsal vessel (Ward and Skeath, 2000). This was confirmed by examination of stage 15 transgenic embryos, where we observed that Tin pericardial cells did not accumulate β-galactosidase (Figure 2A-C, inset). Since Sur is not expressed in pericardial cells (Nasonkin et al., 1999; Lo and Frasch, 2001) our results showing Sur enhancer activity restricted to cardial and not pericardial cells underlines the fidelity of the enhancer in recapitulating Sur expression.
Figure 2. The Sur enhancer is active in the dorsal vessel of Drosophila embryos and is activated by Tinman.

Stage 16 transgenic embryos harboring the Sur-lacZ construct were co-stained for Tin protein (red) and β-galactosidase protein (green) and visualized by confocal microscopy. A-C: In Sur-lacZ embryos, Tin accumulated specifically in the dorsal vessel (A, arrow), and there was also highly specific accumulation of β-galactosidase in the dorsal vessel (B). These images are merged in C. Note that there are some Tin-positive cells on the ventral side of the dorsal vessel which did not accumulate β-galactosidase (arrowhead); these are probably Tin pericardial cells which do not express Sur. Inset: dorsal view of a stage 15 embryo stained as in the main panel. Note that the Tin pericardial cells (arrowhead) do not express Sur-lacZ. D-F: When tin was ectopically expressed throughout the mesoderm of Sur-lacZ embryos (D), β-galactosidase accumulated more broadly in embryonic mesodermal tissues (E, F), particularly in the head region and in ventral somatic muscles (sm). All embryos are oriented with anterior to the left and dorsal side uppermost. Bar, 100μm.
To determine if the activity of the Sur enhancer arose from activation by Tin protein, we determined the effect upon enhancer activity of ectopic expression of tin. When Tin protein was expressed throughout the mesoderm we found that activity of the enhancer was also broadened to include additional mesodermal cells (Figure 2D-F). This effect was most apparent in the head region and in many ventral skeletal muscles, nevertheless a significant number of Tin-positive muscle cells did not show a high accumulation of β-galactosidase protein. We interpret these results to indicate that the Sur enhancer is indeed responsive to activation by Tin, but that additional regulatory factors present in some skeletal muscles cells can influence the function of the enhancer.
2.3 The Sur enhancer shows sustained expression during development in Tin cardiac cells
The data described above indicated that the Sur enhancer could be responsible for controlling Sur expression during embryonic development in Tin-expressing cells. To determine if the Sur-lacZ transgenic construct recapitulated all of the embryonic expression of Sur, we carried out a detailed analysis of enhancer activity during embryogenesis.
Published images showed the earliest expression of endogenous Sur around stage 13 of embryogenesis (Nasonkin et al., 1999). In close agreement with these findings, we first detected Sur-lacZ expression during stage 12 in segmentally-repeating clusters of cells (Figure 3A). To determine if these cells corresponded to cardiac mesoderm, we double-stained animals for β-galactosidase protein plus a number of early developmental markers (Figure 3B-D). We found that early stage 12 expression of Sur-lacZ occurred amongst Tin-expressing cells of the dorsal mesoderm. Furthermore, these cells were not underlying the ectodermal Engrailed expressing cells, but did correspond to the mesodermal cells underlying the ectodermal Wingless signal. Since Wingless signals function at slightly earlier stages of embryogenesis to induce cardiac cell identity (Wu et al., 1995) we interpret these results to indicate that the earliest Sur-lacZ expression is amongst the cells fated to form the dorsal vessel.
Figure 3. The Sur enhancer is active in Tin cells throughout development and shows modulation of activity in a subset of Tin cells.

A: Initial activity of the enhancer was at stage 12, in segmentally-repeating clusters of cells (arrow). B-D: Confocal images demonstrated that initial β-galactosidase accumulation (arrow) was in the dorsal mesoderm (overlapping with Tin, B), and was in presumptive cardiac cells since Sur-lacZ expression did not underlie En-positive ectodermal cells (C) but did underlie Wg-positive cells (D). E: By stage 14, β-galactosidase accumulation was in a continuous line of dorsally-migrating cardiac cells (arrow). F: At stage 16 β-galactosidase was observed in all muscular cardial cells of the dorsal vessel, but levels were slightly lower in the Svp cells (arrowheads). G: Assessment of enhancer-lacZ expression in a stage 15 embryo by in situ hybridization indicated that Sur-lacZ expression was down-regulated in Svp cells (arrowheads), which lack Tin protein. H, H': β-galactosidase (red)/MEF2 (green) double-stain of a third instar Sur-lacZ larva. β-galactosidase was detected in Tin cell nuclei (arrows), but not those of the Svp cells (arrowheads). Note that there is a low level of non-specific fluorescence from the pericardial cells in this preparation. I, I': β-galactosidase (red)/MEF2 (green) double-stain of an Sur-lacZ adult heart. β-galactosidase is detected in Tin cell nuclei (arrows), but not those of the Svp cells (arrowheads). J,J', K, K': In Sur-lacZ larvae stained for β-galactosidase (red) and phalloidin or MEF2 (green), β-galactosidase levels were noticeably lower in Tin cells forming the cardiovascular valve (cv) than in neighboring Tin cells (J, J'), yet were much higher than surrounding cells in part of the anterior aorta (K,K', bracketed). rg, ring gland. L, L': The adult heart also showed an enrichment in β-galactosidase levels in the anterior region (bracketed), which corresponds to the conical chamber (cc). In panels J-L', arrows indicate Tin cell nuclei, and arrowheads indicate Svp cells or nuclei. All samples are oriented with anterior to the left. A-E are sagittal views; F and G are dorsal views; H-L' are ventral views of filleted samples. Bar, 100μm for A, E, F; 30μm for B-D, H-L; 75μm for G.
