

Abstract
Multiple sclerosis (MS) is a chronic autoimmune demyelinating disorder of the central nervous system (CNS) of unknown etiology. Several studies have shown that demyelination in MS is caused by proinflammatory mediators which are released by perivascular infiltrates and/or activated glial cells. To understand if proinflammatory mediators such as IL (interleukin)-1β and TNF (tumor necrosis factor)-α are capable of modulating the expression of myelin-specific genes, we investigated the effect of these cytokines on the expression of myelin basic protein (MBP), 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase), myelin oligodendrocyte glycoprotein (MOG), and proteolipid protein (PLP) in human primary oligodendrocytes. Interestingly, both IL-1β and TNF-α markedly inhibited the expression of MOG, CNPase, and PLP but not MBP, the effect that was blocked by antioxidants such as N-acetylcysteine (NAC) and pyrrolidine dithiocarbamate (PDTC). Consistently, oxidants and prooxidants like H2O2 and diamide also markedly inhibited the expression of MOG, CNPase, and PLP. Furthermore, both IL-1β and TNF-α induced the production of H2O2. Taken together, these studies suggest that proinflammatory cytokines inhibit the expression of myelin genes in human primary oligodendrocytes through the alteration of cellular redox.
Keywords: Human primary oligodendrocytes, Myelin gene expression, Proinflammatory cytokines, Reactive oxygen species
Introduction
Multiple sclerosis (MS) is a chronic human demyelinating disorder of the central nervous system (CNS). From a clinical standpoint, MS symptoms are diverse, ranging from tremor, nystagmus, paralysis, and disturbances in speech and vision; it occurs chiefly in early adult life with characteristic exacerbations and remissions [1]. Pathologically, it can be identified by the presence of diffuse, discrete demyelinated areas, called plaques. Although the etiology of MS is not completely understood, studies of MS patients suggest that the observed demyelination in the CNS is a result of an autoimmune inflammation [1,2]. Consistently, demyelinated areas in the CNS of MS patients are associated with an inflammatory reaction orchestrated by activated T cells, macrophages, and endogenous glial cells (astroglia and microglia). Activated T cells, macrophages, and endogenous glial cells produce a variety of proinflammatory and neurotoxic factors, including proinflammatory cytokines [3]. Among proinflammatory cytokines, primary inflammatory cytokines, such as interleukin (IL)-1β/α and tumor necrosis factor (TNF)α/β, play a predominant role since they are involved at multiple levels of neuroimmune regulation [3-5]. Analysis of cerebrospinal fluid (CSF) from MS patients has shown increased levels of proinflammatory cytokines compared with normal control, and levels of those cytokines in the CSF of MS patients also correlate with disease severity [3,6,7]. Consistently, blockade of proinflammatory cytokine synthesis or function by signaling inhibitors or neutralizing antibodies or gene knockout can also prevent the development of EAE [8-10]. While all these studies suggested an important role of proinflammatory cytokines in pathogenesis of EAE and MS, recent clinical trials with anti-TNF antibodies in patients with MS caused disease exacerbation [11,12]. Therefore, it may not be wise to block the synthesis of proinflammatory cytokines, such as TNF-α, in the inflamed CNS of MS patients. Due to autocrine and paracrine modes of action, overlapped activities, and pleiotropic function, these cytokines may induce the activation of certain factors from activated astroglia or microglia that may eventually support the growth of oligodendrocytes. Because demyelination in the CNS of MS patients is associated with widespread inflammation, and inflammation is also thought to be an exacerbating factor for MS, another logical way would be to block cytokine-mediated toxic signaling pathways in the CNS, if any.
Oligodendrocytes are the myelinating cells of the CNS that extend membranous processes to form multilamellar structures around axons, facilitating conduction of nervous impulses [13,14]. Therefore, we investigated the effect of proinflammatory cytokines on the expression of myelin-specific genes in human primary oligodendrocytes. Here we demonstrate that IL-1β and TNF-α were capable of inhibiting the expression of 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase), myelin oligodendrocyte glycoprotein (MOG) and proteolipid protein (PLP) but not myelin basic protein (MBP). In addition, we also show that proinflammatory cytokine-mediated down-regulation of myelin genes was redox sensitive.
Materials and methods
Reagents
Fetal bovine serum, Hank’s balanced salt solution (HBSS), trypsin and DMEM/F-12 were from Mediatech (USA). Recombinant human TNF-α and IL-1β were obtained from R&D (USA). N-Acetylcysteine, pyrrolidine dithiocarbamate, diamide, and H2O2 were purchased from Sigma.
