

Abstract
HIV-associated dementia, like several other neurodegenerative diseases, is characterized by selective degeneration of neurons amidst survival of glial cells like, astroglia. The molecular basis of such selective susceptibility within the same milieu remains largely unknown. Neurons are rarely infected by the virus. However, they are vulnerable to viral products, like HIV-1 coat protein gp120. Interestingly, gp120 induced oxidative stress in neurons, but not in astroglia. This led us to postulate that astroglia were armed with a more efficient anti-oxidant system than neurons. Here, we report that constitutive level of MnSOD (SOD2), the major cellular anti-oxidant enzyme, is significantly higher in astroglia than in neurons. Furthermore, gp120 treatment enhanced MnSOD level in astroglia but decreased the same in neurons. This increase in astroglial MnSOD was dependent on NF-κB, the crucial transcription factor required for sod2 gene transcription. Blocking NF-κB with p65-antisense, p65-si-RNA or a specific inhibitor, NBD peptide, led to reduced MnSOD level and enhanced vulnerability of astroglia to gp120. Additionally, neurons were found to have a lower constitutive level of NF-κB p65 than astrocytes. Over-expression of p65 increased the level of MnSOD in neurons. This, in turn, elicited greater neuronal resistance to gp120. Taken together, our study suggests that astroglia manifest higher threshold for gp120-induced lethality than neurons due to greater MnSOD availability, which it is able to demonstrate due to greater level of NF-κB p65.
Keywords: Oxidative stress, MnSOD, NF-κB-p65, Neurons, Glia
Introduction
Reactive oxygen species (ROS) are highly unstable oxygen species with reactive unpaired electrons. Cellular ROS, including superoxide radical anion (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (HO•), are generated during endogenous aerobic metabolism and in response to exogenous toxic challenges [1]. When generated in excess of physiological limits, they pose serious threat to cellular homeostasis by oxidizing and damaging cellular macromolecules. Such oxidative stress, manifested by oxidized protein and DNA, peroxidized lipids and peroxynitrite generation, is well recognized as a major cause behind several forms of neurodegeneration [1–4].
Among many other neurodegenerative diseases, HIV-associated dementia (HAD) is also triggered and modulated by oxidative stress [5–7]. Still considered a major crisis in the highly active anti-retroviral therapy (HAART) era [8, 9], HAD includes a spectrum of clinical events ranging from mild neurocognitive deficits (cognitive-motor disorders) to severe and debilitating AIDS dementia complex [10]. These clinical manifestations result due to loss of normal neuronal functionality during HAD when neurons undergo dendritic pruning, loss of synapse, and cell death [9–11].
Interestingly, neurons themselves are rarely infected by the virus. Thus the theory of ‘indirect killing’ has been forwarded by several groups [9, 10] to explain neurodegeneration during HAD. According to this theory, toxins generated by the virus and activated immune-profile in the brain are held responsible for neuronal loss. Among several potential neurotoxic HIV-1 proteins, the structural protein gp120 is considered a major etiological reagent of HAD. The viral gp120 is shed from infected cells in the HAD brain and causes cell death in various neuronal populations, including midbrain dopaminergic neurons, hippocampal neurons, cortical neurons, and cerebellar granule neurons in picomolar concentration range [12] even in absence of glia [13].
One unique feature of HAD, like several neurodegenerative disorders, is selective demise of neurons among all brain cells. While neurons die, astroglia, the other CNS cell type of neuroectodermal origin, endure the neurotoxic microenvironment and undergo astrogliosis [10, 11]. Considering the central role of oxidative stress in HAD pathogenesis, it is interesting to note that neurons, but not astroglia, experience elevated oxidative stress in response to several HIV-1 proteins like, gp120 and Tat [11]. This disparity in endurance of brain cells to oxidative stress within the same niche in brain suggests difference in adeptness of these cells to eliminate ROS and prevent oxidative damage.
Cells prevent oxidative damage by utilizing several anti-oxidant systems present in them, which negate oxidative adversity by eliminating ROS. They include anti-oxidant enzymes like, superoxide dismutase (SOD), glutathione peroxidase and catalase. The SOD system, which converts O2•− to H2O2 and molecular oxygen, constitutes the primary line of anti-ROS defense for cells. Currently, three different isoforms constitute this system [14]. Utilizing Cu or Zn as their prosthetic group, SOD1 (Cu/Zn-SOD) is localized in cytosolic compartments while SOD3 (Ec-SOD) is found in extracellular spaces. On the contrary, SOD2 uses Mn at its active site (MnSOD), and is strictly localized in the mitochondrion [14]. The critical relevance of MnSOD as an anti-oxidant enzyme in CNS is revealed by studies in sod2 (−/−) and sod2 (−/+) animals. Complete knockout of MnSOD results in perinatal lethality with CNS irregularities like mitochondrial vacuolization and deposition of oxidized lipids [15], while sod2 (−/+) animals were more susceptible to seizures and kainite-induced neurodegeneration than wild type counterparts [16]. Additionally, strong induction of MnSOD in surviving neurons of Huntington’s disease brain [17] suggests a role of this enzyme in evading neurodegeneration. Not surprisingly therefore, overexpression of MnSOD can protect neurons from lethal consequences of oxidative damage both in vitro and in vivo[18].
During HAD, significant mitochondrial stress is generated in neurons by HIV-derived products, like Tat and gp120 [19]. Because, neurons, not astroglia, are subjected to oxidative stress during HAD, and because overexpression of MnSOD can protect neurons from oxidative damage [18], we hypothesized that neuron may have insufficient level of MnSOD in comparison to astroglia. This hypothesis was tested in the current project. We hereby report that, in terms of MnSOD functionality and elimination of oxidative stress, neurons are less furnished than astroglia on two fronts. Firstly, they posses lower constitutive level of MnSOD. Secondly, they also have comparatively lower level of NF-κB p65, the key molecule required for MnSOD upregulation in the face of ROS [20, 21].
Materials and methods
Reagents
HIV-1 gp120 protein, expressed in insect CHO cells (strain: HIV-1 MN), was obtained from US Biologicals. Anti-p65, anti-MAP-2, anti-GFAP, and anti-Actin were from SantaCruz Biotechnology, anti-MnSOD was from Abcam, anti-NeuN was from Chemicon, and anti-cleaved Caspase3 was from Cell Signaling. Vectors expressing wild type p65 was kindly provided by Dr. Sankar Ghosh of Yale University. Wild type and mutated NBD peptides were procured from Biomol. All si-RNAs were purchased from Ambion.
Isolation of rat cerebellar granule cells (CGC neurons)
Rat CGC neurons were prepared as described previously [22] with some modifications. In brief, cerebella of 7-day-postnatal Sprague-Drawly rats were dissociated in Versene solution (1:5000, Invitrogen) and were plated in Poly-D-Lysine (Sigma) coated plates or dishes for five minutes after which, the non-adherent cell suspension was removed and used for preparing glial cells. Adherent cells were maintained in MEM (Mediatech) supplemented with heat-inactivated 10% fetal bovine serum (FBS, Atlas Biologicals), 25 mM KCl, 3gm/500 ml glucose (Sigma), and 1% antibiotic-antimycotic solution (Sigma). Cytosine-D-arabinoside (10 μM Ara-C, Sigma) was added to cultures 24 hrs after plating to block the proliferation of non-neuronal cells. Neurons were routinely used during 9–12 days in vitro (DIV).
Isolation of human primary neurons
Human neurons were prepared from second trimester fetal brain (Human Embryology Laboratory, University of Washington, Seattle; approved by institutional review board, IRB number 224-01-FB) as described previously [7]. Both post-natal rat CGC and pre-natal human neuronal cultures were found to be more than 98% immunopositive to the neuronal marker microtubule-associated protein-2 (MAP-2).
Isolation of rat and human astrocytes
Rat astroglia was obtained from the non-adherent cell suspension after plating of neurons as described earlier [23]. This fraction was then plated in poly-D-lysine coated flasks or dishes and kept overnight. Human astroglia was isolated from non-adherent cell suspension as described earlier [24].
Transfection
Primary neurons were transfected with Lipofectamine PLUS® (Invitrogen) and Nupherin-neuron (Biomol) as per manufacturer’s protocol. Briefly, each well of 12-well plate was transfected with 0.25 μg of DNA complexed with Neupherin peptide and Lipofectamine PLUS®. Transfection efficiency for neurons was about 20 ± 2.89 % (Fig. 7a). Astrocytes were transfected with si-RNA using the NeoFX-siPORT transfection reagent from Ambion.
Figure 7. NF-κB p65 overexpression upregulates MnSOD in rat neurons.