By the conclusion of germ band shortening through the end of embryogenesis, β-galactosidase accumulation was continuous in the cardial cells of the dorsal vessel (Figure 3E, F). At these stages, tin expression is normally down-regulated in two bilateral cell pairs per segment, which instead express the COUP-TFII homolog seven-up (svp; Bodmer and Frasch, 1999; Gajewski et al., 2000; Ward and Skeath, 2000; Lo and Frasch, 2001). Why then did we observe β-galactosidase accumulation in Svp cells no longer expressing tin ? We reasoned that the stable β-galactosidase protein perdured in the Svp cells at later stages of development. This was confirmed by analysis of lacZ expression by in situ hybridization in stage 15 transgenic embryos. This experiment showed a pattern of Sur-lacZ expression in Tin cells only; therefore at later stages of embryogenesis the Sur-lacZ also recapitulated expression of the endogenous Sur gene (Figure 3G).
It is also apparent from our in situ hybridization experiments that the levels of Sur-lacZ expression in the dorsal vessel are particularly high in the anterior-most third of the dorsal vessel. We believe this to be a faithful recapitulation of endogenous Sur expression in the region of the anterior aorta. Furthermore, the increased expression by this region of the dorsal vessel appears to be maintained later in development (see below).
Having demonstrated that the Sur enhancer faithfully controlled reporter gene expression during embryogenesis, we also sought to determine if the enhancer showed activity in Tin cells at later stages of development, to more precisely map where Sur is expressed through the life cycle. Enhancer activity was detected at all developmental stages tested, including the Tin cells of the third instar larval dorsal vessel (Figure 3H), and the Tin cells of the adult heart (Figure 3I). In each of these instances, we were able to determine that strong β-galactosidase accumulation was in the Tin cardial cells but not in the Svp cardial cells since the Svp cardial cells can be distinguished based upon the smaller size of their nuclei (Molina and Cripps, 2001).
Interestingly, our studies also identified modulations of enhancer activity during development. In the larval dorsal vessel, we noted that Sur-lacZ expression was down-regulated in a single pair of Tin cells immediately anterior to the posterior heart region (Figure 3J). This pair of cells forms a small cardiovascular valve (Rizki, 1978; Molina and Cripps, 2001). By contrast, Sur-lacZ expression was up-regulated in the most anterior cells of the dorsal vessel, corresponding to the anterior aorta (Figure 3K). These same anterior cells also showed the strongest accumulation of β-galactosidase at the adult stage, where this region of the heart straddles the junction of the abdomen and thorax (Figure 3L).
While we have yet to define the physiological reasons for this modulation of Sur enhancer activity, these findings are important for three reasons. Firstly, this analysis is the first to describe post-embryonic Sur expression on a cell-by-cell basis - such findings confirm previous RT-PCR studies (Nasonkin et al., 1999; Akasaka et al., 2006) but also significantly extend them; secondly, we define for the first time distinct molecular differences between Tin cells which have been previously thought to have equivalent functions; finally our findings showing distinct enhancer activities in distinct cell types provide an opportunity to dissect the specification of different Tin cell types via analyses of enhancer activity in each cell.
2.4 Full activation of the Sur enhancer depends upon Tin binding sites
Having demonstrated that the Sur intronic enhancer is active in Tin cells of the dorsal mesoderm and mature cardiac tube during Drosophila development, we next sought to determine if the two Tin sites that we had characterized were required for enhancer function. Accordingly we mutated both TinA and TinB sites in the context of the 811-bp enhancer, and assayed in parallel wild-type and mutant enhancer-lacZ activity in transgenic animals.
In each of eight independent transgenic lines studied, activity of the mutant Sur-lacZ construct was severely reduced compared to that of the wild-type construct. While the wild-type enhancer initiated lacZ expression during stage 12 (Figure 4A), the mutant enhancer was not active at this stage and was only weakly active during stage 13 (Figure 4B). Later during embryonic development, enhancer activity was observed in the mutant lines, but at drastically lower levels compared to wild-type enhancer-lacZ (Figure 4C, D). These findings supported an important role for Tin in the activation of the Sur enhancer during embryogenesis.
Figure 4. High levels of enhancer activity depend upon the TinA and TinB sites.

Transgenic animals harboring either the wild-type Sur-lacZ (A-C) or a mutant version lacking functional TinA and TinB sites (D-F) were stained for β-galactosidase accumulation (brown stain in A, B, D, E; red stain in C, F; C, F counterstained with phalloidin in green to visualize F-actin). For the wild-type construct, strong β-galactosidase accumulation was observed at late stage 13 (A), and was sustained in stage 16 embryos (B) and L3 larvae (C). By contrast, the mutant enhancer-lacZ was severely delayed in the initial activation of the enhancer and showed muted expression even at stage 13 (D), and accumulated β-galactosidase to much lower levels than animals carrying the wild-type construct (F). All samples are oriented with anterior to the left. A, D are sagittal views; B, E are dorsal views; C, F are ventral views of filleted samples. Bar, 100μm for A, B, D, E; 30μm for C, F.