Isolation of human primary oligodendrocytes
Human fetal tissue was obtained from the Human Embryology Laboratory, University of Washington, Seattle. All of the experimental protocols were reviewed and approved by the Institutional Review Board (IRB Number 224-01-FB) of the University of Nebraska Medical Center. Briefly, 11- to 17-week-old fetal brains obtained from the Human Embryology Laboratory (University of Washington, Seattle, WA) were dissociated by trituration and trypsinization (0.25% trypsin in PBS at 37°C for 15 min). The trypsin was inactivated with 10% heat-inactivated FBS (Mediatech, Washington, DC). The dissociated cells were filtered successively through 380- and 140-μm meshes (Sigma, St. Louis, MO) and pelleted by centrifugation. The resulting suspension was centrifuged for 10 min at 1500 rpm and then resuspended in DMEM supplemented with 20% heat-inactivated FBS. Cells were plated on poly-d-lysine-coated 75 cm2 flasks and incubated at 37°C with 5% CO2 in air. Culture medium was changed after every 3 days. The initial mixed glial cultures, grown for 9 days, were placed on a rotary shaker at 240 rpm at 37°C for 2 h to remove loosely attached microglia. The olgodendrocytes were detached after shaking for 18 h at 200 rpm at 11 days. To purify oligodendrocytes from astrocytes and microglia, the detached cell suspension was plated in tissue culture dishes (2 × 106 cells/100 mm) for 60 min at 37°C. This step was repeated twice for nonadherent cells to minimize the contamination. The nonadhering cells, mostly oligodendrocytes, were seeded into poly-d-lysine-coated 6-well culture plates in complete medium at 37°C with 5% CO2 in air.
Induction of EAE
Specific pathogen-free female SJL/J mice (4–6 weeks old) were purchased from Harlan Sprague Dawley, Inc. (IN). Relapsing-remitting EAE was induced by adoptive transfer of MBP-primed T cells as described earlier [15,16]. Briefly, donor mice were immunized sc with 400 μg bovine MBP (Invitrogen) and 60 μg M. tuberculosis (H37RA; Difco Labs) in IFA (Calbiochem). Animals were killed 10–12 days postimmunization and the draining lymph nodes were harvested. Single cell suspensions were cultured at a concentration of 4–5 × 106 cells/ml in 6-well plates in RPMI 1640 supplemented with 10% FBS, 50 μg/ml MBP, 50 μM 2-ME, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. On Day 4, cells were harvested and resuspended in HBSS. Then 2 × 107 viable cells in a volume of 200 μl were injected into the tail vein of naive mice. Pertussis toxin (150 ng/mouse, Sigma) was injected once via intraperitoneal (ip route on 0 day posttransfer (dpt) of cells. Cells isolated from donor mice immunized with CFA or IFA alone were not viable after 4 days in culture with 50 μg/ml bovine MBP and therefore were not transferred. MBP reactivity of lymph node cells (LNC) was measured by [3H]thymidine (NEN) incorporation assay of parallel microplate cultures [15,16]. Animals were observed daily for clinical symptoms. Experimental animals were scored by a masked investigator as follows: 0, no clinical disease; 0.5, piloerection; 1, tail weakness; 1.5, tail paralysis; 2, hind limb weakness; 3, hind limb paralysis; 3.5, forelimb weakness; 4, forelimb paralysis; 5, moribund or death.
Immunostaining of GalC, CNPase, MOG, PLP, and GFAP
Immunostaining was performed as described earlier [17]. Briefly, coverslips containing 200–300 cells/mm2 were fixed with 4% paraformaldehyde for 15 min, followed by treatment with cold ethanol (−20°C) for 5 min and two rinses in PBS. Samples were blocked with 3% BSA in PBS containing Tween 20 (PBST) for 30 min and incubated in PBST containing 1% BSA and goat anti-GalC (1:50), rabbit anti-GFAP (1:50), goat anti-MOG (1:50), rabbit anti-PLP (1:70) and rabbit anti-CNPase (1:50). After three washes in PBST (15 min each), slides were further incubated with Cy5 (Jackson ImmunoResearch, West Grove, PA). For negative controls, a set of culture slides was incubated under similar conditions without the primary antibodies. The samples were mounted and observed under a Bio-Rad (Hercules, CA) MRC1024ES confocal laser-scanning microscope [16,17].
Assay of hydrogen peroxide (H2O2) production
The production of H2O2 by human primary oligodendrocytes was quantified by a colorimetric H2O2 kit (Assay designs, Inc) as described earlier [18]. Briefly, different amounts of supernatants were allowed to react with 100 μl of a color reagent containing xylenol orange in an acidic solution with sorbitol and ammonium iron sulfate followed by reading of the optical density at 550 nm.
Semiquantitative real-time polymerase chain reaction (RT-PCR) analysis
Total RNA was isolated from human oligodendrocytes and mouse cerebral tissues using RNA-Easy Qiagen kit following manufactures protocol. To remove any contaminating genomic DNA, total RNA was digested with DNase. Semiquantitative RT-PCR was carried out as described earlier [16] using oligo(dT)12 – 18 as primer and MMLV reverse transcriptase (Clontech) in a 20-μl reaction mixture. The resulting cDNA was appropriately diluted, and diluted cDNA was amplified using Titanium Tag polymerase and the following primers.
Primers for human myelin genes
The primers for MBP (284 bp) were sense, 5′-GGA AAC CAC GCA GGC AAA CGA GA-3′; antisense, 5′-GAA AAG AGG CGG ATC AAG TGG GG-3′.
The primers for PLP (343 bp) were sense, 5′-CTT CCC TGG TGG CCA CTG GAT TGT-3; antisense, 5′-TGA TGT TGG CCT CTG GAA CCC CTC-3′.
The primers for MOG (218 bp) were sense, 5′-TCC TCC TCC TCC TCC AAG TGT CT-3′; antisense, 5′-AGT GGG GAT CAA AAG TCC GGT GG-3′.