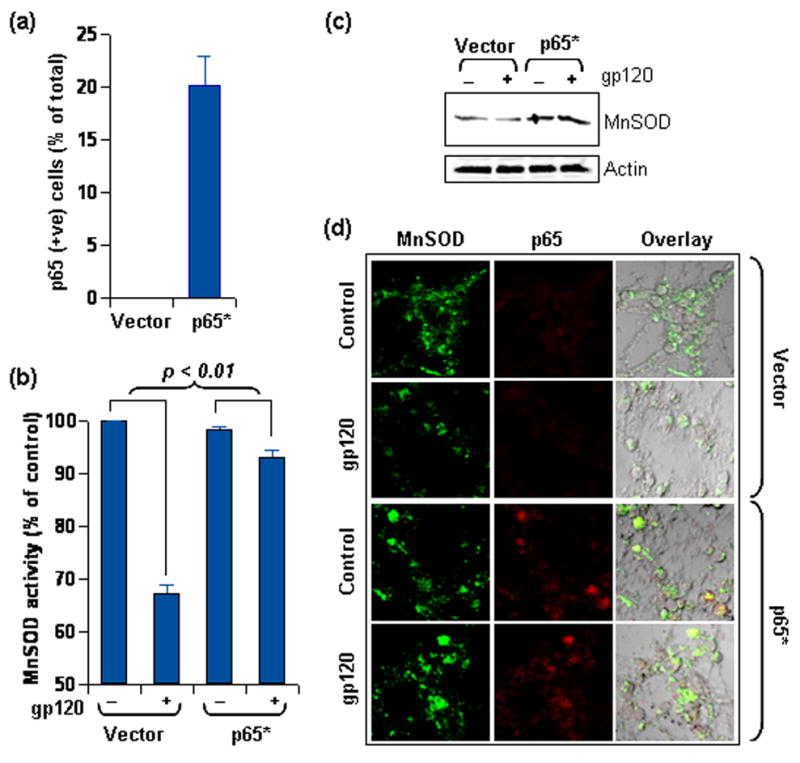
(a) Rat neurons were either transfected with p65 expression vector or with empty vector and transfection efficiency was assessed by immuno-staining cells for p65. Neurons with significantly higher p65 immuno-reactivity than empty vector transfected neurons were considered transfected and were counted by three unrelated investigators from sets of 10 micrographs. Data was tested with t-test statistics. (b) Rat neurons transfected as in (a) were treated with gp120 for 18 hours. Lysate from these cells was used to estimate MnSOD activity after 5M KCN treatment. Data was tested with ANOVA statistics. (c) Lysate of rat neurons similarly transfected and treated as in (b) was separated by electrophoresis and immuno-blotted for MnSOD. Same blots were stripped and reprobed for Actin. (d) Rat neurons, similarly transfected and treated as in (b), were immuno-stained for MnSOD (Cy2) and p65 (Cy5). Settings for confocal microscopy were strictly kept unaltered for detection of both signals for comparative evaluation (60X). Also, detection of p65 (in Cy5 channel) was performed at microscopy settings similar to that of Fig 5d.
Immunoblotting assays
Cells were lysed in RIPA buffer [1X PBS, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS with freshly added 0.5% protease inhibitor cocktail (PIC, Sigma)] in ice. Protein content was estimated by Protein assay dye reagent concentrate (Bio-Rad) using manufacturer’s protocol, and immunoblotting was performed as described previously [24]. Immuno-blots were probed either by chemiluminescence (PerkinElmer) or by fluorescence detection in Odyssey infrared imaging system (LI-COR Biosciences).
Immuno-fluorescence assays
Cells on coverslips were fixed with methanol at −20°C for 5 minutes and washed twice with PBS. Paraffin embedded brain sections were obtained from aged rats after thorough perfusion and subsequent tissue fixation by gradient dehydration. Cells or de-paffinized tissue sections were blocked with 3% BSA-PBS for 1 hour at RT followed by incubation with primary antibodies in 1% BSA-PBS for 3 hours in a thermal rocker at 37°C. Subsequently samples were washed thrice with PBS-Tween solution, incubated with Cy2, Cy3, or Cy5 tagged secondary antibodies (Jackson ImmunoResearch), mounted, and observed under a BioRad MRC1024ES confocal laser-scanning microscope. Negative controls were obtained by treating a set of samples similarly without incubating them with primary antibodies.
Fragment end labeling of DNA (TUNEL)
Fragmented DNA was detected in situ by the terminal deoxynucleotidyl transferase (TdT)-mediated binding of 3′-OH ends of DNA fragments generated in response to apoptotic signals, using a commercially available kit (TdT FragEL) from Calbiochem.
MTT assay
Mitochondrial activity (a measure of cellular viability) was measured with the 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium (MTT) assay (Sigma) as per manufacturer’s protocol as described previously [7].
Superoxide dismutase activity assay
Using the superoxide dismutase activity assay kit (Chemicon, CA), activity of MnSOD was measured as per manufacturer’s protocol. Briefly, cell lysate was obtained by using manufacturer’s recommended harvesting buffer. Total-SOD or MnSOD activity was estimated with a Xanthine/Xanthine oxidase system. For estimation of MnSOD, activity of SOD1 was inhibited by incubation of samples with 5mM KCN for 5 minutes in room temperature.
Native gel superoxide activity assay
Cells were sonicated to obtain protein extracts. Equivalent amount of protein extracts from each cell type was separated on native 8% Tris-Hcl gel. After brief washing, the gels were soaked in Riboflavin-Nitro Blue Tetrazolium (NBT) solution (0.1 mg/mL Riboflavin and 0.25 mg/mL NBT) for 15 minutes in complete darkness. After a brief wash, the gels were then soaked in 0.1% TEMED solution for another 15 minutes in the dark. Gels were then washed and exposed to illumination with shaking until sufficient contrast between clear areas and the purple background was obtained. Gels were subsequently photographed.
Superoxide assay
Superoxide level was detected by LumiMax Superoxide Anion Detection kit (Stratagene) following the manufacturer’s protocol as described previously [7]. Samples were treated with recombinant SOD (2.5 U/mL) to obtain negative controls. Light emission was recorded at regular intervals in a TD-20/20 Luminometer (Turner Designs).
Statistics
TUNEL and p65-positive brain cells were counted by three unrelated investigators from a minimum of ten randomized observations from three independent trials for experiments in figures 5, 7 and 8. Student’s t-test and ANOVA statistics were performed using the software SAS.
Figure 5. Differential level of NF-κB p65 in neuron and glia.
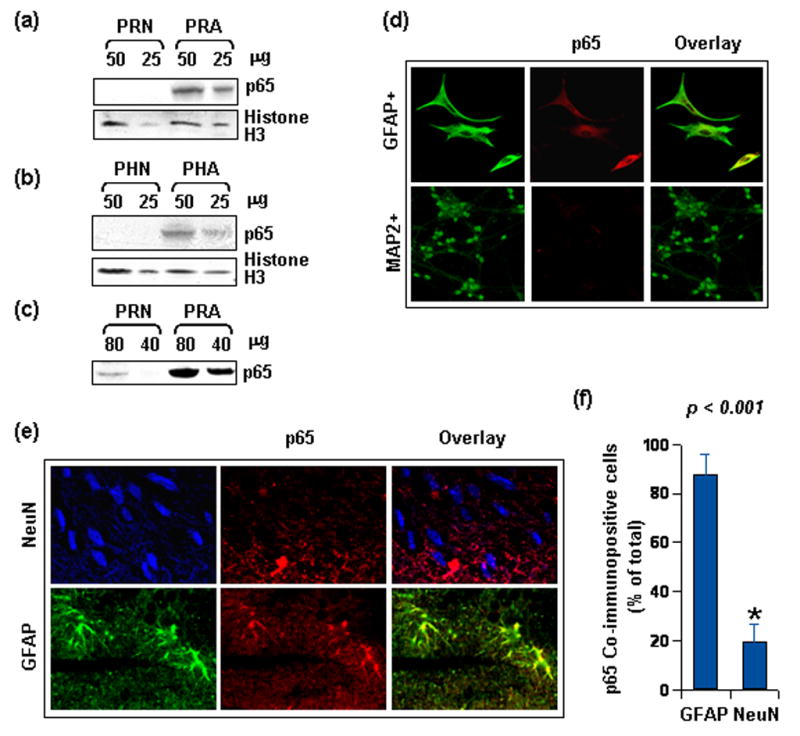
(a) Whole cell lysate was obtained from unstimulated primary rat neuron (PRN) and astroglia (PRA) of same DIV. Indicated protein quantity was separated by electrophoresis and immuno-blotted for p65 with a polyclonal antibody (sc-372). Same blots were stripped and re-probed for Histone3. (b) Same as (a), except lysate was obtained from human neuron (PHN) and astroglia (PHA) and immuno-blot was probed with a monoclonal antibody against p65 (sc-8008). (c) Higher quantity of protein load from similar lysate as in (a) was separated by electrophoresis and immuno-blotted for p65 with the polyclonal antibody (sc-372). (d) Isolated rat neurons (PRN) and astroglia (PRA) were immuno-stained for p65 in addition to MAP-2 and GFAP respectively. Markers were detected in Cy2 (green) channel while NF-κB proteins were detected in Cy5 (red) channel. Setting of the confocal microscope was strictly kept unaltered during whole study. Figures presented are observations at 40X and represent several similar observations. (e) Deparaffinized sections of rat cerebral cortex were probed with NeuN (Cy3 in blue), GFAP (Cy2 in green), and p65 (Cy5 in red). Settings for confocal microscopy were strictly kept unaltered for detection of p65 signal for comparative evaluation (60X). Presence of purple and yellow in the overlay respectively denotes overlapping of NeuN and GFAP with p65 in respective panels. (f) Data representative of strong co-reactivity of neurons (purple) and astrocytes (yellow) with p65 as per observation of at least ten randomized microscopic fields by three unrelated investigators. Data was tested with t-test statistics.