To determine if the Sur enhancer was also Tin-dependent at later stages of development, we studied β-galactosidase accumulation in late third instar larvae transgenic for either the wild-type or mutant enhancer-lacZ. At this stage, the mutant enhancer was barely active in the dorsal vessel compared to the wild-type enhancer (Figure 4E, F). Together, these findings established an important role for the Tin binding sites in the activation of the Sur enhancer in cardiac tissue.
2.5 Additional non-consensus Tin binding sites within the Sur enhancer
While the embryonic Sur enhancer activity was strongly reduced in the absence of TinA and TinB binding sites, some residual activity was still detected in our assays. We reasoned that this result might arise from two possibilities: firstly, additional regulatory factors might impact the Sur enhancer in cardiac tissues and might function independently of Tin to partially activate the enhancer; secondly, perhaps Tin was capable of binding to additional sites in the enhancer, to provide for enhancer activation in the absence of TinA and TinB. For the first possibility, it is quite likely that additional factors impact cardiac enhancer activity for this gene, and future analyses will define the identity of these factors in more detail. Regarding the possibility of additional Tin sites, while this work was in preparation Akasaka et al. (2006) presented data confirming our observations that the Sur enhancer is activated by Tin. In their paper they identified putative Tin sites T1 through T4, two of which were in addition to the ones that we had identified, because Akasaka et al. (2006) used a more permissive consensus sequence of 5′-TNAAGTG (summarized in Figure 5A). Akasaka et al. (2006) also presented DNA binding assays demonstrating that their Tin3 site (our TinB site) could bind to Tin protein.
Figure 5. The Sur enhancer contains non-consensus Tin binding sites.
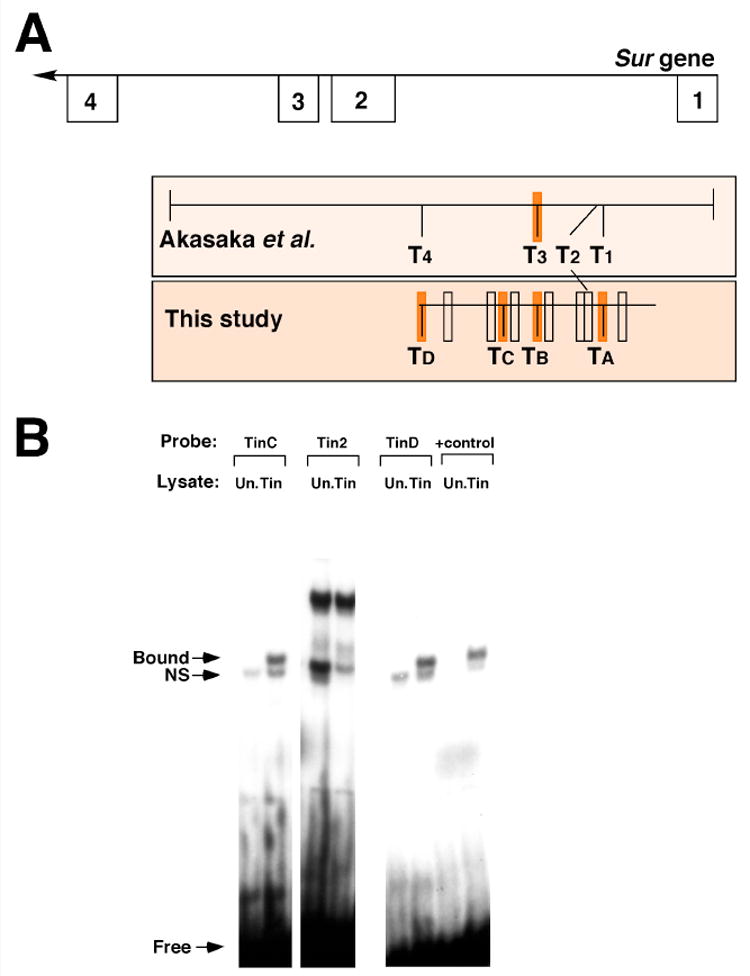
A: Structure of the 5′ end of the Sur gene and enhancer fragments tested for function. The top line defines part of the Sur gene structure with known exons indicated as numbered boxes. Below, the extents of the enhancers tested by Akasaka et al. (2006) and by this study are indicated. Open boxes on the enhancers indicate potential Tin sites which did not bind Tin protein; filled orange boxes indicate sites which were shown either by ourselves or others to bind Tin. B: Binding of Tin protein to putative Tin sites. Binding was only considered to have occurred if a complex was not observed in the reactions with unprogrammed lysate (Un., lacking Tin protein) but was observed in the reactions with programmed Tin lysate. Sites TinC and TinD bound Tin protein effectively, however the region corresponding to T2 of Akasaka et al. (2006) did not show Tin protein binding. Binding of Tin to a positive control sequence (+control) was included in all assays and is shown here for comparison.
To address the possibility that Tin binds to additional sites in the enhancer, we searched for sites using the 5′-TYAAGTG sequence consensus, but permitted one mismatch. This identified 11 sites in total: each of Tin1 (our TinA), Tin2, Tin3 (our TinB) and Tin4 from Akasaka et al. (2006), and seven other sites.
