

Abstract
Human immunodeficiency type-1 (HIV-1) infection is known to cause disorders of the CNS, including HIV-associated dementia (HAD). It is suspected that tumor necrosis factor-α (TNF-α) released by infected microglia and macrophages play a role in neuronal injury seen in HAD patients. Accordingly, studies suggest that the level of TNF-α mRNA increases with increasing severity of dementia in patients, and that inhibitors of TNF-α release reduces neuronal injury in murine model of HAD. However, the exact role of TNF-α in relation to neuronal dysfunction is a matter of ongoing debate. One school of thought hails TNF-α as the inducer and mediator of neurodegeneration and their evidence suggest that TNF-α kill neurons directly by recruiting caspases or may kill indirectly by various means. In sharp contrast to this, another concept theory envisages a role for TNF-α in negotiating neuroprotection during HAD. The current compilation examines these contradictory concepts, and evaluates their efficacy in the light of TNF-α signaling. It also attempts to elaborate the current consensus outlook of TNF-α’s role during HAD.
Keywords: activation of glia, HIV dementia, neurodegeneration, neuroprotection, nuclear factor-kappa B, tumor necrosis factor-α
HIV-1-associated dementia (HAD) refers to the severe form of neurological disability, which is observed in about 20% of patients suffering from acquired immunodeficiency syndrome (AIDS) (Adle-Biassette et al. 1999; Kolson and Gonzalez-Scarano 2000). From a clinical standpoint, HAD is characterized by neurologic impairments manifested by cognitive, behavioral and motor dysfunction (Adle-Biassette et al. 1999). Histopathologically, it is identified by infiltration of inflammatory cells into the central nervous system (CNS), gliosis, pallor of myelin sheaths, abnormalities of dendritic processes and neuronal apoptosis (Adle-Biassette et al. 1995; Gray et al. 1996). However, the virus neither infects neurons directly nor is it neurotoxic. Furthermore, the correlation between the disease severity and the viral load is unconvincing, and little or no virus has been found in AIDS-related vacuolar myelopathy (Petito et al. 1994). On the other hand, neuronal apoptosis correlates with microglial activation (Adle-Biassette et al. 1999). Taken together, these observations suggest that indirect mechanisms possibly play an important role in neuronal loss during HAD. Productive HIV-1 infection in the brain occurs predominantly in macrophages, microglia, and multinucleated giant cells (Griffin 1997). Infection of astrocytes may also occur with restricted virus replication (Canki et al. 1997), suggesting effects of HIV on astrocytes may also be indirect (Glass and Johnson 1996). Because infected and/or activated macrophages, microglia and astrocytes induce the production of a plethora of soluble toxic proinflammatory cytokines, one means by which indirect effects may be exerted upon cells is via these molecules. Consistently, activated macrophages in white matter of brain, a pathological marker of HAD (Tyor et al. 1995), show increased expression of cytokines like interferon (IFN)-α, interleukin (IL)-1β, TNF-α and TNF receptors (Tyor et al. 1992; Sippy et al. 1995). Among these proinflammatory cytokines, mRNA for TNF-α, is markedly present at a higher concentration in brain of HAD patients (Wesselingh et al. 1993). The temporal profile of TNF-α expression also correlates with the onset, progression and severity of HAD (Griffin 1997). Furthermore, in a murine model of HIV-1 encephalitis (HIVE), the pathological equivalent of HAD, TNF-α release inhibitor show marked reduction in brain inflammation and a reduction in neuronal injury (Persidsky and Gendelman 2002). All this evidence convincingly urges us to believe that TNF-α plays a major role in pathogenesis of HAD. However, diverse and contradictory opinions are being currently offered regarding the exact nature of this obligatory role of the cytokine during HAD in context of neurotoxicity. This is in part due to restraints of experimental protocols and designs involving variations such as cytokine concentration and timing of treatment. While conclusive knowledge in this regard is elusive, this article attempts to illuminate such restrains and compile the current set of predilections regarding the role of TNF-α during HAD.
Admission of HIV in the brain
HIV enters immunologically privileged CNS early in the course of infection and establishes its life long niche in the brain. The principal pathway of HIV entry in brain is through the increased influx of infected monocytes into CNS (Kaul et al. 2001; Langford and Masliah 2001). Peripherally infected monocytes, during advanced HIV infection, adhere to normal brain microvasculature endothelium, transmigrate, and translocate the virus into the CNS (Gartner 2000). Increased trafficking of cells across blood–brain barrier (BBB) involves complex interaction of cell adhesion molecules like, intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) with their receptors like leukocyte function associated antigen-1 (LFA-1) and very late antigen-4 (VLA-4) (Gartner 2000). Accordingly, marked over-expression of these contact molecules has been observed in the inflamed atmosphere of HAD brain (Diesing et al. 2002). Apart from adhesion molecules and their receptors, chemokines (Asensio and Campbell 1999) and cytokines (Lee et al. 1998) produced by astrocytes and microglia also regulate migration of monocytes through the BBB, which supposedly could otherwise physically confine entry of the cloaked virus. In the brain, the majority of infected cells are infiltrated monocyte-derived macrophages and brain resident microglia, which gets infected on a secondary basis (Lipton and Gendelman 1995). In addition, astrocytes and capillary endothelial cells have also been found to contain HIV-1 protein and/or DNA (Lawrence and Major 2002).
The role of non-neuronal cells in HAD pathogenesis
According to several studies, HIV-1 is neurovirulent but not neurotropic. Although this concept has been challenged by certain observations, where presence of HIV genome in neuron has been demonstrated (Torres-Munoz et al. 2001), we will abide by the widely accepted hypothesis, considering HIV non-neuroinvasive. Instead, the virus resides in the brain macrophages and resident microglia. This brings us to an apparent lack of proportion between the small number of productively HIV-infected brain cells and the severe functional deficit and death of neurons. This suggests the presence of a functional cellular activation/amplification loop that generates neurotoxins. Indeed, we have ample evidence to deem this true.
Role of microglia and macrophages in HAD
Apoptotic neurons do not colocalize with infected microglia in HAD patients (Shi et al. 1996), although their activation has been widely associated with HAD. Thus, HIV infection must induce release of certain soluble factors from these infected cells, which are deleterious for neurons. Indeed, HIV-infected/immune-stimulated macrophages/microglia produce neurotoxins, which are required for HIV-1-induced neurotoxicity (Giulian et al. 1993). These neurotoxins include arachidonate and its metabolites, excitotoxic substances, viral proteins like gp120, free radicals, chemokines and proinflammatory cytokines like IL-1β and TNF-α (Dreyer et al. 1990; Lipton and Gendelman 1995). Autocrine TNF-α signaling in microglia promotes p38 MAPK activity that, in turn, turns on the expression of TNF-α gene itself along with other genes like iNOS (Bhat et al. 1998). In addition to producing the deleterious agents themselves, activated microglia also amplify excitotoxic glutamate release from astrocytes considerably (Bezzi et al. 2001).