The primers for CNPase (302 bp) were sense, 5′-GGC CAC GCT GCT AGA GTG CAA GAC-3′; antisense, 5′-GGT ACT GGT ACT GGT CGG CCA TTT-3′.
The primers for GAPDH (300 bp) were sense, 5′-GGT GAA GGT CGG AGT CAA CG-3′; antisense, 5′-GTG AAG ACG CCA GTG GAC TC-3′.
Primers for mouse myelin genes
The primers for PLP (280 bp) were sense, 5′-CTTCCCTGGTGGCCACTGGATTGT-3′; antisense, 5′-CCGCAGATGGTGGTCTTGTAGTCG-3′.
The primers for MOG (432 bp) were sense, 5′-CCTCTCCCTTCTCCTCCTTC-3′; antisense, 5′-AGAGTCAGCACACCGGGGTT-3′.
The primers for CNPase (339 bp) were sense, 5′-CTACCCTCCACGAGTGCAAGACGCT-3′; antisense, 5′-AGTCTAGTCGCCACGCTGTCTTGGG-3′.
Primers for mouse proinflammatory cytokine genes
The primers for TNF-α (354 bp) were sense, 5′-TTCTGTCTACTGAACTTCGGGGTGATCGGTCC-3′; antisense, 5′-GTATGAGATAGCAAATCGGCTGACGGTGTGGG-3′.
The primers for IL-1β (563 bp) were sense, 5′-ATGGCAACTGTTCCTGAACTCAACT-3′; antisense, 5′-CAGGACAGGTATAGATTCTTTCCTTT-3′.
The primers for GAPDH (342 bp) were sense, 5′-GGTGAAGGTCGGT GTGAACG-3′; antisense, 5′-TTGGCTCCACCCTTCAAGTG-3′.
Amplified products were electrophoresed on a 1.8% agarose gels and visualized by ethidium bromide staining. Message for the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene was used to ascertain that an equivalent amount of cDNA was synthesized from different samples.
RT-PCR analysis
RT-PCR analysis was performed using theABI-Prism7700 sequence detection system (Applied Biosystems, Foster City, CA) as described earlier [16]. Briefly, it was performed in a 96-well optical reaction plate (Applied Biosystem) on cDNA equivalent to 50 ng DNase-digested RNA in a volume of 25 μl, containing 12.5 μl TaqMan Universal Master Mix and optimized concentrations of FAM-labeled probe, forward and reverse primers following the manufacturer’s protocol. All primers and FAM-labeled probes for human PLP, MOG CNPase, and GAPDH were obtained from Applied Biosytems. The mRNA expression of myelin genes was normalized to the label of GAPDH mRNA. Data were processed by the ABI Sequence Detection System 1.6 software and analyzed by ANOVA.
Results
Expression of different myelin-specific markers (GalC, MOG, PLP, and CNPase) in human fetal oligodendrocytes
Mature oligodendrocytes arise from oligodendrocytes progenitor cells (OP) through a series of developmental stages, e.g., proliferative prooligodendrocyte stage, nonproliferative immature oligodendrocyte stage, and finally nonproliferative mature oligodendrocyte stage [13,14]. These developmental stages are marked by sequential expression of developmental markers such as immature oligodendrocytes express galactocerebroside (GalC) while only mature oligodendrocytes express myelin basic protein and proteolipid protein [13,14]. To investigate if primary oligodendrocytes isolated from 11- to 17-week-old fetal brains were mature and expressed different myelin-specific markers, purified oligodendrocytes were immunostained with antibodies against GalC, MOG, PLP, and CNPase. As shown in Fig. 1A, human primary oligodendrocytes readily expressed GalC, MOG, PLP, and CNPase. These cells also expressed MBP (data not shown). However, the expression of GFAP (a specific marker for astrocytes) was rarely observed (one astrocyte out of hundred oligodendroglia) in this preparation (Fig. 1B). By semiquantitative RT-PCR, we were also unable to detect any GFAP mRNA in purified oligodendrocytes (data not shown). Taken together, these studies suggest that human primary oligodendrocytes were mature and about 99% pure.
Fig. 1. Immunofluorescence analysis of human primary oligodendrocytes for the expression of GalC, MOG, CNPase, PLP, and GFAP.