To determine if any of these sequences were capable of interaction with Tin protein, we generated dsDNA probes for each site, and tested their Tin binding ability. These studies identified two important results (Figure 5B). Firstly, two additional Tin sites were identified and named TinC and TinD; TinD is the same site as Tin4 from Akasaka et al. (2006). These findings provided support for our initial hypothesis that this enhancer contained Tin binding sites additional to the two which we had inactivated in Figure 4, and provided an explanation for the residual low levels of enhancer activity we observed even after TinA and TinB had been mutated. Secondly, we failed to identify a specific bound complex for Tin protein with the Tin2 site of Akasaka et al. (2006), although this analysis was partially complicated by the presence of a DNA binding interaction even in the absence of Tin protein. Extensive modification of our experimental conditions did not effectively abolish the formation of this complex without also attenuating the ability of Tin to bind to positive control sequences. Our inability to demonstrate a specific interaction of Tin with the Tin 2 site was puzzling since the published data clearly showed an important requirement for the Tin2 site in enhancer function. While it is possible that an in vivo interaction might not be fully recapitulated by our in vitro DNA binding studies, our result also raises the possibility that a factor other than Tin interacts with the enhancer via the Tin2 sequence. Nevertheless both our studies and those of Akasaka et al. (2006) are in strong agreement that cardiac-restricted expression of the Sur gene is mediated by a Tin-dependent intronic enhancer, containing multiple Tin binding sites.
3. Discussion
In this manuscript we have identified the Sulphonylurea receptor gene as a direct downstream target of the NK homeodomain transcription factor Tinman in the Drosophila cardiac mesoderm and mature dorsal vessel. These findings bolster an important link between a factor critical to cardiac specification, and a structural gene characteristic of the cardiac tissue in this and many higher organisms. While this manuscript was in preparation, Akasaka et al. (2006) presented findings consistent with those presented here. Together the two studies provide robust evidence for control of Sur expression by Tin.
Further support for our data also comes from a recent paper by Zaffran et al. (2006). In their study, an enhancer controlling late-stage embryonic expression of tin was removed from a genomic rescue construct, and re-introduced to a tin mutant background. These authors demonstrated that ablation of tin expression at the latter stages of embryogenesis was critical for normal cardiac function, and in particular they observed a lack of Sur expression in the mutants. These studies, which place Sur genetically downstream of tin, complement our demonstration of a direct transcriptional connection between Tin protein and the Sur gene.
The regulation of Sur expression is of high interest given the critical importance of this gene to cardiac function. In Drosophila, Akasaka et al. (2006) showed that Sur knockdowns in adults resulted in higher rates of cardiac failure. In humans, mutations in the SUR2 gene are associated with cardiac hypertrophy and ventricular tachycardia (Bienengraeber et al., 2004). Clearly understanding the genetic regulation of this gene in animal systems stands to provide insight into mechanisms of cardiac disease. Since Tin activates Sur in the Drosophila system, it is reasonable to propose that the Tin homolog NKX2.5 might be an activator of mammalian SUR2. Mutations in human NKX2.5 can result in congenital cardiac defects including atrial septal defects and problems with atrioventricular conduction (Schott et al., 1998; Hosoda et al., 1999). Although defects arising from mutations in human SUR2 differ from those described for NKX2.5 mutations, studies have yet to directly address the possible regulation of SUR2 by NKX2.5.
Expression of Sur specifically in the Drosophila cardiac tissue most likely arises from the effects of a number of factors acting in collaboration with Tin. Our data show that enhancer activation is restricted to presumptive cardial cells of the dorsal vessel since there is no β-galactosidase accumulation in trunk visceral muscle, nor pericardial cells, nor dorsal somatic muscles, each of which are derivatives of the Tin-expressing dorsal mesoderm (reviewed in Cripps and Olson, 2002). Furthermore, despite broad expression of tin in the dorsal mesoderm at the time of enhancer activation, Sur-lacZ expression arises in a tightly-defined group of cells underlying the ectodermal Wingless signal. Akasaka et al. (2006) demonstrated an important role for the GATA factor Pannier in Sur expression. Thus it is likely that the Sur enhancer integrates a number of distinct developmental factors and signals to generate cardiac-specific expression. This scenario is similar to the activation of mammalian cardiac genes by suites of regulatory factors associated with NKX2.5 (Chen et al., 1996; Durocher et al., 1996; Sepulveda et al., 1998).
Our previous studies have demonstrated that tin expression is sustained through development to the adult stage, in essentially the same pattern of cardiac cells as are specified in the embryo (Molina and Cripps, 2001). Sur-lacZ expression is also sustained during development, and since the mutated Sur-lacZ shows severely reduced expression in the larval dorsal vessel, this later expression is also likely to result from Tin activation. These studies also establish that Sur is expressed in the cardiac tube during the entire life cycle of the animal. This conclusion is consistent with, and also extends, the observation by Akasaka et al. (2006) that Sur is expressed in the adult heart.