Role of astrocytes in HAD
HIV can infect astrocytes non-productively (Canki et al. 1997), leading to proliferation of activated astrocytes (astrocytosis) and occasional astroglial apoptosis (Kaul et al. 2001). Astrocytes are often portrayed as amplifiers of neurotoxic signals originating from HIV-activated/infected microglia (Bezzi et al. 2001). They produce a constellation of cytokines upon activation, including TNF-α (Wang and Shuaib 2002). In addition, astrocytes contribute by increasing the level of glutamate to excitotoxic levels by enhanced release and restricted uptake of the excitotoxin (Fine et al. 1996). TNF-α plays an indispensable role in this release (Bezzi et al. 2001). Moreover, viral proteins can act on astrocytes to trigger release of other neurotoxic agents. Recently, we have shown that the viral protein, Tat, activates astrocytes and induces the expression of inducible nitric oxide synthase (iNOS) (Liu et al. 2002) leading to release of abnormal amount of nitric oxide (NO), which forms neurotoxic peroxynitrite (ONOO−) after reacting with superoxide anion (O2−).
Modes of neuronal injury and death during HAD
The exclusive outcome of all pathological and immunological events in HIV-infected brain is neuronal injury. Subsequently, most of the injured neurons succumb to apoptosis, the primary mode of neuronal demise in HAD (Adle-Biassette et al. 1995; Talley et al. 1995). However, the exact modus operandi of neurovirulence during HAD is still elusive. Two theories have been forwarded in this regard; the ‘direct injury’ theory and the ‘bystander effect’ theory (Kaul et al. 2001).
The hypothesis of direct injury
This theory, involving various viral proteins (including Tat, gp120 and Vpr) suggests direct interaction of the assassin molecule with the neuron without any intermediate role of non-neuronal cells. To illustrate this, gp120 has been shown to induce the death signal in neurons directly through α-chemokines, like CXCR4 (Zheng et al. 1999), and NMDA receptors (Fontana et al. 1997) present on neuronal surface. Tat, an important regulator of viral transcription, can induce elevation of intracellular calcium and cell death in both human and rodent neurons through NMDA receptors (Magnuson et al. 1995; Nath et al. 1996). Another HIV-1 regulatory protein, vpr, has been shown to induce apoptosis in neuronal cells via the activation of caspase 8 (Patel et al. 2000).
The hypothesis of bystander effect
According to this theory, various diffusible products of non-neuronal cells induce death processes in non-specific innocent bystander neurons. Such diffusible products predominantly include proinflammatory cytokines (TNF-α, IL-1β, etc.), NO, free radicals, various chemokines along with certain HIV proteins and excitotoxic amino acids (EAA) (Kaul et al. 2001). These two processes are not mutually exclusive. However, the general consensus tilts towards the second theory as the key mode of neuronal loss during HAD.
Although neurons die mainly by undergoing apoptosis, the alternative death process of necrosis also prevails during HAD. It is suggested that neurons undergo apoptosis or necrosis depending on the intensity of the initial insult (Bonfoco et al. 1995). If the initial challenge is severe, loss of ionic homeostasis in neurons lead to necrosis, marked by acute swelling and lysis. Alternatively, milder abuse triggers apoptotic signals via p38 MAPK (detailed subsequently), and programmed cell death is brought about in a caspase dependent fashion (Bonfoco et al. 1995).
Involvement of TNF-α in neuronal demise during HAD
TNF-α and TNF-α receptors (TNF-R) are expressed at a nominal level in all kinds of brain cells under normal conditions, suggesting a set of physiological roles in brain functioning for this cytokine (Perry et al. 2002). However, we have strong evidence to suggest involvement of TNF-α and its receptors in the pathogenesis of HAD and other neurodegenerative disorders such as Parkinson’s disease (Nagatsu et al. 2000), Alzheimer’s disease (Perry et al. 2000, 2002), Prion disease (Veerhuis et al. 2002), neurotrauma and stroke (Barone and Parsons 2000). Although the presence of TNF-α as a major component of brain inflammatory response during all these abnormal conditions is not questioned, we still do not have enough data to confine our opinion regarding its exact role in neurodegeneration. Throughout this text, we will correlate findings in all these diseases with observations in HAD studies to generate an overall picture about the role adopted by TNF-α during neurodegeneration.
During HAD, microglia, macrophages and infiltrated monocytes show increased expression of both TNF-α and TNF-Rs (Tyor et al. 1992). Studies have shown that TNF-α levels in vulnerable brain regions correlate with neurologic disease severity in HIV patients (Gelbard 1999). Assessment of mRNA in autopsy-collected brain tissue of HAD patients by semiquantitative RT-PCR reveals elevated levels of TNF-α (Wesselingh et al. 1993). Interestingly, the same study showed diminished levels of IL-1β mRNA expression (another much acclaimed proinflammatory cytokine related to dementia) in demented compared with non-demented HIV patients. Additionally, soluble TNF-α levels in cerebrospinal fluid dropped significantly after treatment of a HAD patient with highly active antiretroviral therapy (HAART) (Gendelman et al. 1998). The cytokine level fell in conjunction with a drop in viral load and marked improvement in the neurological function of the patient. Interestingly, while levels of TNF-α mRNA increase with increasing severity of dementia, no differences have been observed for IL-6, transforming growth factor (TGF)-β and IFN-γ mRNA in demented and non-demented HIV patients (Griffin 1997). This dementia-associated increase of TNF-α expression against the backdrop of unaltered and diminished expression of other cytokines suggests an emblematic up-regulation of this cytokine during HAD.
Moreover, several polymorphisms in promoter of the TNF-α gene have been found to alter the cytokine expression and therefore, have been associated with various inflammatory diseases. Such polymorphisms have been associated with Alzheimer’s disease (see Perry et al. 2002 for review on the topic). While looking for any genetic predisposition for development of HAD among HIV-infected individuals, it was found that the TNF-α-308 A allele had a much greater frequency in HAD patients (Quasney et al. 2001). Such polymorphism is known to positively influence the amount of cytokine produced in response to inflammatory stimuli.
These obvious demarcating differences in genomic background and at protein level in HAD patients strongly assert TNF-α as a major proctor of HAD consequences. However, the exact role of TNF-α in relation to neuronal abnormalities is still obscure. Is it responsible for inducing death or does it actually resist neuronal loss? From the signaling point of view, the TNF-α ligand binding to its receptors can induce apoptotic as well as survival pathways (this will be elaborated on later in this article). Not surprisingly, therefore, on the one hand, evidence suggests a neurocidal role of TNF-α during HAD, but, on the other hand, there are numerous testaments in the literature to insinuate a neuroprotective role for the cytokine.