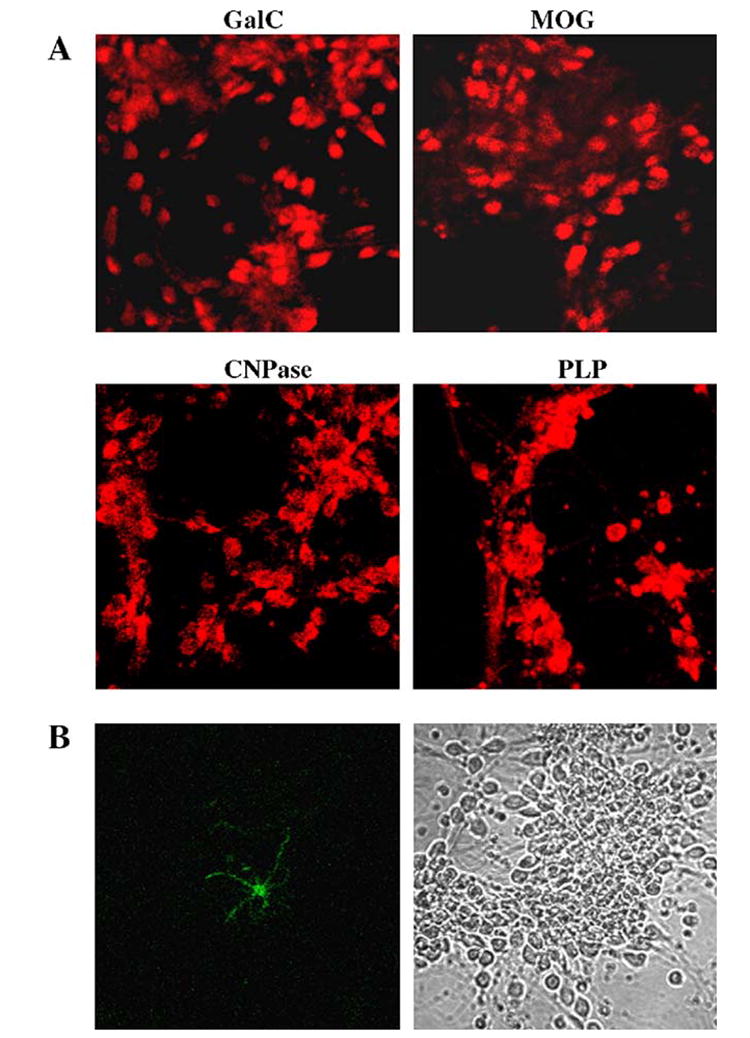
(A) Differentiated oligodendrocytes were immunostained with antibodies against GalC, MOG, CNPase, and PLP as described under Materials and methods. (B) To examine the purity of oligodendrocytes, cells were also immunostained for astroglial marker GFAP (left panel, GFAP; right panel, phase contrast).
Effect of TNF-α and IL-1β on the expression of myelin-specific genes in human primary oligodendrocytes
To investigate the effect of proinflammatory cytokines on the expression of myelin-specific genes in primary oligodendrocytes, cells were treated with different concentrations of TNF-α and IL-1β for 18 h. The expression of MBP, MOG, PLP, and CNPase was analyzed by semiquantitative RT-PCR. It is clearly evident from Fig. 2 that both TNF-α and IL-1β dose dependently inhibited the expression MOG, PLP, and CNPase. Marked decrease in the expression of MOG mRNA was observed at 20 ng/ml of TNF-α and IL-1β while down-regulation of CNPase mRNA expression was evident at 30 ng/ml of these cytokines. On the other hand, the expression of PLP mRNA was affected moderately by treatment with TNF-α and IL-1β (Fig. 2). Although MBP is a major constituent of both peripheral and CNS myelin sheaths and plays a major role in myelin membrane formation, the expression of MBP remained unaltered under the same treatment condition (Fig. 2). These results suggest that the decreased expression of MOG, PLP, and CNPase in cytokine-challenged oligodendrocytes is not due to cell death. To further confirm the effect of proinflammatory cytokines on the expression of myelin genes, we treated primary oligodendrocytes with 30 ng/ml of either TNF-α or IL-1β for different time periods, and the relative expression of MOG, PLP, and CNPase was determined by quantitative RT-PCR. The results revealed a gradual decrease in the expression of MOG, PLP, and CNPase mRNAs with increase in time of incubation (Fig. 3). The decrease in myelin gene expression was evident even at 9 h of incubation (Fig. 3). This effect was specifically striking for MOG that decreased by about 50 to 55% after 9 h and by about 70 to 80% after 18 h of treatment with proinflammatory cytokines (Fig. 3). However, TNF-α was more potent than IL-1β in inhibiting the expression of different myelin genes.
Fig. 2. Effect of TNF-α and IL-1β on the expression of myelin-specific genes in human primary oligodendrocytes.

(A) Differentiated oligodendrocytes were treated with different concentrations (10, 20, and 30 ng/ml) of either TNF-α or IL-1β. After 18 h of treatment, total RNA was analyzed for the expression of MBP, MOG, PLP, and CNPase by semiquantitative RT-PCR as described under Materials and methods. (B) The relative mRNA expression of myelin genes (MBP, MOG, PLP, or CNPase/GAPDH) after TNF-α and IL-1β treatment was measured after scanning the bands with a Fluor Chem 8800 Imaging System (Alpha Innotech Corporation). Results are means ± SD of three different experiments.
Fig. 3. Real-time PCR analysis of different myelin-specific genes in TNF-α and IL-1β-treated human primary oligodendrocytes.

Differentiated oligodendrocytes were treated with 30 ng/ml of either TNF-α or IL-1β. After different hours of treatment, total RNA was analyzed for the expression of MOG, PLP, and CNPase by quantitative real-time PCR analysis as described under Materials and methods. Results are means ± SD of three different experiments.
Next we determined the effect of these proinflammatory cytokines on the expression of MOG, PLP, and CNPase proteins in primary oligodendrocytes. After treatment with 30 ng/ml of either TNF-α or IL-1β for 18 h, cells were immunostained with antibodies against MOG, PLP, and CNPase. Consistent to the inhibition of mRNA expression, both TNF-α and IL-1β markedly reduced the protein expression of MOG, PLP, and CNPase as evident from decreased immunoreactivity of MOG, PLP, and CNPase in cytokine-treated cells compared to untreated cells (Fig. 4). Taken together, these results suggest that proinflammatory cytokines are capable of inhibiting the expression of MOG, PLP, and CNPase in human primary oligodendrocytes at both mRNA and protein levels.