Figure 8. NF-κB p65 overexpression protects rat neurons from gp120 toxicity.
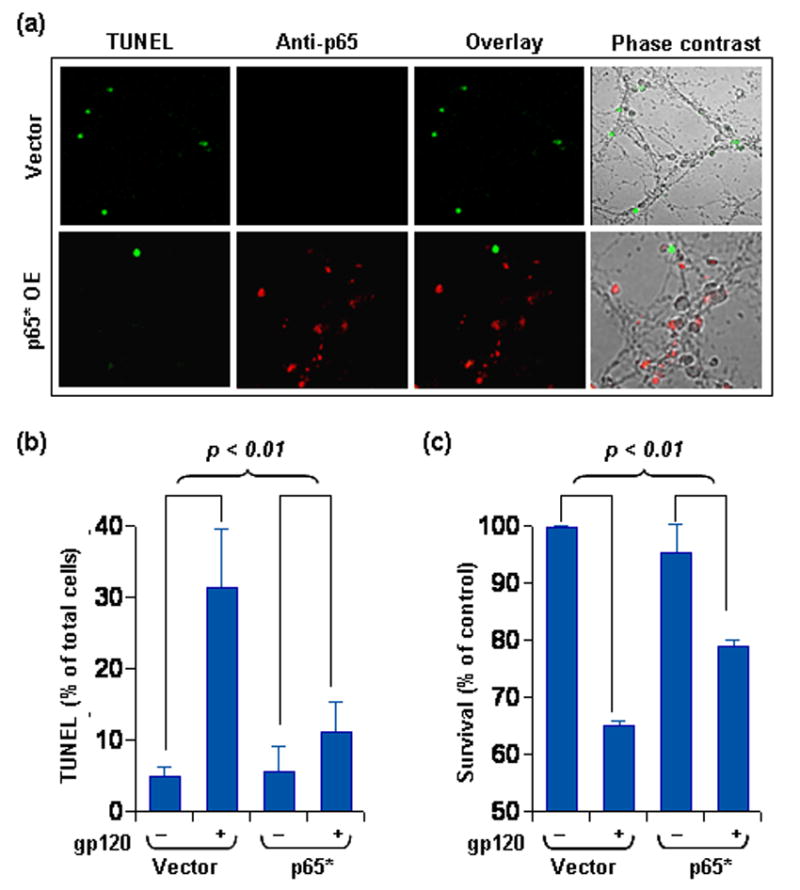
(a) Rat neurons were either transfected with p65 expression vector or empty vector and were then treated with 400pM gp120 for 24 hours. Subsequently, cells were fixed, were TUNEL labeled for fragmented DNA, and were immuno-stained with anti-p65 antibody. Cells were observed under confocal microscope for TUNEL signal (green fluorescence) and p65 immuno-staining (Cy5). Upper panel of the represented figure are observations at 40X, while those in lower panel are at 60X. (b) TUNEL positive cells were counted from sets of 10 micrographs from three different trials by three unrelated investigators to obtain the represented data which was tested with ANOVA statistics. (c) Neurons transfected and treated as in (a) were used to perform MTT assay. Data was tested by ANOVA statistics.
Results
HIV-1 gp120 is toxic to neurons but not to astroglia
HIV-1 envelope protein gp120 is one of the prime etiological reagents of HAD and has been shown to induce injury and apoptosis in primary rodent and human neurons both in vitro and in vivo [7, 12, 25]. Agreeably, in our experiments, gp120 induced degeneracy in rat CGC neurons (more than 98% pure), but not astroglia, in a dose dependent fashion (Fig. 1a). As revealed by the MTT assay, treatment with gp120 caused mitochondrial dysfunction and death in about 30% neurons at 400pM concentration. This corroborates well with similar findings of a previous study [13]. However, heat-denatured gp120 was unable to cause any neuronal death (data not shown). Because 400pM gp120 showed significant neurotoxicity without altering astroglial survival, we conducted most of our subsequent experiments with this dose.
Figure 1. Differential effect of gp120 on rat neuron and glia.
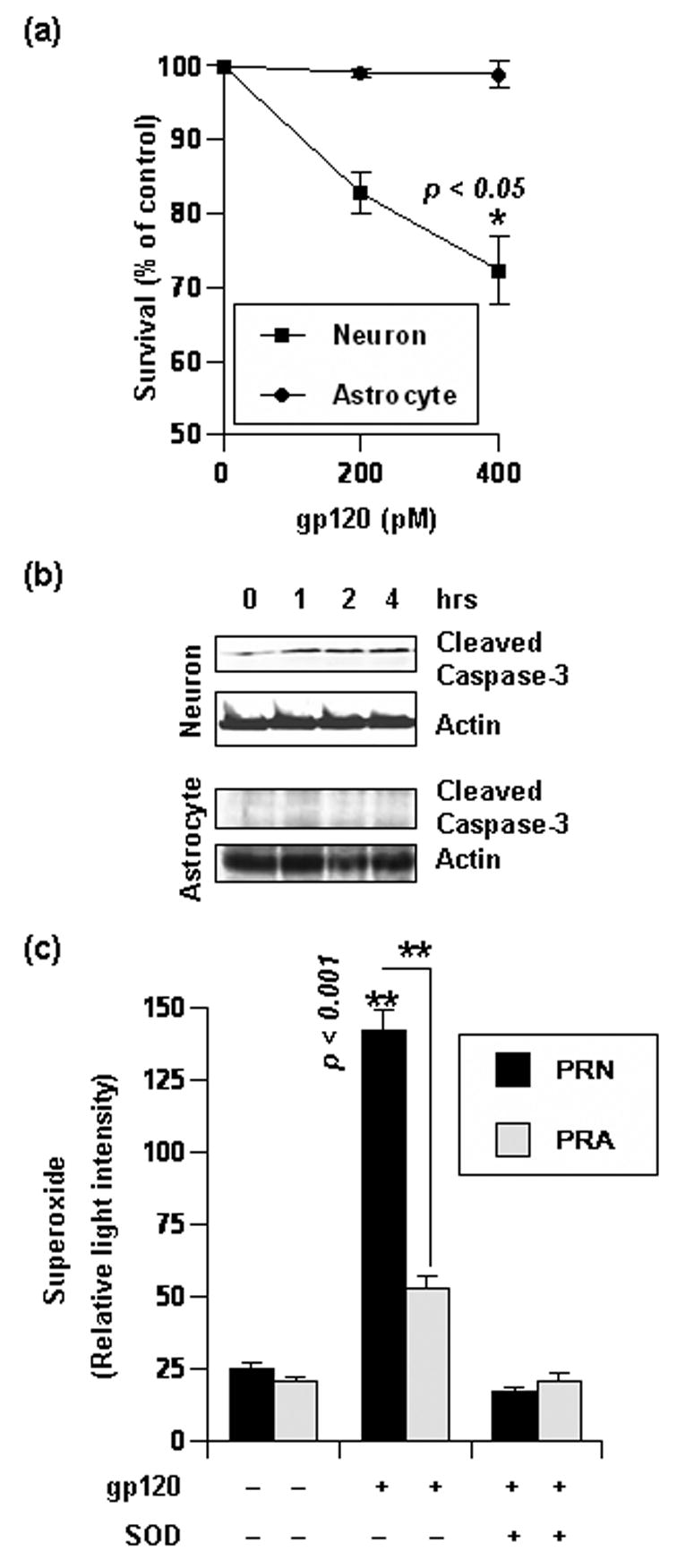
(a) Rat Neurons and glia of same DIV were treated with indicated amount of gp120 for 24 hours. Subsequently, cell vitality was assessed by MTT assay. Data was tested with t-test statistics and is expressed as mean ± SD of three separate experiments. (b) Western blots of whole cell lysate obtained from rat neurons and glia treated with 400pM gp120 for indicated time. Blots were probed for presence of cleaved Caspase-3 and Actin. (c) Primary rat neurons (PRN) and astroglia (PRA) of same DIV were treated with indicated amount of gp120 for 30 minutes. Then superoxide release in cellular media was assayed either in presence or absence of recombinant SOD. Data was tested with t-test statistics and is expressed as mean ± SD of three separate experiments.