Neurotoxic nature of TNF-α
It is postulated that overproduction of TNF-α for an extended time period during the diseased condition brings about indirectly on neurons to bring about their extermination. Serious outcomes due to elevation in cytokine level during viral infection are summarized in Fig. 1(a).
Fig. 1. Comparative display of potential modulation of cell death or protection by TNF-α during HAD.
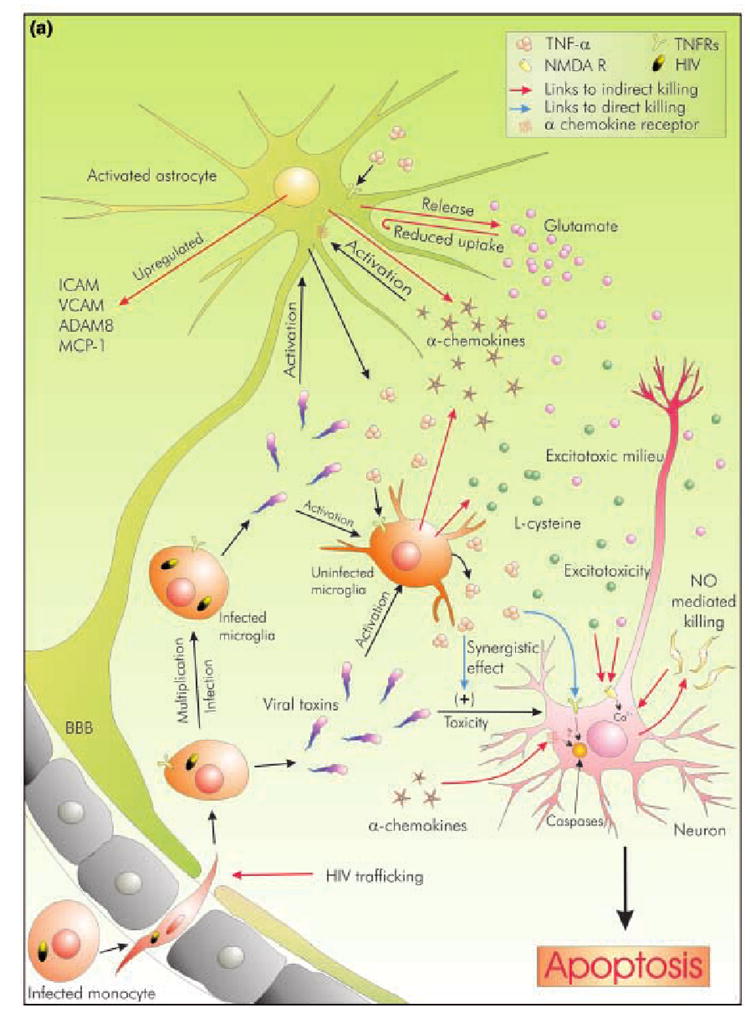
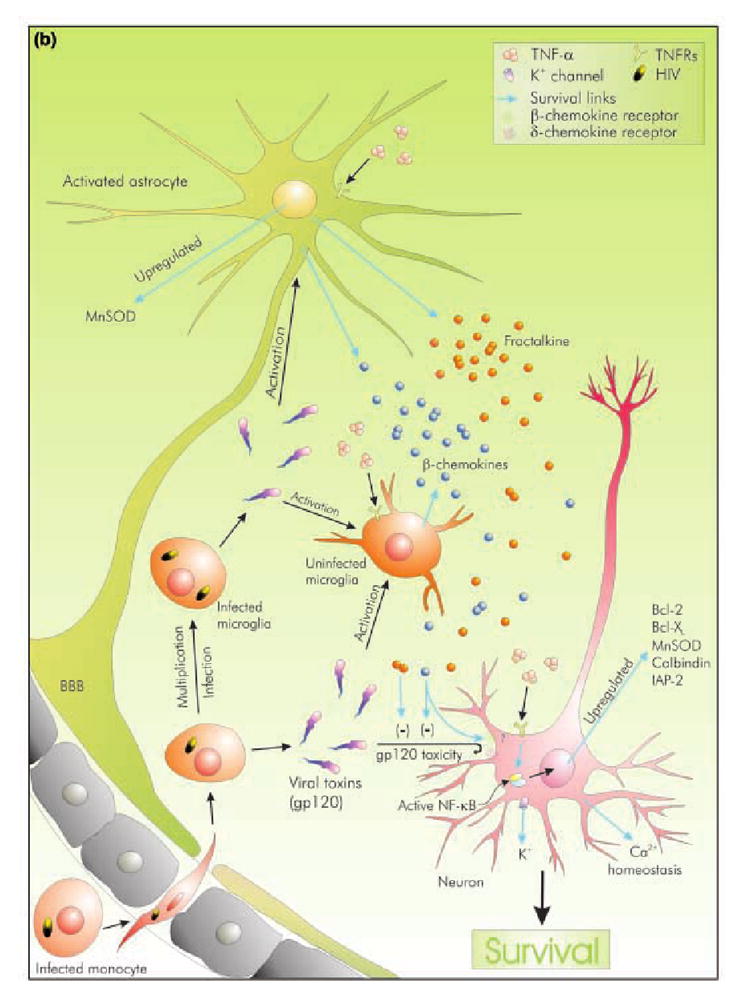
The microenvironment of a neuron during HAD is represented by viral entry and subsequent activation of glial cells. Viral toxins can directly induce neuronal death or may trigger gliosis, which ultimately leads to neuronal damage. TNF-α is significantly present in this environment although its exact role is debatable. (a) Represents possible roles of TNF-α as an inducer of neurotoxicity and degeneration. It may directly kill neurons through its receptors to activate caspases, or may synergistically enhance toxicity of viral toxins like gp120. Indirect modes of killing include NO-mediated peroxynitrite toxicity, and up-regulation of gene products, which are either directly neurotoxic (α-chemokines) or may indirectly induce neuronal injury (e.g. up-regulation of ICAM and VCAM aid in HIV-1 trafficking across BBB). Moreover, it induces the production of excitotoxins from glial cells, like glutamate and l-cysteine, thus offering the neuron a lethal excitotoxic environment. (b) represents possible roles of TNF-α as a neuroprotector. TNF-α acts via NF-κB to up-regulate several pro-survival gene products. Pre-treatment with TNF-α leads to Ca2+ and K+ homeostasis. In addition, TNF-α induces the production of neuroprotective β- and δ- chemokines. These chemokines significantly attenuate toxicity mediated by viral proteins like gp120. Moreover, β-chemokines may protect neurons by interacting with their receptors.