Fig. 4. Immunofluorescence analysis of MOG, PLP, and CNPase in TNF-α- and IL-1β-treated human primary oligodendrocytes.

Differentiated oligodendrocytes were treated with 30 ng/ml of either TNF-α or IL-1β, and after 24 h of treatment, cells were immunostained with antibodies against MOG, PLP, and CNPase.
Down-regulation of myelin-specific genes in cerebral tissues of mice with experimental allergic encephalomyelitis (EAE)
Because proinflammatory cytokines play a role in the pathogenesis of EAE [8-10] and these cytokines inhibited the expression of MOG, PLP, and CNPase in human primary oligodendrocytes, we investigated the mRNA expression of these myelin genes in cerebral tissues of mice with relapsing-remitting EAE. On 18 days posttransfer (dpt), EAE animals exhibiting clinical symptoms of grade 3 (hind limb paralysis) or higher [15,16] were sacrificed and cerebral tissues were analyzed for mRNA expression of proinflammatory cyto-kines and myelin genes. Consistent with proinflammatory cytokine-mediated down-regulation of myelin genes in primary oligodendroglia, Fig. 5 clearly shows a marked decrease in mRNA expression of MOG, PLP, and CNPase in cerebellum of EAE mice compared to that of control mice without EAE. On the other hand, the mRNA expression of proinflammatory cytokine genes inversely correlated to that of myelin-specific genes in cerebellum. Such as, the mRNA expression of TNF-α and IL-1β in cerebellar tissues of EAE mice was higher than that of control mice (Fig. 5).
Fig. 5. Expression of myelin-specific genes and proinflammatory cytokines in cerebellum of RR-EAE mice.

RR-EAE was induced in female SJL/J mice by adoptive transfer of MBP-primed T cells as described under Materials and methods. On 18 dpt, cerebellar tissues were taken out and total RNA was analyzed for the expression of myelin-specific genes (MOG, PLP, and CNPase) and proinflammatory cytokine genes (TNF-α and IL-1β) by semiquantitative RT-PCR. Results are representative of three separate experiments.
Antioxidants block TNF-α- and IL-1β-mediated down-regulation of myelin genes in human primary oligodendrocytes
Because reactive oxygen species (ROS) play many roles as signaling and effector molecules during inflammation [19,20], we were prompted to investigate if ROS were involved in proinflammatory cytokine-mediated down-regulation of myelin genes. We examined the effect of N-acetylcysteine (NAC) and pyrrolidine dithiocarbamate (PDTC), two common antioxidants, on cytokine-mediated down-regulation of myelin genes. Oligodendrocytes preincubated with different concentrations of NAC and PDTC for 1 h were treated with TNF-α and IL-1β for 18 h followed by semiquantitative RT-PCR analysis of myelin genes. As observed in Fig. 6, both NAC and PDTC dose dependently prevented TNF-α- and IL-1β-mediated down-regulation of myelin gene expression, suggesting that proinflammatory cytokines inhibit the expression of myelin genes in human primary oligodendrocytes via ROS.
Fig. 6. N-Acetylcysteine (NAC) and pyrrolidine dithiocarbamate (PDTC) block cytokine-induced down-regulation of myelin genes in human primary oligodendrocytes.

Cells preincubated with different concentrations of NAC and PDTC for 1 h were treated with either TNF-α (A) or IL-1β (B). After 18 h of treatment, total RNA was analyzed for the expression of MOG, PLP, and CNPase by semiquantitative RT-PCR. The relative mRNA expression of myelin genes (MOG, PLP, or CNPase/GAPDH) was measured after scanning the bands with a Fluor Chem 8800 Imaging System (Alpha Innotech Corporation). Results are means ± SD of three different experiments.
H2O2 down-regulates the expression of myelin genes in human primary oligodendrocytes
At first, we considered if H2O2 might be the factor behind proinflammatory cytokine-mediated suppression of myelin genes. Therefore, we measured the level of H2O2 in oligodendrocytes after TNF-α and IL-1β treatment. Fig. 7 shows that the production of H2O2 started within 15 min of stimulation with either TNF-α or IL-1β. However, the production of H2O2 peaked at 30 min of stimulation with TNF-α and at 60 min of stimulation with IL-1β (Fig. 7). Although there was a decrease in H2O2 production afterward, the levels were always higher than control for a prolonged time period (Fig. 7).
Fig. 7. TNF-α and IL-1β induced the production of H2O2 in human primary oligodendrocytes.

Differentiated oligodendrocytes were treated with 30 ng/ml of either TNF-α or IL-1β. At different minute intervals, the level of H2O2 was measured in supernatant as described under Materials and methods. The control group served as 100% and data from other groups were expressed as percentage of control. Results are means of two separate experiments.