Since gp120 has been previously reported to activate caspase cascades [26, 27], we next tested if gp120-induced mitochondrial dysfunction reflected on apoptotic onset in neurons. Consistent with the MTT data, gp120 treatment in CGC neurons induced cleavage of Caspase-3, an apoptotic hallmark, within 1 hour of gp120 treatment (Fig 1b). In contrast, astrocytes did not undergo caspase cleavage after similar treatment (Fig 1b). Together, these results approve selective sensitivity of neurons to gp120.
In light of the involvement of oxidative stress in HAD, we next investigated the ROS level in neuron and astroglia. Since, gp120 induced caspase activation within an hour of treatment (Fig. 1b), we detected superoxide level after half of the hour. As seen in figure 1c, gp120 treatment induced significantly greater amount of superoxide in CGC neurons in comparison to glia. This induction was absent in similar samples treated with recombinant SOD (Fig 1c). This discrepancy in superoxide level may serve to explain selective vulnerability of neurons to gp120.
Differential constitutive level of MnSOD in neuron and astroglia
Discrepancy in gp120-induced superoxide level in these cells hints at differential activity of superoxide dismutase system in these cells. Thus, we next comparatively assessed the constitutive activity of MnSOD in unstimulated rat or human neurons and astroglia. The Constitutive MnSOD activity in rat astroglia was 2.8 times than that in neurons (data not shown). Similarly, we estimated MnSOD activity in human neurons and astroglia. As represented in figure 2a, a significant difference in constitutive MnSOD activity was recorded in these cells. MnSOD activity in human astrocytes was found to be 2.5 times more than human neurons. To confirm further, we next assayed the MnSOD activity in gel in the presence of 5mM KCN (to confirm the presence of MnSOD). As seen in figure. 2b, extracts from rat astrocytes showed greater superoxide scavenging ability than equivalent amount of rat neuronal extracts. Next, we checked the constitutive protein level of MnSOD in unstimulated human neurons and glia. In accordance with its differential constitutive activity in these cells, MnSOD protein was found to be more abundant in astroglia than neurons (Fig. 2c). Because these are mononuclear cells with equivalent chromatin mass per cell, we verified the loading equivalence of our immunoblots by stripping and re-probing the membrane with anti-histone3 antibody (Fig. 2c).
Figure 2. Differential MnSOD activity and level in human neuron and glia.
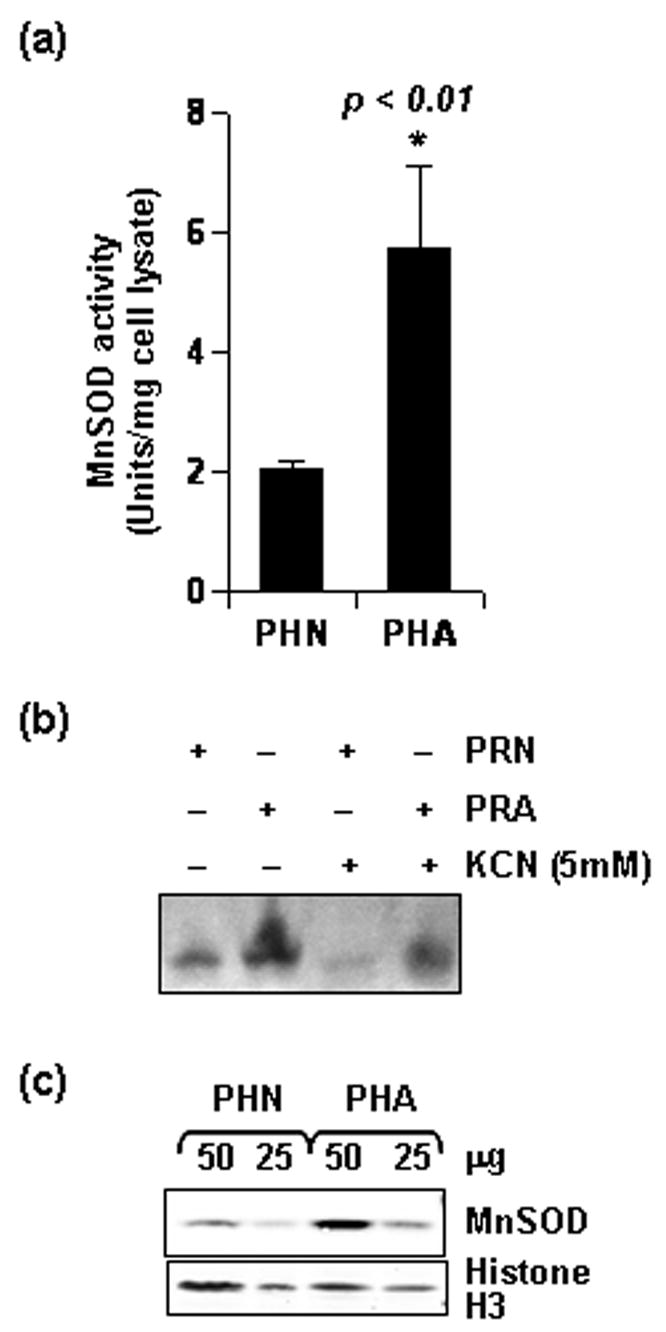
(a) Cell lysate was obtained from untreated primary human neurons (PHN) and astroglia (PHA) of same DIV and MnSOD activity was assayed after 5mM KCN treatment. Data was tested with t-test statistics and is expressed as mean ± SD of three separate experiments. (b) Equal amount of rat neuronal (PRN) and astroglial (PRA) lysate were separated by gel electrophoresis and MnSOD activity in the gel was detected in presence of 5mM KCN, a concentration which completely blocks CuZnSOD activity (c) Whole cell lysate was obtained from untreated human neurons (PHN) and astrocytes (PHA) of same DIV and indicated protein quantity was separated by electrophoresis and immuno-blotted for MnSOD. The same blot was stripped and re-probed for Histone3.
Differential effect of gp120 treatment on neuronal and astroglial MnSOD
Considering the contrasting level of MnSOD in neurons and astroglia, we next examined the effect of gp120 on MnSOD activity and level. When treated with gp120, MnSOD activity decreased dose dependently in CGC neurons (Fig. 3a). To substantiate further, we tested level of MnSOD protein by immunostaining in neurons after gp120 treatment. As seen in Figure 3b, gp120-treated neurons were less immunoreactive to anti-MnSOD antibody in comparison to untreated neurons suggesting a gp120-responsive loss of MnSOD in neurons. In contrast, gp120 increased MnSOD activity in astroglia (Fig. 3c). Additionally, gp120 treatment resulted in modest increase of MnSOD protein level in astrocytes (Fig. 3d). Taken together, such opposite effect of gp120 on MnSOD activity and level in these cells suggest that signal-dependent loss of MnSOD in neurons may compromise their resistance against superoxide. On the other hand, upregulation of MnSOD in astroglia may serve to eliminate superoxide and its deleterious consequences in these cells.
Figure 3. Differential effect of HIV-1 gp120 on rat neuron and glia.
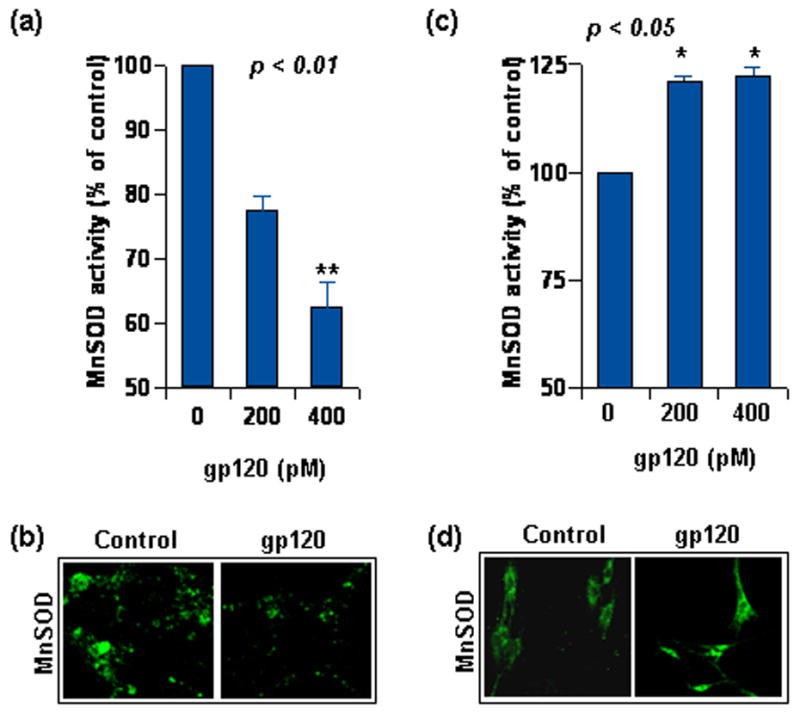
(a) Rat neurons were treated with indicated dose of gp120 for 18 hours. Lysate from these cells was used to estimate MnSOD activity after 5 mM KCN treatment. (b) Rat neurons, treated as in (a), were immuno-stained for MnSOD expression level and observed under confocal microscope (60X). (c) Rat astrocytes were treated with indicated dose of gp120 for 18 hours. Lysate from these cells was used to estimate MnSOD activity after 5 mM KCN treatment. (d) Rat neurons, treated as in (c), were immuno-stained for MnSOD expression level and observed under confocal microscope (40X). Confocal settings for (b) and (d) were different. Quantitative data in (a) and (b) was tested with t-test statistics and is expressed as mean ± SD of three separate experiments.