Aiding virus trafficking
Astrocytic processes abutting cerebrovascular endothelium as a part of the BBB release TNF-α (Hallenbeck 2002), which is known to alter the permeability of the barrier and promotes infiltration of infected monocytes. Moreover, TNF-α increases ICAM-1 (Lee et al. 1998) and VCAM-1 (Winkler et al. 1998) expression by astrocytes. This facilitates infected monocyte adhesion and extravasation through BBB. It also opens a paracellular route for HIV to cross the BBB (Fiala et al. 1997).
Lethal blow via NMDA receptors
TNF-α helps in release of l-cysteine, an EAA, from microglia and macrophages (Yeh et al. 2000). It also releases glutamate from astrocytes (Bezzi et al. 2001). Moreover, the cytokine helps in elevating free glutamate concentration in the vicinity of neurons by restricting glutamate uptake by astrocytes (Fine et al. 1996). This leads to the accumulation of excessive amounts of these excitatory molecules in the vicinity of neurons, which exorbitantly activates N-methyl-d-aspartate (NMDA) receptor-operated channels on neuronal surface, impelling unwarranted Ca2+ influx (Lipton 1998). Subsequently, loss of cellular homeostasis occurs, which can either lead to acute swelling and lysis of the cell, or culminate in apoptosis (Foos and Wu 2002). Ca2+ influx-mediated killing appears to be universal in all forms of neurodegeneration and at least in hippocampal neurons, involves calcineurin-dependent dephosphorylation and activation of BAD, the pro-apoptotic member of the Bcl-2 family (Wang et al. 1999).
Co-stimulatory effects
TNF-α has been shown to act synergistically with many toxic elements to enhance the toxicity of the microenvironment. TNF-α has been accused of elevating the neurotoxic effect of excitotoxic glutamate in cerebral cortex neurons (Chaparro-Huerta et al. 2002). It is known to conspire with the stromal-derived factor-1 (SDF-1, an α chemokine), to enhance glutamate release from astrocytes co-cultured with microglia (Bezzi et al. 2001). TNF-α also teams up with HIV proteins, like Tat (Shi et al. 1998) and gp120 (Kast 2002-2003) for example, to promote neuronal death, brought about by oxidative stress. Moreover, it has been reported to boost neuronal damage induced by other cytokines in the inflammatory soup (Chao et al. 1995).
Carnage by altering glial function
TNF-α activates astrocytes and microglia, which leads to remodeling of the extracellular matrix (Chen and Strickland 1997), shedding of cytokines, cytokine receptors (Rose-John and Heinrich 1994), and further proliferation (Barna et al. 1990). TNF-α, being the prototype inducer of NF-κB (described shortly in the following section), recruits this transcription factor during up-regulation of many proinflammatory genes in activated glial cells (Han et al. 2001a). Extensive glial activation leads to the accumulation of these proinflammatory elements in the neuronal vicinity, causing adverse effects to the latter. Consistently, studies in the ischemic model of neurodegeneration have shown the involvement of NF-κB activation in neuronal death (Schneider et al. 1999; Xu et al. 2002).
Additionally, TNF-α costimulates the expression of iNOS in astrocytes in conjunction with other cytokines (Pahan et al. 2000); this may involve CD23, the low affinity IgE transmembrane receptor. The expression of CD23 is up-regulated in glial cells during combined treatment of TNF-α, IFN-γ and IL-1β (Hunot et al. 1999). Upon ligation, CD23 induces iNOS expression in glial cells (Hunot et al. 1999; Dugas et al. 2001). This leads to enhanced peroxynitrite toxicity. Excess NO also enhances glutamate release from astrocytes (Bal-Price et al. 2002) and prohibits glutamate uptake by astrocytes (Hu et al. 2000) thus adding to excitotoxic levels of the EAA in the neuronal milieu. Taking another example from microglia, cultured human microglia releases platelet-activating factor (PAF) upon induction by TNF-α. Exposure to PAF, an ether phospholipid, leads to neuronal apoptosis (Pulliam et al. 1998).
Hitting a direct blow?
In addition to all these third party mediated killing, a section of researchers have postulated that TNF-α can kill neurons directly by recruiting the apoptotic caspase cascade in them after binding to its receptor TNF-R1 (Garden 2002; Garden et al. 2002). The genesis of this postulation involves the seaming of two independent observations. First, inhibiting TNF-α activity can attenuate gp120-induced neuronal injury (Bezzi et al. 2001) and, second, inhibition of caspase-8 prevents TNF-α induced neurotoxicity in mixed cerebellar cultures (Garden et al. 2002). In summation, it is suggested that gp120 induces TNF-α production in non-neuronal cells, which subsequently activates caspase-mediated death in neurons through its receptor. In another study, involvement of caspases in the cell death of differentiated PC-12 cells in response to TNF-α insult has been demonstrated (Reimann-Philipp et al. 2001). However, in this study, cell death was assessed by lactate dehydrogenase (LDH) release, which does not conclusively prove an apoptotic mode of death in these cells.
Neuroprotective role of TNF-α
In extreme contrast to the theme of evidence presented above, another set of observations recommend a protective role for the cytokine. Although these data lack a sense of completeness, appear hyphenated and are not always related to studies in HAD, they strongly suggest the neuroprotective side of the Janus-faced cytokine (Fig. 1b). In this section, we will consider evidence offered by other studies conducted in other forms of dementia, as the commonness of neuronal killing pathway in various forms of neurodegeneration has already been pointed out (Lipton 1998). Episodes mentioned in the following lines are not mutually exclusive, and may form subparts of neuronal response to elevated level of TNF-α in HAD brain. If stitched together rationally, they indeed accredit TNF-α with a neuroprotective role.
TNF-α protects by maintaining Ca2+ homeostasis
In a pioneering study, Mark P. Mattson and his team demonstrated that pretreatment of neurons with TNF-α could protect hippocampal, cortical and septal neurons from glucose-deprivation neurotoxicity (Cheng et al. 1994). They showed that TNF-α attenuated the elevation in Ca2+ level due to glucose deprivation by inducing an increase in calcium binding protein calbindin-D28k. Similar effects have also been documented in β-amyloid toxicity model, where TNF-α protects neurons by attenuation of Ca2+, and peroxide, accumulation in cytosol (Barger et al. 1995). This urges us to believe that neuroprotection by TNF-α preconditioning is universal for different forms of dementia.
Alliance with NF-κB and up-regulation of survival protein
The NF-κB transcription factor consists of dimeric complexes belonging to the Rel family, which include p50, p52, p65 (Rel A), p68 (Rel B) and p75 (c-Rel) in mammals (Ghosh et al. 1998). TNF-Rs can activate NF-κB via several pathways, which essentially utilize phosphorylation-based ubiquitination and degradation of arresting molecules [inhibitory κB (IκB), p100, or p105] and subsequent release of active NF-κB dimer in cytoplasm for translocation into the nucleus. Inhibitory kappa B kinase (IKK), the upstream kinase that phosphorylates IκB, is recruited directly to the TNF-R1 in immediate response to TNF-α signal. This pathway is known to trigger the NF-κB cascade within minutes (Chen and Goeddel 2002). Another pathway involves the pro-survival kinase, Akt (Protein kinase B), whose role has been well documented in conveying the signal from TNF-Rs to the activation of NF-κB (Diem et al. 2001).