Because TNF-α and IL-1β induced the production of H2O2 in oligodendrocytes, next we examined the possibility if H2O2 alone could inhibit the expression of myelin genes in oligodendrocytes. Cells were treated with different concentrations of H2O2 for 18 h followed by quantitative RT-PCR analysis of myelin genes. As shown in Fig. 8A, H2O2 dose dependently inhibited the expression of myelin genes in oligodendrocytes. Even at a concentration of 5 μM, H2O2 inhibited the expression of MOG, PLP, and CNPase by 50 to 75% (Fig. 8A). At a higher concentrations, such as 25 μM, H2O2 inhibited the expression of myelin genes by 70–86% (Fig. 8A). These results suggest that H2O2 and/or H2O2-derived oxidants are sufficient to attenuate the expression of myelin genes in human primary oligodendrocytes.
Fig. 8. H2O2 and diamide inhibit the expression of myelin genes in human primary oligodendrocytes.

Cells were treated with different concentrations (5, 10, and 20 μM) of either H2O2 (A) or diamide (B). After 18 h treatment, total RNA was analyzed for the expression of MOG, PLP, and CNPase by quantitative real-time PCR analysis. Results are means ± SD of three different experiments.
Diamide, a thiol-depleting agent, inhibits the expression of myelin genes in human primary oligodendrocytes
Because NAC, a thiol antioxidant, blocked cytokine-mediated down-regulation of myelin genes, we were prompted to investigate the effect of thiol-depleting agent (diamide) on the expression of myelin genes. Diamide is known to reduce the intracellular level of GSH by its oxidation to GSSG [21]. Similar to H2O2, treatment of human primary oligodendrocytes with diamide also resulted in marked decrease in myelin gene expression (Fig. 8B). Although diamide at 5 μM concentration was not very efficient in inhibiting the expression of CNPase, the expression of MOG and PLP was inhibited by 55 to 60% at the same concentration (Fig. 8B). However, at 25 μM concentration of diamide, the expression of all three myelin-specific genes was inhibited by 70 to 87% (Fig. 8B). These results suggest that the intracellular level of GSH is an important regulator of myelin gene expression in human primary oligodendrocytes.
Discussion
It has been shown that the expression of myelin-specific genes decreases in the CNS of MS patients and EAE animals, and an increase in expression of myelin-specific genes is a prerequisite to an increase in CNS remyelination [22-24]. However, despite extensive research on the pathogenesis of MS, no effective therapy is available to halt demyelination and/or stimulate remyelination. Several studies have implicated proinflammatory cytokines in the observed demyelination in MS patients [1-7]. Here we investigated if proinflammatory cytokines were capable of altering the expression of myelin-specific genes in human primary oligodendrocytes. Results presented in this paper clearly demonstrate that proinflammatory cytokines specifically down-regulate the expression of MOG, CNPase, and PLP in human mature oligodendrocytes. Although MBP is one of the most important proteins of the myelin sheath, proinflammatory cytokines were unable to down-regulate the expression of MBP. PLP, a hydrophobic integral membrane protein, is the most abundant protein of CNS myelin. It has been reported that it is involved in the formation of the intraperiod line of myelin [25], and the results from the knockout and transgenic mice also suggest an important role of PLP in the interaction of oligodendrocyte and axon [25,26]. On the other hand, MOG is a quantitatively minor component of the CNS myelin that represents only ~0.05% of the total protein content of myelin [27]. It is located at the extracellular surface of myelin sheaths and oligodendrocyte processes and is believed to regulate oligodendrocyte microtubule stability and mediate interactions between the myelin and the immune system [28]. CNPase is a phosphodiesterase that catalyzes the conversion of nucleoside 2,3-cyclic phosphate to nucleoside 2-phosphate [29]. Although the natural substrate of CNPase in myelin is currently unknown, it is highly enriched in myelin and is probably required for the support of axons in vivo [30]. Therefore, by decreasing the expression of MOG, PLP, and CNPase in oligodendrocytes, proinflammatory cytokines may reduce myelination in CNS inflammatory disorders.
ROS have been identified as important second messenger molecules that carry out many of the signaling steps transduced by proinflammatory cytokines. Several reports indicate that oxidative stress plays a major role in the pathogenesis of MS [31]. ROS, leading to oxidative stress, have been implicated as mediators of demyelination and axonal damage in both MS and EAE [31]. Consistently, the concentration of reduced sulfhydryls in the serum of MS patients was lower than in healthy controls and the treatment with interferon-β1a (IFN-β1a) raised the levels approaching the values of healthy controls [32]. Vladimirova et al. [33] have shown that the activation of protein kinase C induces higher production of reactive oxygen species by mononuclear cells of patients with MS than in controls and suggested that the ROS-producing subpopulation preferentially migrates into the CNS during a relapse. ROS are potentially toxic to neurons and oligodendrocytes that may mediate toxicity by damaging lipids, proteins, and nucleic acids of cells and mitochondria. Oligodendrocytes are more sensitive to oxidative stress in vitro than are astrocytes and microglia, seemingly due to a diminished capacity for antioxidant defense, and the presence of raised risk factors, including a high iron content [34]. Here we show that ROS play a crucial role in cytokine-induced down-regulation of myelin genes in human primary oligodendrocytes. First, antioxidants like N-acetylcysteine and pyrrolidine dithiocarbamate blocked cytokine-mediated down-regulation of myelin genes (MOG, CNPase, and PLP). Second, IL-1β and TNF-α induced an increase in H2O2 level in oligodendrocytes. Consistently, the addition of exogenous H2O2 alone caused down-regulation of myelin genes. Fourth, depletion of endogenous glutathione by diamide alone also inhibited the expression of myelin genes, suggesting that cellular redox is an important regulator of myelin gene expression in human oligodendrocytes.