Expression of astroglial MnSOD is critical for astroglial resistance
To corroborate the importance of upregulated MnSOD in astroglial resistance to gp120, we next knocked down MnSOD by using siRNA against SOD2 (si-SOD2). As seen in figure 4a, we obtained a significant reduction in MnSOD level after using si-SOD2 at 100nM concentration. This concentration was used for all subsequent studies. Next, we assayed the survival efficiency of MnSOD knocked-down astrocytes. Astrocytes transfected with either si-C (non-targeted control siRNA) or si-SOD2 were treated with gp120 for 24 hours. Subsequent MTT analysis demonstrated a significant reduction in survival efficiency of astrocytes treated with si-SOD2, but not si-C (Fig. 4b). This suggests that MnSOD is an integral part of astroglial defense against gp120.
Figure 4. Astroglial MnSOD upregulation by gp120 is dependent on NF-κB.
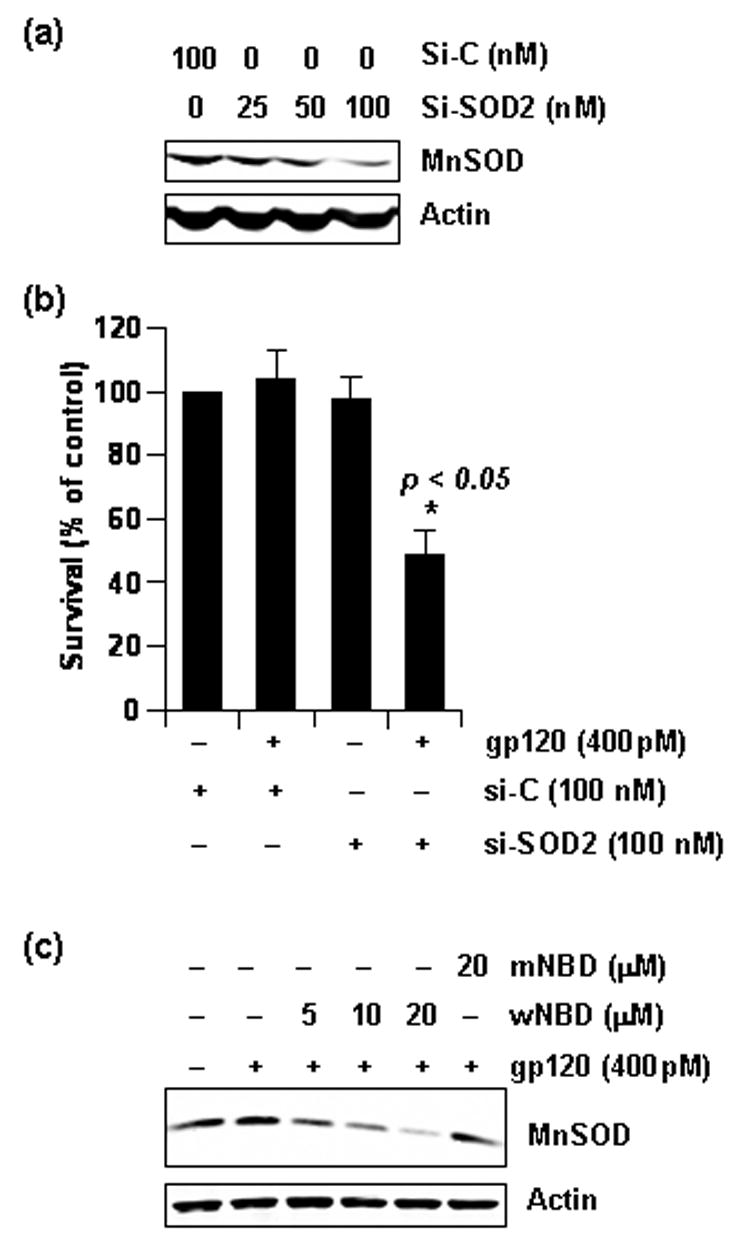
(a) Astrocytes were transfected with indicated doses of si-C or si-SOD2 and whole cell lysate was obtained after 48 hours. Then, samples were separated by electrophoresis and immuno-blotted for MnSOD to test efficiency of the si-SOD2. The blot was re-probed for Actin. (b) Cells were transfected with 100nM si-C or si-SOD2 for 48 hours and then were treated with 400 pM gp120 for another 24 hours. MTT assay was subsequently conducted. Data was tested with t-test statistics and is expressed as mean ± SD of three separate experiments. (c) Rat astrocytes were pretreated with indicated dose of either wild type NBD peptide (wNBD) or mutated NBD peptide (mNBD) for 2 hours prior to gp120 treatment for another 18 hours. Both treatments were in serum free media. Lysate was obtained from these cells and was separated by electrophoresis and immuno-blotted for MnSOD. The same blot was stripped and re-probed for Actin.
HIV gp120-mediated upregulation of astroglial MnSOD is dependent on NF-κB
Although the signaling events for the induction of MnSOD are not completely established so far, different stimuli induce the expression of MnSOD via NF-κB activation. The presence of a consensus sequence in the promoter region of MnSOD for the binding of NF-κB and the inhibition of MnSOD expression with the inhibition of NF-κB activation in various cell types including brain cells establish an essential role of NF-κB activation in the induction of MnSOD. HIV-1 gp120 is also known to activate NF-κB [28, 29]. Therefore, we next verified if NF-κB is required for gp120-induced upregulation of MnSOD in primary astroglia. In order to do so, we pre-treated astroglia with various doses of NEMO-binding domain (NBD) peptides, a specific inhibitor of NF-κB [30]. These cells were then treated with gp120 and MnSOD level was assessed by immunoblotting. As seen in figure 4c, the gp120-induced compensatory expression of MnSOD was lost dramatically in a dose-dependent fashion when cells were pre-treated with wild type, but not the mutated NBD peptide.
Profiling constitutive level of NF-κB p65 in neurons and glia
Because NF-κB was found to be critical for astroglial increase in MnSOD and because neurons were vulnerable to gp120, a possibility remained that level of NF-κB p65 was not adequate in neurons. To test this hypothesis, we compared the constitutive level of p65 in unstimulated rat CGC neurons and astroglia. Equivalent lysate of each cell type was immuno-probed with a polyclonal anti-p65 antibody. Interestingly, constitutive expression of p65 was significantly greater in astrocytes than neurons (Fig. 5a). Next we examined the constitutive level of p65 in human primary astroglia and neurons. Immunoblots were probed with the same polyclonal (data not shown) or a monoclonal anti-p65 antibody (Fig. 5b). It is clearly evident from figure 5b that the level of p65 was more in human astroglia than neurons. Histone-3 level was used as loading control in these cases.
Apparently, figure 5a and 5b may suggest that p65 is absent in neurons. However, that is not true. NF-κB p65 in neurons is in comparatively lower quantity than astrocytes, but not absent. We could detect p65 in rat neuronal lysate, when 80 μg or more protein was loaded (Fig. 5c). To support the immunoblot data further, comparative analysis was done by immunofluorescence studies. Unstimulated cultures of rat neurons and astroglia were immuno-stained for p65 and either MAP-2 or GFAP, the cell type markers for neurons and astroglia respectively. Under identical settings of the microscope, GFAP positive cells showed greater overlap with p65 signals in comparison MAP-2 positive cells (Fig. 5d). This trend, in compliance with immunoblot data, strongly suggests the presence of a higher level of NF-κB p65 in astroglia than in neurons in vitro.
Many times, in vitro observations do not truly reflect situations in vivo. Therefore, we investigated constitutive level of p65 in adult rat brain in vivo. Brain was obtained from normal old rats of post-reproductive age and constitutive level of p65 was assessed in tissue sections by double-labeled immunofluorescence. Astroglia were labeled with GFAP and neurons were labeled with NeuN, which selectively stains neuronal perikarya and nuclei and is often used to detect neurons in brain sections [31]. In our study, the differential expression pattern of p65 was distinctly observed in adult brain cells, where astroglia were more p65 positive than neurons (Fig. 5e). Quantification of p65 positive cells by three unrelated investigators (by counting color defined cells from several micrographs) revealed a significant difference in immuno-reactivity of neuronal and astroglial population for p65 (Fig. 5f). Together, this validates the difference in constitutive level of p65 also as an in vivo phenomenon.