In neurons, NF-κB activity is essentially protective in nature (Mattson et al. 2000). Once activated, NF-κB dimers enter into the nucleus and bind to target DNA. This influences the expression/repression of a complex array of gene products, which are vital for neuronal responses to various forms of insult (Mattson and Camandola 2001). Several studies have shown that certain subunits of NF-κB are more critical in neuronal survival during challenges simulating HAD milieu. The lack of a p50 subunit increases vulnerability of hippocampal neurons to excitotoxic injury (Yu et al. 1999) and striatal neurons in Huntington’s disease model (Yu et al. 2000). Similar lack of the p52 subunit has been responsible for neurotoxic effects of microglial-derived TNF-α in mouse cerebellar neurons, suggesting the engrossing role of this subunit in TNF-α-mediated survival conditioning of neurons (Nicholas et al. 2001).
How does TNF-α -induced NF-κB underwrite neuronal protection? Studies in survival after TNF-α treatment in hypoxia or NO-mediated model of neurodegeneration have unveiled expression of anti-apoptotic proteins in primary hippocampal neurons (Tamatani et al. 1999). This NF-κB-mediated up-regulation of Bcl-2 and Bcl-Xl has been shown to be partially responsible for cell survival, suggesting that TNF-α–NF-κB pathway may trigger expression of more pro-survival proteins than just two of these (Table 1). Similarly, studies conducted in Alzheimer disease and vascular dementia patients displayed higher intrathecal level of the cytokine, which up-regulated expression of bcl-2 in neuronal cells by three times (Tarkowski et al. 1999).
Table 1.
A list of TNF-α-induced neuroprotective and neurodegenerative molecules
| Gene product | Modulation | Cell type | Reference | Effect on neurons | Remark |
|---|---|---|---|---|---|
| Bcl-2 | Up-regulated | Neuron | Tamatani et al. (1999) | Neuroprotection | Enhances cell survival by inhibiting apoptosis |
| Bcl-XL | Up-regulated | Neuron | Tamatani et al. (1999) | Neuroprotection | |
| MnSOD | Up-regulated | Neuron/Astrocytes | Bruce-Kellar et al. (1999) | Neuroprotection | Reduces reactive oxygen species toxicity by superoxide scavenging |
| Calbindin | Up-regulated | Neuron | Cheng et al. (1994) | Neuroprotection | Binds Ca2+, maintains Ca2+ homeostasis |
| c-IAP-2 | Up-regulated | Spinal cord | Kim et al. 2001 | Neuroprotection | Induces survival signal from TNF receptors |
| Fractalkine (CX3CL1) | Up-regulated | Astrocyte | Yoshida et al. (2001) | Neuroprotection | Inhibits gp120 toxicity |
| MIP-1β | Up-regulated | Astrocyte | Guo et al. (1998) | Neuroprotection (?) | Related with preservation of cognitive function at higher conc. |
| RANTES | Microglia | McManus et al. (1998) | |||
| CXCR4 | Down-regulated | Astrocyte | Han et al. (2001) | Neuroprotection | Delimits astrogliosis |
| GluR1 (AMPA- receptor subunit) | Up-regulated | NT2-N neurons | Yu et al.2002 | Neurodegeneration | Increases susceptibility to EAA toxicity |
| iNOS | Up-regulated | Neuron | Combs et al. 2001 | Neurodegeneration | Peroxynitrite toxicity |
| ICAM-1 | Up-regulated | Astrocyte | Lee et al. (1998) | Neuronal injury | Helps viral trafficking across BBB |
| VCAM-1 | Up-regulated | Astrocyte | Winkler and Benveniste (1998) | Neuronal injury | Enhances astrogliosis, EAA accumulation. |
| TNF-R2 | Up-regulated | Astrocyte | Lung et al. 2001 | Neuronal injury | Modulates microglial activation |
| MCP-1 | Up-regulated | Astrocyte | Andjelkovic et al. (1999) | Neuronal damage | |
| ADAM8 | Up-regulated | Neuron, astroglia, microglia | Schlomann et al. 2000 | Neuronal damage (?) | Neuro-glial interaction during inflammation (MMP) |
Manipulating chemokine activity
As already mentioned, α-chemokines can act via their receptors on astrocytes and induce them to generate more EAAs. In this context, it has been shown that TNF-α down regulates CXCR4 (receptor for α-chemokine SDF-1 and viral protein gp-120) expression in astrocytes in vitro (Han et al. 2001b). If true in vivo, this definitely narrows down the scope for viral protein/chemokine induced astrogliosis during HAD. Additionally, β-chemokines are known to protect neurons from gp120 toxicity (Kaul and Lipton 1999). Higher concentration of these cytokines is associated with preservation of cognitive function (Letendre et al. 1999). In this regard, TNF-α is known to induce production of β-chemokines in activated glial cells (Guo et al. 1998; Garden 2002), suggesting yet another neuroprotective facet of this cytokine. Moreover, TNF-α induces expression of CX3CL1 (fractalkine) expression in astrocytes (Yoshida et al. 2001). This δ-chemokine protects neurons from gp120-mediated toxicity and inhibits HIV-1 entry into microglial cells, expressing its receptor (CX3CR1) (Cotter et al. 2002).
Neuroprotection by increasing potassium current
The opening of potassium channels limits and rectifies neuronal excitability, which is theorized to play a role in neuronal defense at the face of excitotoxic insult (Sapolsky 2001). It has been shown that TNF-α induces increase in transient outward potassium current in cortical cells, which, if blocked, attenuates TNF-α-mediated neuroprotection (Houzen et al. 1997).
Neuroprotective enforcements of TNF-α are mediated by direct interaction with its receptors
Following ischemic and excitotoxic brain injuries, genetically altered mice lacking TNF-α receptors exhibit more susceptibility of neurons in CNS than their normal counterparts (Bruce et al. 1996; Gary et al. 1998). They have shown here that, between its two receptors (detailed below), TNF-R1 is more suitable as the candidate to carry out the neuroprotective effects of TNF-α. In yet another study (Yang et al. 2002), it has been shown that TNF-α has little effect on hippocampal neurons in which TNF-R1 was absent; whereas neurons from TNF-R2 knockout mice are vulnerable to TNF-α, even at low doses. Moreover, NF-κB nuclear translocation was inhibited in these mice. These studies imply the role of TNF-R2 in neuroprotective activities of this cytokine.