In summary, we have demonstrated that proinflammatory cytokines inhibit the expression of MOG, CNPase, and PLP but not MBP in human primary oligodendrocytes via redoxsensitive mechanisms. Although the in vitro situation of human fetal oligodendrocytes in culture does not truly resemble the complex in vivo situation of oligodendrocytes in the CNS of MS patients, our results suggest that the use of antioxidants may be beneficial in restoring the cellular redox and thus attenuating proinflammatory cytokine-mediated inhibition of myelin genes in the inflamed CNS of MS patients.
Acknowledgments
This study was supported by grants from National Multiple Sclerosis Society (RG3422A1/1) and National Institutes of Health (NS39940 and P20RR18759).
Abbreviations
- MS
multiple sclerosis
- CNS
central nervous system
- IL
interleukin
- TNF
tumor necrosis factor
- CSF
cerebrospinal fluid
- CNPase
2′,3′-cyclic nucleotide 3′-phosphodiesterase
- MOG
myelin oligodendrocyte glycoprotein
- PLP
proteolipid protein
- MBP
myelin basic protein
- HBSS
Hank’s balanced salt solution
- DMEM
Dulbecco’s modified Eagle’s medium
- PBS
phosphate-buffered saline
- FBS
fetal bovine serum
- LNC
lymph node cells
- NEN
[3H]thymidine
- RT-PCR
real-time polymerase chain reaction
- EAE
experimental allergic encephalomyelitis
- ROS
reactive oxygen species
- NAC
N-acetylcysteine
- PDTC
pyrrolidine dithiocarbamate
References
- 1.Martin R, McFarland HF, McFarlin DE. Immunological aspects of demyelinating diseases. Annu Rev Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 2.Liu H, MacKenzie-Graham AJ, Palaszynski K, Liva S, Voskuhl RR. Mice resistant to experimental autoimmune encephalomyelitis have increased thymic expression of myelin basic protein and increased MBP specific T cell tolerance. J Neuroimmunol. 2001;116:83–93. doi: 10.1016/s0165-5728(01)00269-7. [DOI] [PubMed] [Google Scholar]
- 3.Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med. 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- 4.Saha RN, Pahan K. TNF-α at the crossroad of neuronal life and death during HIV-associated dementia: a review. J Neurochem. 2003;86:1057–1071. doi: 10.1046/j.1471-4159.2003.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brosnan CF, Raine CS. Mechanisms of immune injury in multiple sclerosis. Brain Pathol. 1996;6:243–257. doi: 10.1111/j.1750-3639.1996.tb00853.x. [DOI] [PubMed] [Google Scholar]
- 6.Sharief MK, Hentges R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med. 1991;325:467–472. doi: 10.1056/NEJM199108153250704. [DOI] [PubMed] [Google Scholar]
- 7.Maimone D, Gregory S, Arnason BG, Reder AT. Cytokine levels in the cerebrospinal fluid and serum of patients with multiple sclerosis. J Neuroimmunol. 1991;32:67–74. doi: 10.1016/0165-5728(91)90073-g. [DOI] [PubMed] [Google Scholar]
- 8.Ruddle NH, Bergman CM, McGrath KM, Lingenheld EG, Grunnet ML, Padula SJ, Clark RB. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J Exp Med. 1990;172:1193–1200. doi: 10.1084/jem.172.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samoilova EB, Horton JL, Hilliard B, Liu TS, Chen Y. IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells. J Immunol. 1998;161:6480–6486. [PubMed] [Google Scholar]
- 10.Esiri MM, Gay D. In: Multiple sclerosis: clinical and pathogenetic basis. Raine CS, McFarland HF, Tourtellotte WW, editors. Chapman and Hall; London: 1997. pp. 173–186. [Google Scholar]
- 11.The Lenercept Multiple Sclerosis Study Group. TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. Neurology. 1999;53:457–465. [PubMed] [Google Scholar]
- 12.Robinson WH, Genovese MC, Moreland LW. Demyelinating and neurologic events reported in association with tumor necrosis factor alpha antagonism: by what mechanisms could tumor necrosis factor alpha antagonists improve rheumatoid arthritis but exacerbate multiple sclerosis? Arthritis Rheum. 2001;44:1977–1983. doi: 10.1002/1529-0131(200109)44:9<1977::AID-ART345>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 13.Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-k. [DOI] [PubMed] [Google Scholar]
- 14.Miller RH. Regulation of oligodendrocyte development in the vertebrate CNS. Prog Neurobiol. 2002;67:451–467. doi: 10.1016/s0301-0082(02)00058-8. [DOI] [PubMed] [Google Scholar]
- 15.Dasgupta S, Jana M, Zhou Y, Banik NL, Pahan K. Sodiumphenylacetate inhibits the adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice at multiple steps. J Immunol. 2003;170:3874–3882. doi: 10.4049/jimmunol.170.7.3874. [DOI] [PubMed] [Google Scholar]