Astrocytes are resistant to gp120 due to adequate level of NF-κB
Astroglia, when treated with NF-κB inhibitor NBD peptide, showed dramatic loss of MnSOD (Fig. 4c). In order to correlate this MnSOD depletion with mitochondrial activity and survival efficiency in these cells, we performed MTT assay. As revealed by the MTT assay in figure 6a, astroglia pretreated with wild type, but not the mutated NBD peptide, failed to resist gp120 challenge. This indicates an important role of gp120-induced MnSOD in mediating the astroglial resistance to the viral protein.
Figure 6. NF-κB p65 level is critical for astroglial resistance to gp120.
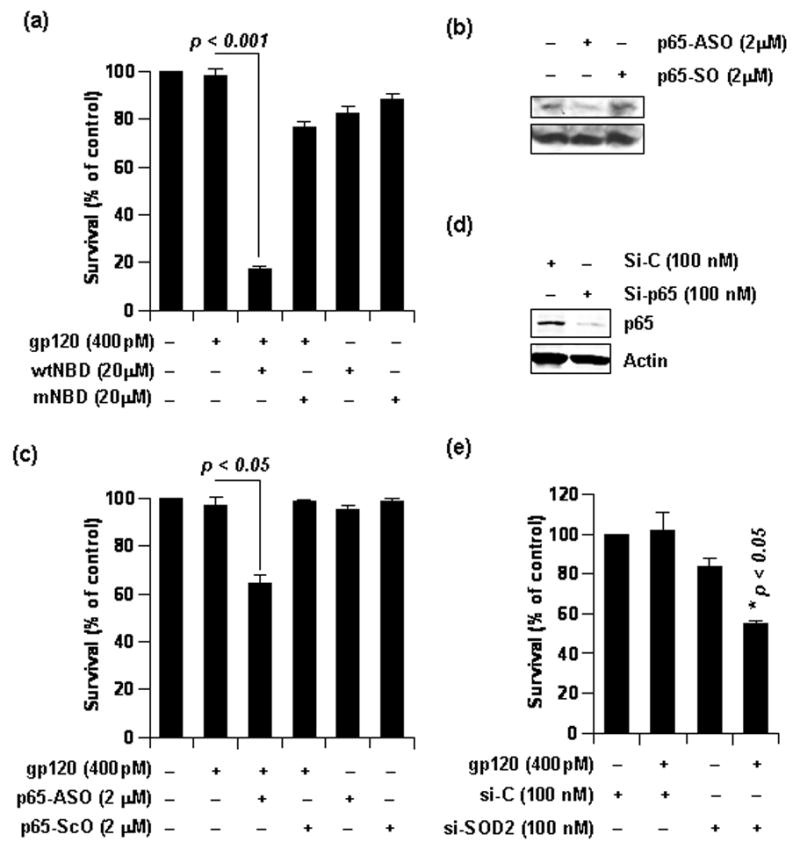
(a) Rat astrocytes were pretreated with indicated dose of either wild type NBD peptide (wNBD) or mutated NBD peptide (mNBD) for 2 hours prior to gp120 treatment for another 24 hours. Both treatments were in serum free media. MTT assay was subsequently conducted. Data was tested with t-test statistics and is expressed as mean ± SD of three separate experiments. (b) Rat astrocytes were treated with either p65-antisense oligonucleotides or p65-sense oligonucleotides for 48 hours in complete media. Then, lysate was obtained from these cells and was separated by electrophoresis and immuno-blotted for p65 to test efficiency of the antisense oligonucleotide. The same blot was stripped and re-probed for Actin. (c) Rat astrocytes, pre-treated as in (b), were subjected to further treatment with gp120 for 24 hours in serum free media. MTT assay was subsequently conducted. Data was tested with t-test statistics and is expressed as mean ± SD of three separate experiments. (d) Astrocytes were transfected with 100nM si-C or si-p65 and whole cell lysate was obtained after 48 hours. Then, samples were separated by electrophoresis and immuno-blotted for MnSOD to test efficiency of si-p65. The blot was re-probed for Actin. (b) Cells were transfected with 100nM si-C or si-SOD2 for 48 hours and then were treated with 400 pM gp120 for 24 hours. MTT assay was subsequently conducted. Data was tested with t-test statistics and is expressed as mean ± SD of three separate experiments.
Correlating contradistinctive level of p65 in neurons and astroglia and the differential gp120-responsive survival rate of these cells, we next hypothesized that greater presence of p65 renders astroglia with additional survival advantage. In order to further explore this possibility, we knocked down p65 level in astroglia by using p65 antisense oligonucleotides [32]. Phosphorothioate-labeled antisense oligonucleotides (ASO: 5′-GGG GAA CAG TTC GTC CAT GGC-3′) and sense oligonucleotides (SO: 5′-GCC ATG GAC GAA CTG TTC CCC-3′), obtained from Invitrogen, were used to treat astroglia for 48 hours. It is evident from figure 6b that p65-ASO, but not p65-SO, reduced the constitutive expression of p65 protein. Utilizing this antisense, we next tested the survival efficiency of p65 knocked-down astroglia. After 48 hours of treatment with either p65-ASO or p65-SO, astrocytes were challenged with gp120 for 24 hours followed by MTT assay. Although gp120 was unable to kill untreated or p65-SO-treated astroglia, it induced significant mitochondrial dysfunction and cell death in p65-ASO-treated astroglia (Fig. 6c). To confirm further, we knocked-down p65 level by using specific siRNA against p65 (si-p65). After 48 hours of transfection, si-p65, but not si-C, reduced the level of p65 in astrocytes (Fig. 6d). When these cells were assessed for survival efficiency in response to gp120 challenge, si-p65-transfected cells exhibited greater vulnerability to the toxin than si-C-transfected cells (Fig 6e). Taken together, these observations indicate that astroglia are able to resist gp120 due to adequate level of NF-κB p65.
Over-expression of p65 in neurons elevates MnSOD, and renders them greater survival efficiency
Considering that lower MnSOD level in neurons (Fig. 2) is coupled with lower level of p65 in comparison to astroglia (Fig. 5), we next asked if neurons could generate greater level of MnSOD if provided with additional p65. To test this hypothesis, p65 was over-expressed in rat CGC neurons by transfecting them with wild type p65 expression plasmid utilizing the peptide formulation Nupherin-neuron. It has been shown that Nupherin-neuron serves as an enhancer of plasmid transfer and their expression in lipofected post-mitotic neurons by catalyzing the rate-limiting step of nuclear import in these cells [33]. Post-transfection, about 20% cells were detected to be p65 positive (Fig 7a). Next, MnSOD activity was assessed in these neurons. As seen in figure 7b, neurons transfected with the empty vector exhibited similar loss of MnSOD activity in response to gp120 treatment (Fig. 3a). However, p65-transfected neurons showed significant restoration of MnSOD activity (Fig. 7b). Along same lines, p65-transfected neurons showed higher MnSOD level than vector-transfected neurons (lane-1 and lane-3 in Fig. 7c). When challenged with gp120, p65-transfected neurons did not undergo MnSOD loss like vector-transfected neurons (lane-2 and lane-4 in Fig. 7c). To further verify the immunoblot findings, we performed immunofluorescence studies with vector- and p65-transfected neurons. A prominent increase in MnSOD immuno-reactivity was observed in cells showing detectable p65 expression (Pair of bottom panels in Fig. 7d). Also, gp120 challenge induced the loss of MnSOD in vector-transfected neurons, but not in p65-transfected neurons (MnSOD panel, Fig. 7d). Taken together, these observations suggest that overexpression of p65 suffices to enhance MnSOD level and activity in neurons and prevents the loss of this anti-oxidant enzyme in response to gp120.
Considering the compromised astroglial resistance to gp120 in the absence of optimum p65 level (Fig. 6b) and the coupling of neuronal vulnerability to lower constitutive level of p65, our next question was as follows: Would overexpression of p65 protect neurons from gp120 toxicity? Vector- and p65-transfected neurons were challenged with gp120 and the onset of apoptosis was assessed by TUNEL. Neurons with positive immuno-staining for p65 (over-expressed) were never found to overlap with TUNEL-positive apoptotic neurons (Fig. 8a). Also, as revealed in figure 8a, gp120-responsive survival efficiency improved in p65-transfected neurons in comparison to vector-transfected ones. A quantitative estimation of this increased survival efficiency was done by counting TUNEL positive cells from random microscope-fields. As revealed in figure 8b, over-expression of p65 significantly decreased gp120-mediated formation of TUNEL bodies in neurons compared to vector-transfected cells with their respective controls. This was further corroborated with MTT assessment of similar treatments (Fig. 8c), where we found significant increase in mitochondrial activity and survival of p65-expressing neurons in comparison to vector-transfected counterparts in response to gp120 insult.