Several groups of cardiac cells in the larva and adult of our Sur-lacZ lines show modulations of β-galactosidase accumulation. While these modulations might result from unique physiologies of the cells in question affecting β-galactosidase stability, it is more likely that they result from specific effects either upon Tin transcriptional activity or upon enhancer activation. Interestingly, β-galactosidase accumulation is reduced in the pair of Tin cells that form a small cardiovascular valve. The cardiovascular valve separates the posterior heart region from the aorta, and is thought to prevent backflow of hemolymph during normal circulation (Rizki, 1978; Molina and Cripps, 2001). Very little is known of the specification and development of this valve, since no molecular markers have been described which show specific expression in the cardiovascular valve. We speculate that further dissection of the Sur enhancer will provide insight into the formation of this important structure.
Our findings provide additional support for the important role played by Tin in the control of cardiac development and function, via the direct activation of a number of genes with distinct functions in the developing or mature cardiac tissue. A common feature of Drosophila cardiac genes is the activation by Tin via enhancers containing multiple Tin binding sites. Indeed, the presence of clustered Tin sites has been used as a criterion for the identification of cardiac enhancers, and was the method used both by ourselves and Akasaka et al. (2006) to define the Sur enhancer. The ability to easily and successfully identify cardiac enhancers based upon the presence of the Tin sites stands to greatly enhance our understanding of transcriptional regulatory networks in cardiac development.
Enhancers with only two Tin sites can be effectively inactivated by mutation of one of the sites (Gajewski et al., 1997; Cripps et al., 1999; Kremser et al., 1999), suggesting that at least two sites need to be occupied by Tin for enhancer activation. The ability of Tin molecules to dimerize (Zaffran and Frasch, 2005) might therefore be a mechanism to load Tin protein onto enhancers containing two Tin sites. It is also reasonable to expect that other cardiac factors perform a similar function upon enhancers with a single Tin site, such as that of the Toll gene (Wang et al., 2005). Our identification of additional Tin sites in the Sur enhancer provide an explanation for why mutation of TinA and TinB did not completely abrogate enhancer function.
More difficult to explain, however, is the observation by Akasaka et al. (2006) that mutation of Tin2 and Tin3 completely prevented Sur-lacZ activity; in this instance our studies would argue that three Tin sites remained intact. One explanation for this is that the Tin2 site might be a target for a cardiac factor other than Tin, whose binding to the enhancer is essential for enhancer activity. Some support for this argument comes from our own observations that Tin does not effectively bind to the Tin2 site in the assays that we have carried out.
We conclude that while the Sur enhancer is clearly an effective target of Tin activation during cardiac development and function, a number of additional critical regulators of this gene remain to be identified.
4. Material and Methods
4.1 Bioinformatics
Sequence of the Sur gene and surrounding DNA was obtained from FlyBase (Grumbling et al., 2006; url: http://flybase.bio.indiana.edu), as was the GBrowse image of the Sur gene structure shown in Figure 1A. This sequence was mined for the presence of Tin sites with the consensus 5′-TYAAGTG sequence using online software at Genomatix (url: http://www.genomatix.de).
4.2 Drosophila stocks and crosses
Flies were maintained on Carpenter's medium (Carpenter, 1950) at 25°C unless indicated. The 24B-Gal4 driver line (Brand and Perrimon, 1993) was obtained from the Bloomington Drosophila Stock Center. Generation of UAS-tin flies was described in Ryan et al. (2007). Transgenic lines were generated according to the methods of Rubin and Spradling (1982) using the Delta2-3 helper plasmid (Robertson et al., 1988). Flies of the genotype yw were injected with transforming plasmids and transgenic lines were isolated in the G1 generation as those individuals showing a rescue of the white eye phenotype. At least three lines were analyzed for each construct.
To determine the effects of ectopic expression of tin protein upon enhancer activity, we first crossed homozygous Sur-lacZ animals to the mesodermal driver 24B-gal4 (Brand and Perrimon, 1993). Progeny of the genotype Sur-lacZ / +; 24B-gal4 / + were then crossed to homozygous UAS-tin adults at 29°C. Embryos generated from this cross were harvested and subjected to dual staining for β-galactosidase protein (to identify transgenic embryos) and Tin protein (to identify those over-expressing tin).
4.3 Immunohistochemistry and in situ hybridization
Antibody stains of embryos were performed as described by Patel (1994). Stains of filleted larvae and adults followed Molina and Cripps (2001). Primary antibodies were: mouse anti-β-galactosidase (Promega, 1:1000); rabbit anti-β-galactosidase (Immunology Consultants Laboratory, Newberg, OR, 1:500); mouse anti-Wg (University of Iowa Developmental Studies Hybridoma Bank [DSHB], 1:25); mouse anti-En (DSHB, 1:10); rabbit anti-Tin, 1:500 (Yin et al., 1997). Non-fluorescent antibody detection was done using Vectastain Elite kit and diaminobenzidine (DAB) staining (Vector Laboratories, Burlingame, CA). Fluorescent anti-mouse or anti-rabbit secondary antibodies were Alexa-488 or Alexa-568 conjugated (Molecular Probes, Seattle, WA; 1:2000).
In situ hybridization was carried out using a lacZ antisense riboprobe labeled with Digoxigenin (Roche Molecular Biochemicals, Indianapolis, IN), according to the method of O'Neill and Bier (1994).