Receptor knockout studies have also been conducted in the MPTP model of Parkinson’s disease (Rousselet et al. 2002; Sriram et al. 2002). However, these two separate studies offer contrasting observations. Sriram et al. (2002) observed amelioration of MPTP-induced dopamine decrease in double TNF-R knockout mice (DKO) along with prevention of tyrosine hydroxylase immuno-reactive nerve terminal loss, as observed in wild-type littermates. On the contrary, Rousselet et al. (2002) observed that striatal dopamine level was more reduced by MPTP in the DKO group than in the other groups of mice. Such contrasting observations do not let us conclusively ascertain the actual role played by the cytokine receptors in the MPTP model and this indeed serves as a classic example of confusing observations arising out of experimental limitations.
Explaining diametrical roles of TNF-α; insights into TNFR signaling pathways
The findings reviewed above underline the array of heterogeneous roles a single pleiotropic molecule and two of its receptors are capable of eliciting. What might contribute to such paradoxical effects? At least partially, the answer lies in multifaceted signaling pathways originating from two receptors of this cytokine, TNF-R1 (p55TNF-R, p60, CD120a) and TNF-R2 (p75TNF-R, p80, CD120b) (MacEwan 2002). While TNF-R1 is more generously expressed in basal conditions, both receptors are expressed in brain cells upon TNF-α challenge (Nadeau and Rivest 1999). Binding of the self-assembled non-covalent TNF-α trimer to TNF-R1 and TNF-R2 leads to receptor trimerization and recruitment of specific adaptor proteins to the cytoplasmic domain of the receptors (Chen and Goeddel 2002). The signal is transduced downstream via these adapter proteins.
Signaling via TNF-R1
Owing to the presence of death domains, TNF-R1 recruits TRADD (TNF-R1-associated death domain protein). This protein can recruit either RIP1 (receptor interacting protein) or FADD (Fas-associated death domain protein) among many other interacting proteins (Fig. 2). It is the recruited protein at this juncture, which further dictates whether the cell will live or die (Baud and Karin 2001).
Fig. 2. Pathways originating from TNF-R1 and TNF-R2 leading to cell survival or death.
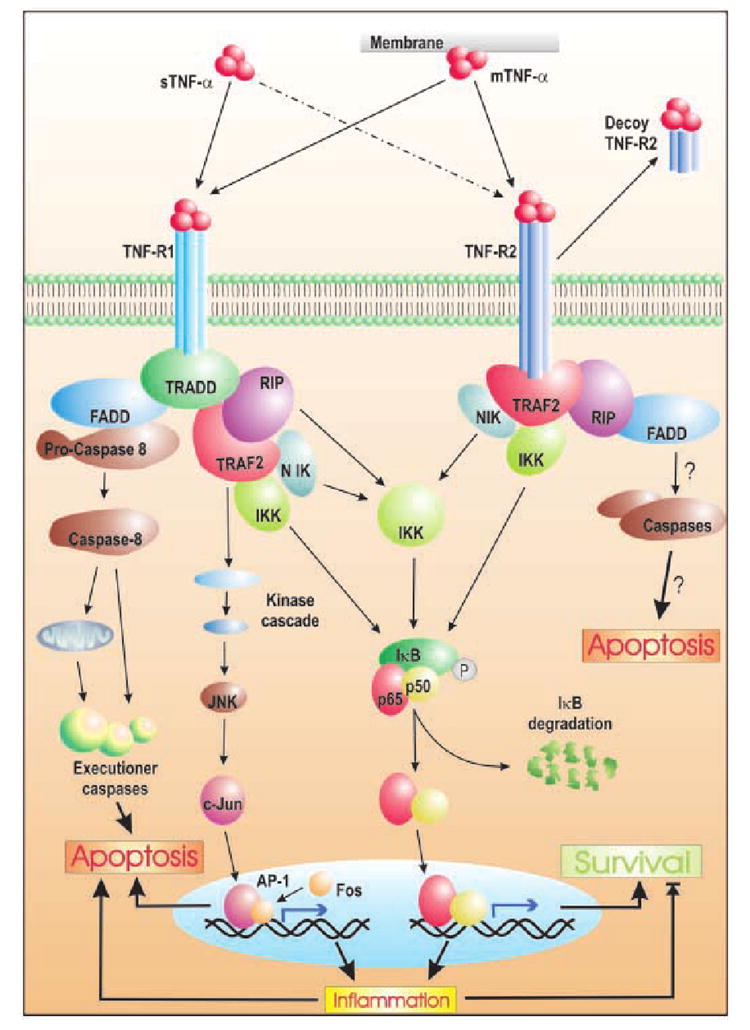
While the membrane-bound form of TNF-α can bind to both receptors with similar affinity, the soluble form binds TNF-R2 with lesser affinity. Binding of the trimeric cytokine ligand leads to receptor trimerization. This triggers intracellular signal transduction, although activated TNF-R2 may be cleaved by metalloproteinases to be released as TNF-bound-soluble receptor. Once activated, both receptors recruit adaptor molecules. TNF-R1 binds to TRADD through its death domain and recruits FADD or RIP. Binding of FADD deploys the caspase train leading to apoptosis via mitochondria-dependent and independent pathways. Alternately, RIP recruits TRAF2 to the receptor complex. TRAF2 activates the upstream kinases of different MAP kinase pathways that may lead to inflammation and cell death via AP-1-activated genes. On the contrary, TRAF2 can induce survival signal by activating IKK by binding to its NEMO subunit. It may also activate IKK via other upstream molecules like RIP1 and NIK. Activated IKK phosphorylates IκB, setting it up for ubiquitination-dependent degradation, which then releases p65–p50 heterodimer of NF-κB to translocate into the nucleus and primarily activate survival genes. NF-κB may also induce inflammatory genes, which could then tilt the balance towards apopto-sis. TNF-R2 also utilizes the same pathway for up-regulating survival gene products via NF-κB. However, it is also postulated to induce death via FADD-dependent caspase activation.
Survival signal
Recruitment of RIP1 to TNF-R1
Upon binding to TRADD, RIP1 binds to TRAF2. This protein then engages two anti-apoptotic proteins, cellular inhibitor of apoptosis-1 (cIAP-1) and cIAP-2. The exact pathway of cIAP triggered survival signal pathway is still largely elusive. TRAF2 also activates mitogen-activated protein kinase kinase kinases (MAPKKK) such as extracellular signal-regulated kinase kinase1 (MEKK1). Thus aroused, the MAP kinase pathways end up mobilizing transcription factors like NF-κB or AP-1. In the absence of NF-κB, cellular susceptibility to apoptosis increases, suggesting a pro-survival role for the transcription factor in this signaling pathway (Chen and Goeddel 2002), while activation of AP-1 generally proves deleterious (MacEwan 2002). Moreover, TNF-R1 also directly employs IKK (IκB kinase) complex, which, when activated, can finally release NF-κB as the effector molecule. RIP-1 is known to play a major part in this radial arm of TNF-R1 signaling (Chen and Goeddel 2002).