Taken together, this set of observation reveals the level of p65 in neurons as a critical factor in shaping their response to gp120. Adequate p65 results in adequate expression of MnSOD, which in turn renders greater survival fitness to neurons.
Discussion
Other than being an important ROS itself, superoxide radical anion is also the precursor to production exceeds the rate at which endogenous superoxide several other harmful ROS. If O2•− dismutases can scavenge them, or if there is not enough endogenous dismutase scavengers, then excess O2•− may either reduce transition metals, which in turn can react with H2O2 to generate the lethal OH•, or react with NO• to generate deleterious peroxynitrite [34]. In turn, cellular proteins, lipid and nucleic acids may be oxidized leading to deterioration of cellular architecture dismutation holds significant importance for a cell trying to defend itself and death. Thus, O2•− against any oxidative stress-inducing toxin. In order to survive, the dismutase machinery of a cell production rate. Therefore, only those cells can survive oxidative insults has to outrun the O2•− which possess most effective anti-oxidant capacity. This cascade of logic, when applied to neurodegenerative situations, immediately suggest more efficient anti-oxidant abilities of astroglia in comparison to neurons, as most forms of neurodegeneration are manifested by selective death of neurons, not astroglia.
In brain cells, the main source of O2•− production is the electron transport chains in mitochondria [3]. Also, mitochondrial dysfunction has been intimately linked with neurodegeneration [35]. Thus, in order to verify our hypothesis, we focused on the mitochondrial form of SOD, MnSOD. Our studies reveal a significant difference in steady-state MnSOD activity and protein level in neurons and astroglia. Thus, to begin with, we found the neuronal dismutase machinery to be quantitatively and functionally less efficient than that of astroglia.
The situation worsenes when cells face gp120 challenge. Quite surprisingly, neurons undergo loss of MnSOD, which is known to be a ROS target and is nitrated in response to neurodegenerative stimuli [36]. Since, oxidized and nitrated proteins are tagged for proteosomal degradation [3], neuronal MnSOD may very well undergo the same fate. Subsequently, in the absence of optimum p65 level, any compensatory replenishment of MnSOD does not occur in neurons resulting in the visible loss of the enzyme.
On the other hand, astrocytes demonstrate an enhancement in MnSOD activity and level in response to gp120 treatment. This observation may serve to explain greater resistance of astroglia to gp120. Since this increase in MnSOD was signal-dependent, we were prompted to investigate the involvement of NF-κB in the process. NF-κB has been previously shown to induce MnSOD in astroglia in response to a different inducer [20] and is also considered indispensable for the purpose [37]. Both, murine and human sod2 houses critical κB-sites in their 5′-proximal promoter region and in TNF-responsive element in second intron [38]. We wondered if gp120 also utilized NF-κB in primary astrocytes. Indeed, as indicated by our NBD peptide data, NF-κB activity was required for MnSOD upregulation in astrocytes.
Considering that astrocytes could upregulate MnSOD protein level utilizing NF-κB, the next question was as follows: Why could the neurons not do so? To answer this question, we compared the protein level of NF-κB p65 in these cells. Quite interestingly, as revealed by our data, steady-state p65 level in neurons was significantly less in comparison to astroglia. Such limited availability of p65 may serve to explain failure of gp120-treated neurons to induce MnSOD. Also, it serves as a basis for overall low constitutive level of MnSOD in neurons as over-expression of p65 led to greater level of MnSOD in neurons. Interestingly, this excess MnSOD in p65-transfected neurons appeared more stable in terms of quantity and activity in the face of gp120 challenge. Since enhanced level of MnSOD has been shown to confer greater survival efficiency to neurons [18, 20], it was hardly surprising to observe p65-transfected neurons manifest improved survival efficiency than vector-transfected ones. Also, specific blockage of NF-κB by NBD peptide negated MnSOD compensation in astrocytes, thereby rendering them vulnerable to gp120 like neurons. Taken together, these observations reveal an important role of NF-κB p65 level in maintaining the level of MnSOD in neurons and astroglia, which in turn dictates their survival competence to gp120.
At this point, our observations stand in apparent contradiction with recent reports suggesting that p65 is deleterious for neurons and upregulation of MnSOD is contingent on c-Rel, the other NF-κB subunit [39, 40]. With gp120, over-expression of p65 has led to neuronal protection in our study, while anti-sense knock down of p65 led to amelioration of glutamate-induced cell death in the previous study [40]. Since NF-κB has been previously shown to play either a pro- or an anti-apoptotic role in neurons depending solely on the stimulus [41], this difference could result from different inducers used in these two studies. Also, functional redundancy and compensation among various five subunits of the NF-κB family is well recognized [42]. Thus increase of MnSOD level in p65-transfected neurons is possible as is regulation of MnSOD by c-Rel in response to mGlu5 receptor agonist [39].
In summary, we have demonstrated that higher constitutive level of MnSOD in astroglia arm them with greater survival advantage in comparison to neurons during gp120 toxicity. Such difference in MnSOD level may be attributed, at least in part, to greater level of NF-κB p65 in astrocytes, which also allows them to generate more of the anti-oxidant enzyme when challenged with gp120. Neurons on the other hand, with lower level of p65, lack adequate constitutive MnSOD level and also fail to compensate it when treated with gp120. Cumulatively, they succumb to lethal effects of gp120 (Fig. 9). These revelations proffer the therapeutic possibility of strengthening neuronal anti-oxidant machinery by exogenously enhancing neuronal p65 level in HIV infected patients.
Figure 9. Overview.
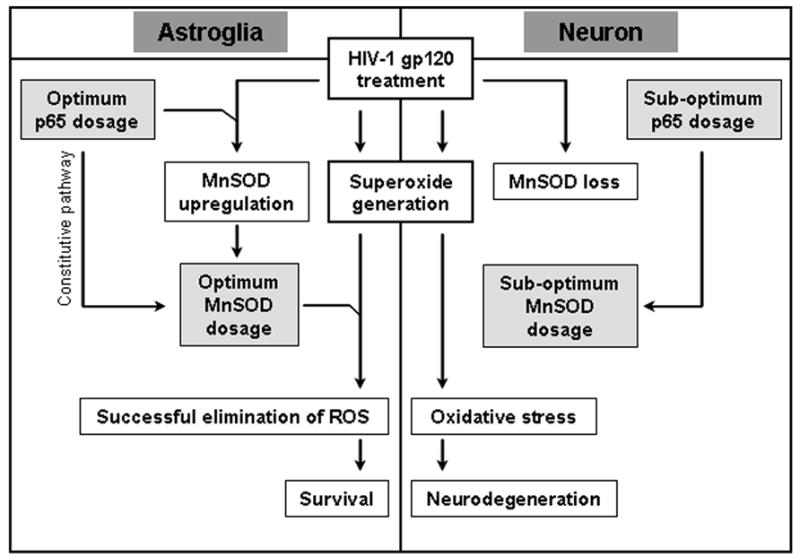
HIV-1 gp120 generates superoxide in both astroglia and neurons. However, only neurons are vulnerable to gp120. Due to an optimum constitutive level of MnSOD, astroglia can eliminate the oxidative threat and survive. Neurons fail to do so due to sub-optimum level of MnSOD in them. Also, gp120 treatment upregulates MnSOD expression in astroglia but leads to a loss of MnSOD in neurons. In astrocytes, higher constitutive MnSOD level and its upregulation in response to gp120 is possible due to a higher level of NF-κB p65 in them. Neurons are vulnerable to gp120 as they have a lower level of NF-κB p65 in them and as a consequence, lack adequate level of MnSOD to eliminate superoxide stress generated by the viral toxin.