All samples were prepared as whole mounts for photography. Embryos were mounted in 80% (v/v) glycerol/1XPBS and larval/adult stains were mounted in SloFade Mounting Medium (Molecular Probes). DAB-stained embryos and larval/adult samples were studied and documented using an Olympus BX51 microscope with Nomarski or fluorescent optics. Confocal microscopy of fluorescently-labeled samples was performed using a BioRad MRC-600 confocal laser-scanning microscope using 488/568 nm excitation in the dual-channel mode (T1/T2A filter cubes, BioRad, Hercules, CA). All figures were assembled using Adobe Photoshop. Some of the larval figure panels represent montages of images taken at slightly different focal planes to generate a complete documentation of expression patterns.
4.4 Molecular biology methods
DNA methods were essentially as described by Sambrook et al. (1998). Generation of the 811-bp Sur intron fragment for enhancer testing was by PCR of genomic DNA using the primers Surint+2: 5′-GGCGGCCGCGATGTCCAGTTTACTCCTTGC and Surint-2: 5′-GCTCGAGCTCATATGGAACCACTTCAGC. Underlined sequences represent those added to the primers to facilitate subsequent cloning steps. Mutagenesis of this enhancer was carried out according to Horton (1995). Tin A and Tin B sites were changed from 5′-TTAAGTG to 5′-GGATCCG and 5′-TCAAGTG to 5′-CCCGGGG respectively. Constructs were cloned in to the pCaSpeR hs-AUG-β-Gal (CHAB; Thummell and Pirrotta, 1992) transformation vector to generate transgenic lines.
Electrophoretic mobility shift assays were carried out using radioactively labeled probe DNA and Tin protein synthesized in vitro. Probes were generated by annealing two complementary oligonucleotides corresponding to each putative Tin site. Annealed DNAs contained 5′GG overhangs at each end. Filling in these sticky ends using Klenow enzyme (New England Biolabs, Beverly, MA) and 32P-dCTP generated radioactive probes. Top strand sequences for the sites tested were as follows: TinA: 5′-GGAACATATTTTAAGTGCTTAATTT; TinB: 5′-GGTCCGTACTTCAAGTGCATTGAAA; TinC: 5′-GGGTTGCAGAACCAATTGAGATTTACCCA; TinD: 5′-GGCATATGGAACCACTTCAGCATAATAAA; 162/168: 5′-GGACTAGGGAGCTCAAGTTTGTTACCTCA; 262/272[Tin2]: 5′-GGCTTTTGCACGCACTTTAAGTCATTTGCATAC; 409/415: 5′-GGTCCGGAAACTCACTCAAAATCCCATAA; 631/637: 5′-GGGGGATGTATCTCAAATGGGTAAATCTC; 673/679: 5′-GGTAAGAATCCGCCCTTAAAGCAAAGTGC; 748/754: 5′-GGATTTCTATAATTAACTGCAGTGTAAAT. Note that the two 5′ G nucleotides added for labeling purposes are included. Putative Tin binding sites are underlined. Tin protein was synthesized from pKS-Tin (Bodmer, 1993) using the Promega TNT coupled reticulocyte lysate kit (Promega Corp.) and T7 RNA polymerase (Roche Molecular Biochemicals). Unprogrammed lysate was generated in the same manner except lacking the plasmid DNA. Binding reactions contained polydI.dC (1μg), Tin or unprogrammed lysate (3 μl), 5X buffer (2μl; Gossett et al., 1989), competitor DNA (100x molar ratio), probe, and water to bring the volume to 10μl. Reactions were incubated at room temperature for 25 minutes and then electrophoresed on a 5% (w/v) non-denaturing polyacrylamide gel. The gel was dried for one hour and exposed to autoradiography film overnight.
Acknowledgments
We are grateful to the following individuals and organizations who provided us with essential reagents for our work: Manfred Frasch; the Bloomington Drosophila Stock Center; the University of Iowa Developmental Studies Hybridoma Bank. This work was supported by HL080545 from the NIH awarded to RMC. We acknowledge technical support from the Department of Biology's Molecular Biology Facility, supported by NIH grant number 1P20RR18754 from the Institute Development Award (IdeA) Program of the National Center for Research Resources.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, IV, Boyd AE, III, Gonzalez G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Akasaka T, Klinedinst S, Ocorr K, Bustamante EL, Kim SK, Bodmer R. The ATP-sensitive potassium (KATP) channel-encoded dSUR gene is required for Drosophila heart function and is regulated by Tinman. Proc Natl Acad Sci USA. 2006;103:11999–12004. doi: 10.1073/pnas.0603098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azpiazu N, Frasch M. tinman and bagpipe: Two homeobox genes that determine cell fates in the dorsal mesoderm of Drosophila. Genes and Development. 1993;7:1325–1340. doi: 10.1101/gad.7.7b.1325. [DOI] [PubMed] [Google Scholar]
- Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O'Cochlain F, Gao F, Karger AB, Ballew JD, Hodgson DM, Zingman LV, Pang YP, Alekseev AE, Terzic A. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic K(ATP) channel gating. Nature Genet. 2004;36:382–387. doi: 10.1038/ng1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer R. The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development. 1993;118:719–729. doi: 10.1242/dev.118.3.719. [DOI] [PubMed] [Google Scholar]