- 16.Dasgupta S, Jana M, Zhou Y, Fung YK, Ghosh S, Pahan K. Anti-neuroinflammatory effect of NEMO-binding domain peptides in the adoptive transfer model of experimental allergic encephalomyelitis. J Immunol. 2004;173:1344–1354. doi: 10.4049/jimmunol.173.2.1344. [DOI] [PubMed] [Google Scholar]
- 17.Jana A, Pahan K. HIV-1 gp120 induces apoptosis in human primary neurons through redox-regulated activation of neutral sphingomyelinase. J Neurosci. 2004;24:9531–9540. doi: 10.1523/JNEUROSCI.3085-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jana A, Pahan K. Fibrillar amyloid-β peptides kill human primary neurons via NADPH oxidase-mediated activation of neutral sphingomyelinase: implications for Alzheimer’s disease. J Biol Chem. 2004;279:51451–51459. doi: 10.1074/jbc.M404635200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Floyd RA. Neuroinflammatory processes are important in neurodegenerative diseases: an hypothesis to explain the increased formation of reactive oxygen and nitrogen species as major factors involved in neurodegenerative disease development. Free Radic Biol Med. 1999;26:1346–1355. doi: 10.1016/s0891-5849(98)00293-7. [DOI] [PubMed] [Google Scholar]
- 20.Garg AK, Aggarwal BB. Reactive oxygen intermediates in TNF signaling. Mol Immunol. 2002;39:509–517. doi: 10.1016/s0161-5890(02)00207-9. [DOI] [PubMed] [Google Scholar]
- 21.Akamatsu Y, Ohno T, Hirota K, Kagoshima H, Yodoi J, Shigesada K. Redox regulation of the DNA binding activity in transcription factor PEBP2. The roles of two conserved cysteine residues. J Biol Chem. 1997;272:14497–14500. doi: 10.1074/jbc.272.23.14497. [DOI] [PubMed] [Google Scholar]
- 22.Yao DL, Liu X, Hudson LD, Webster HD. Insulin-like growth factor I treatment reduces demyelination and up-regulates gene expression of myelin-related proteins in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 1995;92:6190–6194. doi: 10.1073/pnas.92.13.6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salvati S, D’Urso D, Conti Devirgiliis L, Serlupi Crescenzi G. Biochemical changes in central nervous system membranes in experimental allergic encephalomyelitis. J Neurochem. 1986;47:239–244. doi: 10.1111/j.1471-4159.1986.tb02855.x. [DOI] [PubMed] [Google Scholar]
- 24.Hartung HP, Rieckmann P. Pathogenesis of immune-mediated demyelination in the CNS. J Neural Transm. 1997;50:173–181. doi: 10.1007/978-3-7091-6842-4_17. [DOI] [PubMed] [Google Scholar]
- 25.Greer JM, Lees MB. Myelin proteolipid protein—The first 50 years. Int J Biochem Cell Biol. 2002;34:211–215. doi: 10.1016/s1357-2725(01)00136-4. [DOI] [PubMed] [Google Scholar]
- 26.Jurevics H, Hostettler J, Sammond DW, Nave KA, Toews AD, Morell P. Normal metabolism but different physical properties of myelin from mice deficient in proteolipid protein. J Neurosci Res. 2003;71:826–834. doi: 10.1002/jnr.10544. [DOI] [PubMed] [Google Scholar]
- 27.Amiguet P, Gardinier MV, Zanetta J, Matthieu J-M. Purification and partial structural and functional characterization of mouse myelin/oligodendrocyte glycoprotein. J Neurochem. 1992;58:1676–1682. doi: 10.1111/j.1471-4159.1992.tb10040.x. [DOI] [PubMed] [Google Scholar]
- 28.Brunner C, Lassmann H, Waehneldt TV, Matthieu J, Linington C. Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2′,3′-cyclic nucleotide 3′-phosphodiesterase in the CNS of adult rats. J Neurochem. 1989;52:296–304. doi: 10.1111/j.1471-4159.1989.tb10930.x. [DOI] [PubMed] [Google Scholar]
- 29.Sprinkle TJ. 2′,3′-Cyclic nucleotide 3′-phosphodiesterase, an oligodendrocyte-Schwann cell and myelin-associated enzyme of the nervous system. Crit Rev Neurobiol. 1989;4:235–301. [PubMed] [Google Scholar]
- 30.Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- 31.Gilgun-Sherki Y, Melamed E, Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J Neurol. 2004;251:261–268. doi: 10.1007/s00415-004-0348-9. [DOI] [PubMed] [Google Scholar]
- 32.Lucas M, Rodriguez MC, Gata JM, Zayas MD, Solano F, Izquierdo G. Regulation by interferon beta-1a of reactive oxygen metabolites production by lymphocytes and monocytes and serum sulfhydryls in relapsing multiple sclerosis patients. Neurochem Int. 2003;42:67–71. doi: 10.1016/s0197-0186(02)00057-8. [DOI] [PubMed] [Google Scholar]
- 33.Vladimirova O, Lu FM, Shawver L, Kalman B. The activation of protein kinase C induces higher production of reactive oxygen species by mononuclear cells in patients with multiple sclerosis than in controls. Inflamm Res. 1999;48:412–416. doi: 10.1007/s000110050480. [DOI] [PubMed] [Google Scholar]
- 34.Smith KJ, Kapoor R, Felts PA. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 1999;9:69–92. doi: 10.1111/j.1750-3639.1999.tb00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]