Acknowledgments
This study was supported by grants from NIH (NS39940 and NS48923), National Multiple Sclerosis Society (RG3422A1/1) and Michael J. Fox Foundation for Parkinson Research to KP and UNMC graduate research assistantship to RNS. The authors wish to thank Terry Fangman and Dr. You Zhou (University of Nebraska at Lincoln) for their assistance with microscopy, Marian Schmid (University of Nebraska Medical Center) for excellent animal care. Authors also acknowledge Pahan lab members who helped in data quantification.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Klein JA, Ackerman SL. Oxidative stress, cell cycle, and neurodegeneration. The Journal of clinical investigation. 2003;111:785–793. doi: 10.1172/JCI18182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krantic S, Mechawar N, Reix S, Quirion R. Molecular basis of programmed cell death involved in neurodegeneration. Trends in neurosciences. 2005;28:670–676. doi: 10.1016/j.tins.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Halliwell B. Oxidative stress and neurodegeneration: where are we now? Journal of neurochemistry. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 4.Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? The Journal of clinical investigation. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mollace V, Nottet HS, Clayette P, Turco MC, Muscoli C, Salvemini D, Perno CF. Oxidative stress and neuroAIDS: triggers, modulators and novel antioxidants. Trends in neurosciences. 2001;24:411–416. doi: 10.1016/s0166-2236(00)01819-1. [DOI] [PubMed] [Google Scholar]
- 6.Pocernich CB, Sultana R, Mohmmad-Abdul H, Nath A, Butterfield DA. HIV-dementia, Tat-induced oxidative stress, and antioxidant therapeutic considerations. Brain research. 2005;50:14–26. doi: 10.1016/j.brainresrev.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Jana A, Pahan K. Human immunodeficiency virus type 1 gp120 induces apoptosis in human primary neurons through redox-regulated activation of neutral sphingomyelinase. J Neurosci. 2004;24:9531–9540. doi: 10.1523/JNEUROSCI.3085-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nath A, Sacktor N. Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Current opinion in neurology. 2006;19:358–361. doi: 10.1097/01.wco.0000236614.51592.ca. [DOI] [PubMed] [Google Scholar]
- 9.Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA. HIV-1 infection and AIDS: consequences for the central nervous system. Cell death and differentiation. 2005;12(Suppl 1):878–892. doi: 10.1038/sj.cdd.4401623. [DOI] [PubMed] [Google Scholar]
- 10.Saha RN, Pahan K. Tumor necrosis factor-alpha at the crossroads of neuronal life and death during HIV-associated dementia. Journal of neurochemistry. 2003;86:1057–1071. doi: 10.1046/j.1471-4159.2003.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mattson MP, Haughey NJ, Nath A. Cell death in HIV dementia. Cell death and differentiation. 2005;12(Suppl 1):893–904. doi: 10.1038/sj.cdd.4401577. [DOI] [PubMed] [Google Scholar]
- 12.Dawson TM, Dawson VL. gp120 neurotoxicity in primary cortical cultures. Advances in neuroimmunology. 1994;4:167–173. doi: 10.1016/s0960-5428(06)80253-6. [DOI] [PubMed] [Google Scholar]
- 13.Bachis A, Major EO, Mocchetti I. Brain-derived neurotrophic factor inhibits human immunodeficiency virus-1/gp120-mediated cerebellar granule cell death by preventing gp120 internalization. J Neurosci. 2003;23:5715–5722. doi: 10.1523/JNEUROSCI.23-13-05715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free radical biology & medicine. 2002;33:337–349. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]
- 15.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang LP, Patel M. Mitochondrial oxidative stress and increased seizure susceptibility in Sod2(−/+) mice. Free radical biology & medicine. 2004;36:542–554. doi: 10.1016/j.freeradbiomed.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 17.Browne SE, Ferrante RJ, Beal MF. Oxidative stress in Huntington’s disease. Brain pathology (Zurich, Switzerland) 1999;9:147–163. doi: 10.1111/j.1750-3639.1999.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valcour V, Shiramizu B. HIV-associated dementia, mitochondrial dysfunction, and oxidative stress. Mitochondrion. 2004;4:119–129. doi: 10.1016/j.mito.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 20.Bruce-Keller AJ, Geddes JW, Knapp PE, McFall RW, Keller JN, Holtsberg FW, Parthasarathy S, Steiner SM, Mattson MP. Anti-death properties of TNF against metabolic poisoning: mitochondrial stabilization by MnSOD. Journal of neuroimmunology. 1999;93:53–71. doi: 10.1016/s0165-5728(98)00190-8. [DOI] [PubMed] [Google Scholar]
- 21.Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-kappaB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. Journal of neuroscience research. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 22.Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21:6480–6491. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saha RN, Liu XJ, Pahan K. Up-regulation of BDNF in Astrocytes by TNF-a: A Case for the Neuroprotective Role of Cytokine. J Neuroimmune Pharmacol. 2006;1:212–222. doi: 10.1007/s11481-006-9020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jana M, Anderson JA, Saha RN, Liu X, Pahan K. Regulation of inducible nitric oxide synthase in proinflammatory cytokine-stimulated human primary astrocytes. Free radical biology & medicine. 2005;38:655–664. doi: 10.1016/j.freeradbiomed.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 25.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- 26.Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D’Emilia DM, Friedlander RM, Yuan J, Masliah E, Lipton SA. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J Neurosci. 2002;22:4015–4024. doi: 10.1523/JNEUROSCI.22-10-04015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh IN, Goody RJ, Dean C, Ahmad NM, Lutz SE, Knapp PE, Nath A, Hauser KF. Apoptotic death of striatal neurons induced by human immunodeficiency virus-1 Tat and gp120: Differential involvement of caspase-3 and endonuclease G. Journal of neurovirology. 2004;10:141–151. doi: 10.1080/13550280490441103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bossis G, Salinas S, Cartier C, Devaux C, Briant L. NF-kappaB activation upon interaction of HIV-1 envelope glycoproteins with cell surface CD4 involves IkappaB kinases. FEBS letters. 2002;516:257–264. doi: 10.1016/s0014-5793(02)02566-8. [DOI] [PubMed] [Google Scholar]
- 29.Briant L, Robert-Hebmann V, Acquaviva C, Pelchen-Matthews A, Marsh M, Devaux C. The protein tyrosine kinase p56lck is required for triggering NF-kappaB activation upon interaction of human immunodeficiency virus type 1 envelope glycoprotein gp120 with cell surface CD4. Journal of virology. 1998;72:6207–6214. doi: 10.1128/jvi.72.7.6207-6214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strickland I, Ghosh S. Use of cell permeable NBD peptides for suppression of inflammation. Annals of the rheumatic diseases. 2006;65(Suppl 3):iii75–iii82. doi: 10.1136/ard.2006.058438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development (Cambridge, England) 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 32.Autieri MV, Yue TL, Ferstein GZ, Ohlstein E. Antisense oligonucleotides to the p65 subunit of NF-kB inhibit human vascular smooth muscle cell adherence and proliferation and prevent neointima formation in rat carotid arteries. Biochemical and biophysical research communications. 1995;213:827–836. doi: 10.1006/bbrc.1995.2204. [DOI] [PubMed] [Google Scholar]
- 33.Subramanian A, Ranganathan P, Diamond SL. Nuclear targeting peptide scaffolds for lipofection of nondividing mammalian cells. Nature biotechnology. 1999;17:873–877. doi: 10.1038/12860. [DOI] [PubMed] [Google Scholar]
- 34.Turrens JF. Mitochondrial formation of reactive oxygen species. The Journal of physiology. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeevalk GD, Bernard LP, Song C, Gluck M, Ehrhart J. Mitochondrial inhibition and oxidative stress: reciprocating players in neurodegeneration. Antioxidants & redox signaling. 2005;7:1117–1139. doi: 10.1089/ars.2005.7.1117. [DOI] [PubMed] [Google Scholar]
- 36.Anantharaman M, Tangpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK, St Clair DK. Beta-amyloid mediated nitration of manganese superoxide dismutase: implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. The American journal of pathology. 2006;168:1608–1618. doi: 10.2353/ajpath.2006.051223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maehara K, Hasegawa T, Isobe KI. A NF-kappaB p65 subunit is indispensable for activating manganese superoxide: dismutase gene transcription mediated by tumor necrosis factor-alpha. Journal of cellular biochemistry. 2000;77:474–486. doi: 10.1002/(sici)1097-4644(20000601)77:3<474::aid-jcb12>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 38.Guo Z, Boekhoudt GH, Boss JM. Role of the intronic enhancer in tumor necrosis factor-mediated induction of manganous superoxide dismutase. The Journal of biological chemistry. 2003;278:23570–23578. doi: 10.1074/jbc.M303431200. [DOI] [PubMed] [Google Scholar]
- 39.Pizzi M, Sarnico I, Boroni F, Benarese M, Steimberg N, Mazzoleni G, Dietz GP, Bahr M, Liou HC, Spano PF. NF-kappaB factor c-Rel mediates neuroprotection elicited by mGlu5 receptor agonists against amyloid beta-peptide toxicity. Cell death and differentiation. 2005;12:761–772. doi: 10.1038/sj.cdd.4401598. [DOI] [PubMed] [Google Scholar]
- 40.Pizzi M, Goffi F, Boroni F, Benarese M, Perkins SE, Liou HC, Spano P. Opposing roles for NF-kappa B/Rel factors p65 and c-Rel in the modulation of neuron survival elicited by glutamate and interleukin-1beta. The Journal of biological chemistry. 2002;277:20717–20723. doi: 10.1074/jbc.M201014200. [DOI] [PubMed] [Google Scholar]
- 41.Kaltschmidt B, Heinrich M, Kaltschmidt C. Stimulus-dependent activation of NF-kappaB specifies apoptosis or neuroprotection in cerebellar granule cells. Neuromolecular medicine. 2002;2:299–309. doi: 10.1385/NMM:2:3:299. [DOI] [PubMed] [Google Scholar]
- 42.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nature reviews. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]