- Bodmer R, Frasch M. Genetic determination of the Drosophila heart development. In: Rosenthal N, Harvey R, editors. Heart Development. Academic Press; New York: 1999. pp. 65–90. [Google Scholar]
- Brand AH, Perrimon N. Targeting gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brand T. Heart development: molecular insights into cardiac specification and early morphogenesis. Developmental Biology. 2003;258:1–19. doi: 10.1016/s0012-1606(03)00112-x. [DOI] [PubMed] [Google Scholar]
- Carpenter JM. A new semisynthetic food medium for Drosophila. Drosophila Information Services. 1950;24:96–97. [Google Scholar]
- Chen CY, Schwartz RJ. Identification of novel DNA binding targets and regulatory domains of a murine Tinman homeodomain factor nkx-2.5. Journal of Biological Chemistry. 1995;270:15628–15633. doi: 10.1074/jbc.270.26.15628. [DOI] [PubMed] [Google Scholar]
- Chen CY, Croissant J, Majesky M, Topouzis S, McQuinn T, Frankovsky MJ, Schwartz RJ. Activation of the cardiac alpha-actin promoter depends upon serum response factor, tinman homologue, Nkx-2.5, and intact serum response elements. Dev Genet. 1996;19:119–130. doi: 10.1002/(SICI)1520-6408(1996)19:2<119::AID-DVG3>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Chutkow WA, Simon MC, Le Beau MM, Burant CF. Cloning, tissue expression, and chromosomal localization of SUR2, the putative drug-binding subunit of cardiac, skeletal muscle, and vascular K-ATP channels. Diabetes. 1996;45:1439–1445. doi: 10.2337/diab.45.10.1439. [DOI] [PubMed] [Google Scholar]
- Cripps RM, Olson EN. Control of cardiac development by an evolutionarily conserved transcriptional network. Developmental Biology. 2002;246:14–28. doi: 10.1006/dbio.2002.0666. [DOI] [PubMed] [Google Scholar]
- Cripps RM, Zhao B, Olson EN. Transcription of the myogenic regulatory gene Mef2 in cardiac, somatic, and visceral muscle cell lineages is regulated by a Tinman-dependent core enhancer. Developmental Biology. 1999;214:420–430. doi: 10.1006/dbio.1999.9446. [DOI] [PubMed] [Google Scholar]
- Durocher D, Chen CY, Ardati A, Schwartz RJ, Nemer M. The atrial natriuretic factor promoter is a downstream target for Nkx-2.5 in the myocardium. Mol Cell Biol. 1996;16:4648–4655. doi: 10.1128/mcb.16.9.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski K, Kim Y, Lee YM, Olson EN, Schulz RA. D-Mef2 is a target for tinman activation during Drosphila heart development. EMBO Journal. 1997;16:515–522. doi: 10.1093/emboj/16.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski K, Kim Y, Choi CY, Schulz RA. Combinatorial control of Drosophila Mef2 gene expression in cardiac and somatic muscle cell lineages. Dev Genes Evol. 1998;208:382–392. doi: 10.1007/s004270050194. [DOI] [PubMed] [Google Scholar]
- Gajewski K, Choi CY, Kim Y, Schulz RA. Genetically distinct cardial cells within the Drosophila dorsal vessel. Genesis. 2000;28:36–43. doi: 10.1002/1526-968x(200009)28:1<36::aid-gene50>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Gajewski K, Zhang Q, Choi CY, Fossett N, Dang A, Kim YH, Schulz RA. Pannier is a transcriptional target and partner of Tinman during Drosophila cardiogenesis. Developmental Biology. 2001;233:425–436. doi: 10.1006/dbio.2001.0220. [DOI] [PubMed] [Google Scholar]
- Gossett LA, Kelvin DJ, Sternberg EA, Olson EN. A new myocyte-specific enhancer-binding factor that recognizes a conserved element associated with multiple muscle-specific genes. Molecular and Cellular Biochemistry. 1989;9:5022–5033. doi: 10.1128/mcb.9.11.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grow MW, Krieg PA. Tinman function is essential for vertebrate heart development: Elimination of cardiac differentiation by dominant inhibitory mutants of the tinman-related genes, XNkx2-3 and XNkx2-5. Dev Biol. 1998;204:187–196. doi: 10.1006/dbio.1998.9080. [DOI] [PubMed] [Google Scholar]
- Grumbling G, Strelets V, The Flybase Consortium Flybase: anatomical data, images, and queries. Nucleic Acids Research. 2006;34:D484–D488. doi: 10.1093/nar/gkj068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Olson EN. Hand is a direct target of Tinman and GATA factors during Drosophila cardiogenesis and hematopoiesis. Development. 2005;132:3525–3536. doi: 10.1242/dev.01899. [DOI] [PubMed] [Google Scholar]
- Hernandez Sanchez C, Wood TL, LeRoith D. Developmental and tissue-specific sulfonylurea receptor gene expression. Endocrinology. 1997;138:705–711. doi: 10.1210/endo.138.2.4954. [DOI] [PubMed] [Google Scholar]
- Horton RM. PCR-mediated recombination and mutagenesis. Molecular Biotechnology. 1995;3:93–99. doi: 10.1007/BF02789105. [DOI] [PubMed] [Google Scholar]