Death signaling
Recruitment of FADD to TNF-R1
Association of FADD to TNF-R1 bound TRADD recruits pro-caspase 8 by protein–protein interaction via homologous death effector domain (DED). This forms the death-inducing signal complex (DISC), during which pro-caspase 8 is autolytically cleaved to liberate active caspase 8 from the complex. This triggers the caspase cascade, involving the activation of downstream effector caspases (3, 6 and 7), which leads to execution of programmed cell death (Fotin-Mleczek et al. 2002). In addition, TNF-R1 also induces activation of neutral and acidic sphingomyelinases for the degradation of sphingomyelin to phosphocholine and ceramide (Segui et al. 2001). The latter product, ceramide, has emerged as an important lipid second messenger molecule that is considered to mimic some of the cytotoxic as well as cytoprotective effects of TNF-α (Pahan et al. 1999; Cutler and Mattson 2001).
However, there is one basic question that poses itself at this juncture of our current knowledge about TNF-R1 signaling. As the receptor can bind to both TRAF2 and FADD, how does it select one of them in lieu of its engagement with the TNF-α ligand at a certain point? The involvement of certain regulatory proteins appears to be a viable possibility in postreceptor context under these circumstances.
Signaling via TNF-R2
This receptor lacks the cytoplasmic death domain. This implies an anti-apoptotic role for this receptor, which is manifested by the recruitment of TRAF2 by this receptor upon activation (Gupta 2002). Utilization of TRAF2 by both receptor subtypes hints at the activation of common downstream signal transduction pathways (Fig. 2), leading to NF-κB nuclear translocation. On the contrary, TNF-R2 may also aid in apoptosis by depleting TRAF2 and c-IAP proteins thereby accelerating TNF-R1-dependent activation of caspase 8 (Fotin-Mleczek et al. 2002). It has also been suggested to mediate TNF-induced cell death via RIP, as was found in RIP-deficient Jurkat cells (Pimentel-Muinos and Seed 1999).
However, certain limitations have been encountered in investigating signal transduction through TNF-R2. Once activated, TNF-R2 is readily cleaved by metalloproteases into a soluble form (decoy), which still remains adept at binding the ligand cytokine (MacEwan 2002). If extrapolated to HAD brain conditions, similar decoy TNF-R2 may engage substantial amounts of the cytokine away from the cell surface itself, thus limiting the latter’s activity. Moreover, the soluble form of TNF-α (available commercially or present in the CSF of HAD brain) has a much lower binding affinity with TNF-R2 than with the membrane-bound form of the cytokine (Grell et al. 1995). Thus, signaling via TNF-R2 in vivo may actually rely more on cell–cell contact instead of being regulated by soluble cytokine secreted by a distant cell.
Role of TNF-Rs in neurons
In order to explain its diametric activities in neurons, it is commonly postulated that TNF-α binds TNF receptor subtypes in neurons with distinct affinities and thereby dictates neuronal death or survival through different signal transduction pathways, some of which have been described above. This hypothesis has been attested by studies on TNF-R knockout mice. It has been shown that TNF-α has little effect on hippocampal neurons in which TNF-R1 was absent (TNF-R1 −/−), whereas neurons from TNF-R2 knockouts are vulnerable to the cytokine, even at low doses (Yang et al. 2002). These observations recommend TNF-R2 as the modulator responsible for inducing the survival signal, which it may do either in membrane-bound form, thus inducing pro-survival signals, or in decoy form, where it may inhibit death signal by arresting excess TNF-α in vicinity of neurons.
Current opinions on the role of TNF-α in neuronal dysfunction during HAD
The debate regarding the neuroprotective versus neurodegenerative role of TNF-α is almost a decade old, extends considerably in other forms of neurological disorders, like Parkinson’s disease and Alzheimer’s disease, and, as proved by recent literature, is far from over. Most of the experiments to date have been carried out either in vitro or in brain tissue of deceased patients. Either way, the best of our experimental designs has and will always fall short of mimicking the exact in vivo conditions. Thus, extreme opinions (neurotoxic or neurotropic) must be interpreted in the context of the limitations of the experimental paradigm. With diabolic evidence arguing equally for both ends, the exact role of TNF-α during HAD seems elusive. However, certain opinions regarding the part played by this cytokine in neuronal dysfunction are reaching a consensus among workers and are poised between the two extremes. An update follows:
Cell type, receptor specificity and cross-talk dictate TNF-α action
Although TNF-Rs form homotrimers but not heterotrimers (Moosmayer et al. 1994) upon activation by TNF-α ligand, the TNF-R1 : TNF-R2 protein ratio expressed by a certain cell is known to predetermine the response of the cell to the cytokine signal (Vandenabeele et al. 1995; Medvedev et al. 1996). Thus different neuronal subcell types can react differently to TNF-α insult depending primarily on the repertoire of TNF-Rs expressed by them and eventually may resolve their fate depending substantially on intraneural TNF-R cross-talk. Owing to utilization of similar downstream signaling messengers, such cross-talk could also exist between TNF-Rs and other tropic receptors like nerve growth factor receptor (Haviv and Stein 1999) and insulin growth factor receptor (Wilde et al. 2000) (see Perry et al. 2002 for a review on interneuronal receptor cross-talk). Thus, the mere presence of TNF-α in a high dose may not affect neurons at all, if they do not express the required set of receptors to undergo the effect. However, this does not rule out an indirect mode of neuronal injury by TNF-α via NMDA receptor activation by excitotoxic elements.
At this juncture, we are inclined to comment that although role of TNF-R2 in neuroprotection has been substantiated (Bruce et al. 2002; Yang et al. 1996), there is as yet a paucity of adequate proof to suggest such a bias of TNF-R1 in dictating either the death or the survival of neurons. On the one hand, indirect evidence portrays this receptor as the employer of the caspase-mediated programmed cell death (Garden 2002) and, on the other hand, this receptor triggers transcription factor NF-κB in neurons, which is known to transactivate pro-survival genes (Tamatani et al. 1999).
Timing and duration of TNF-α exposure
Most studies revealing the neuroprotective role of TNF-α have involved preconditioning of neurons with the cytokine for prolonged periods (24–48 h) (Cheng et al. 1994). This long-term preconditioning seems to offer neuroprotection by decreasing currents induced by glutamate, NMDA, AMPA and kainate excitotoxic challenges in NF-κB-dependent mechanisms (Furukawa and Mattson 1998). In addition, pro-survival genes may be induced, or deleterious genes may be suppressed, during the pre-exposure by TNF-α (Table 1), which may lead to cellular defense. Hence, temporal magnitude of exposure to the cytokine certainly hasinfluence over the ultimate outcome.