- Hosoda T, Komuor I, Shiojima I, Hiroi Y, Harada M, Murakawa Y, Hirata Y, Yazaki Y. Familial atrial septal defect and atrioventricular conduction disturbance associated with a point mutation in the cardiac homeobox gene CSX/NKX2-5 in a Japanese patient. Jpn Circ J. 1999;63:425–426. doi: 10.1253/jcj.63.425. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of I(KATP): an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1169. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Kremser T, Gajewski K, Schulz RA, Renkawitz-Pohl R. Tinman regulates the transcription of the beta-3 tubulin gene (βTub60D) in the dorsal vessel of Drosophila. Developmental Biology. 1999;216:327–339. doi: 10.1006/dbio.1999.9425. [DOI] [PubMed] [Google Scholar]
- Lo P, Frasch M. A role for the COUP-TF-related gene seven-up in the diversification of cardioblast identites in the dorsal vessel of Drosophila. Mechanisms of Development. 2001;104:49–60. doi: 10.1016/s0925-4773(01)00361-6. [DOI] [PubMed] [Google Scholar]
- Lyons I, Parsons M, Hartley L, LI R, Andrews JE, Harvey RP. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeobox gene Nkx2-5. Genes Dev. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- Molina MR, Cripps RM. Ostia, the inflow tracts of the Drosphila heart, develop from a genetically distict subset of cardial cells. Development. 2001;109:51–59. doi: 10.1016/s0925-4773(01)00509-3. [DOI] [PubMed] [Google Scholar]
- Nasonkin I, Alikasifoglu A, Ambrose C, Cahill P, Cheng M, Sarniak A, Egan M, Thomas PM. A novel sulfonylurea receptor family member expressed in the embryonic Drosophila dorsal vessel and tracheal system. J Biol Chem. 1999;274:29420–29425. doi: 10.1074/jbc.274.41.29420. [DOI] [PubMed] [Google Scholar]
- O'Neill JV, Bier E. Double-label in situ hybridization using biotin and digoxigenin-tagged RNA probes. Biotechniques. 1994;17:874–875. [PubMed] [Google Scholar]
- Patel N. Imaging neuronal subsets and other cell types in whole-mount Drosophila embryos and larvae using antibody probes. In: Fyrberg EA, Golstein LSB, editors. Methods in Cell Biology. Vol. 44. Academic Press; Boston, MA: 1994. pp. 445–487. [DOI] [PubMed] [Google Scholar]
- Rizki TM. The circulatory system and associated cells and tissues. In: Ashburner G, Wright TRF, editors. The genetics and biology of Drosophila. 2b. Academic Press; New York: 1978. pp. 397–452. [Google Scholar]
- Robertson HM, Preston CR, Phillis RW, Johnson-Schlitz DM, B WK, Engels WR. A stable genomic source of P element transposase in Drosophila melanogaster. Genetics. 1988;118:461–470. doi: 10.1093/genetics/118.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin GM, Spradling GM. Genetic-transformation of Drosophila with transposable element vectors. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- Ryan KM, Hendren JD, Helander LA, Cripps RM. The NK homeodomain transcription factor Tinman is a direct activator of seven-up in the Drosophila dorsal vessel. Dev Biol. 2007 doi: 10.1016/j.ydbio.2006.10.025. in press. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1998. [Google Scholar]
- Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused by mutations in the transcription factor Nkx2-5. Science. 1998;281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- Sepulveda JL, Belaguli N, Nigam V, Chen CY, Nemer M, Schwartz RJ. GATA-4 and Nkx-2.5 coactivate Nkx-2 DNA binding targets: role for regulating early cardiac gene expression. Mol Cell Biol. 1998;18:3405–3415. doi: 10.1128/mcb.18.6.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel CS, Pirrotta V. New pCaSpeR P element vectors. Drosophila Information Services. 1992;71:150. [Google Scholar]
- Wang J, Tao Y, Reim I, Gajewski K, Frasch M, Schultz RA. Expression, regulation, and requirement of the Toll transmembrane protein during dorsal vessel formation in Drosophila melanogaster. Molecular and Cellular Biology. 2005;25:4200–4210. doi: 10.1128/MCB.25.10.4200-4210.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward E, Skeath J. Characterization of a novel subset of cardiac cells and their progenitors in the Drosophila embryo. Development. 2000;127:4959–49. doi: 10.1242/dev.127.22.4959. [DOI] [PubMed] [Google Scholar]
- Wu X, Golden K, Bodmer R. Heart development in Drosophila requires the segment polarity gene wingless. developmental Biology. 1995;169:619–628. doi: 10.1006/dbio.1995.1174. [DOI] [PubMed] [Google Scholar]
- Xu X, Yin Z, Hudson JB, Ferguson EL, Frasch M. Smad proteins act in combination with synergistic and antagonistic regulators to target Dpp responses to the Drosophila mesoderm. Genes and Development. 1998;12:2354–2370. doi: 10.1101/gad.12.15.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin ZZ, Xu XL, Frasch M. Regulation of the Twist target gene tinman by modular cis-regulatory elements during early mesoderm. Development. 1997;124:4971–4982. doi: 10.1242/dev.124.24.4971. [DOI] [PubMed] [Google Scholar]
- Zaffran S, Frasch M. The homeodomain of Tinman mediates homo- and heterodimerization of NK proteins. Bichem Biophys Res Comm. 2005;334:361–369. doi: 10.1016/j.bbrc.2005.06.090. [DOI] [PubMed] [Google Scholar]