Another study involving hippocampal organotypic slices, stresses on this opinion where TNF-α acts in a neuroprotective fashion when applied prior to ischemic stress but becomes neurotoxic when applied after similar ischemic insult (Venters et al. 1999). Other studies involving acute and chronic responses of TNF-α-deficient mice to experimental brain injury suggested that TNF-α could be deleterious during acute post-traumatic period, but that it facilitated long-term behavioral recovery and histological repair (Wilde et al. 2000). These examples advocate the importance of timing and the duration of TNF-α exposure in shaping up the cytokine’s effect in that particular case. However, conjoining spatiotemporal concepts of the cytokine exposure with in vivo atmosphere during HAD may be comparatively more perplexing than doing so with experimental conditions in vitro.
Synergistic modulation
The extent of TNF-α neurotoxicity (or neuroprotection) is affected by other cytokines and toxic elements present in diseased brain. As seen in the case of iNOS gene expression in glial cells, TNF-α provokes a significant expression of the gene when acting in combination with IL-1β or IFN-γ (Pahan et al. 2000), two other cytokines found in plenty in diseased brain. However, it has a very limited effect on the expression of the same gene when applied alone (Pahan et al. 2001). Thus, effects of TNF-α observed in vitro by treating cells with the cytokine alone or in recombination with a few other selected cytokines may not reflect every possible reaction and response in HAD brain where numerous cytokines and chemokines can insult neurons in numerous combinations. Thus, predicting a combinatorial effect of all possible permutations with these toxic molecules with in vitro tools is practically impossible.
Additionally, the additive effect of two particular molecules could vary from cell type to cell type. For example, TNF-α is accused of enhancing the neurotoxic effect of excitotoxic glutamate in cerebral cortex neurons (Chaparro-Huerta et al. 2002). We have, on the contrary, documented the modest neuroprotective effect of TNF-α, without preconditioning in glutamate-insulted rat cerebellar neurons (unpublished data). Thus, glutamate plus TNF-α, or any other combination of effectors, may not always bring about the same results, especially if cell types are different.
Gene selection by the specificity of transcription machinery
The welter of opposing effects brought about by TNF-α apparently appears to be manipulated by the interplay of different signaling pathways originating from two TNF-α receptors. Although this theory is quite credible and well documented, a recently conducted study offers another avenue to explain such pleiotropism, which apparently involves targeting genes based on the composition of the transcription initiation complex deployed by the cytokine into the nucleus (Ginis et al. 2002). In this study, it has been demonstrated that TNF-α-induced phosphorylated p65 subunit of classical NF-κB (p65–p50) binds to p300 coactivator protein and induces ICAM-1 expression in astrocytes. However, in sublethal ischemic preconditioned cells, this transcriptional activity remains suspended. Alternatively, unphosphorylated p65 NF-κB up-regulates expression of Mn-SOD, the scavenging cyto-protector, even after preconditioning. This transcriptional activity is p300 independent. These findings propose a regulatory knob at the level of transcription initiation for TNF-α target genes. In this model, the presence of certain coactivator proteins (like p300) may direct the binding of NF-κB (or similar transactivators) to certain promoters. Thus, depending on the bound coactivator, NF-κB may induce different set of genes under varying sets of condition. This selective binding may be guided by the phosphorylation status of the transcription factor.
Die to let others live
Apoptosis of irreversibly injured neurons is viewed by a segment of workers as a defense strategy with which to prevent further damage to brain cells. In this context, the neurodegenerative action of TNF-α becomes neuroprotective for adjacent healthy cells (Sapolsky 2001). However, there is little justification in generating such high levels of the cytokine in the immunological defense soup to kill few injured neurons unless we attribute it to uncontrolled immune response.
Conclusion
Lessons from the current literature of HAD and other forms of brain diseases urge us to consider TNF-α as an integral part of the inflammatory and excitatory milieu responsible for injury and death of neurons, although the exact role of this cytokine still remains elusive. Instead of holding on to extreme dogmatic views about its role, the complexity of TNF-α signaling has to be appreciated in wider contexts. The timing and extent of TNF-α exposure and its costimulatory role in the presence of other signaling cues have been registered to drive different outcomes in neurons. The basis of these opposing eventualities needs to be satisfactorily explained so as to generate effective therapeutic strategies involving TNF-α to combat HAD. In order to do so, more information is needed concerning the mode of interaction of TNF-α with its own receptors and its cross-talk with others. In addition, we also need to probe deeper into the intracellular signaling of TNF-Rs paying extra attention to synergistic or antagonistic modulation of the same by various other intra- and extracellular components present in the inflamed atmosphere.
Acknowledgments
This study was supported by a Public Health Service Grant NS39940 from the National Institutes of Health.
Abbreviations used
- BBB
blood–brain barrier
- DED
death effector domain
- EAA
excitotoxic amino acid
- HIV-1
human immunodeficiency type-1
- HAD
HIV-associated dementia
- IκB
inhibitory κB
- IKK
Inhibitory kappa B kinase
- IL
interleukin
- PAF
platelet-activating factor
- SDF-1
stromal-derived factor-1
- TNF-α
tumor necrosis factor-α
References
- Adle-Biassette H, Levy Y, Colombel M, Poron F, Natchev S, Keohane C, Gray F. Neuronal apoptosis in HIV infection in adults. Neurpathol Appl Neurobiol. 1995;21:218–227. doi: 10.1111/j.1365-2990.1995.tb01053.x. [DOI] [PubMed] [Google Scholar]
- Adle-Biassette H, Chretien F, Wingertsmann L, Hery C, Ereau T, Scaravilli F, Tardieu M, Gray F. Neuronal apoptosis does not correlate with dementia in HIV infection but is related to microglial activation and axonal damage. Neuropathol Appl Neurobiol. 1999;25:123–133. doi: 10.1046/j.1365-2990.1999.00167.x. [DOI] [PubMed] [Google Scholar]
- Andjelkovic AV, Kerkovich D, Shanley J, Pulliam L, Pachter JS. Expression of binding sites for β chemokines on human astrocytes. Glia. 1999;28:225–235. [PubMed] [Google Scholar]
- Asensio VC, Campbell IL. Chemokines in the CNS: plurifunctional mediators in diverse states. Trends Neurosci. 1999;22:504–512. doi: 10.1016/s0166-2236(99)01453-8. [DOI] [PubMed] [Google Scholar]
- Bal-Price A, Moneer Z, Brown GC. Nitric oxide induces rapid, calcium dependent release of vesicular glutamate and ATP from cultured rat astrocytes. Glia. 2002;40:312–323. doi: 10.1002/glia.10124. [DOI] [PubMed] [Google Scholar]
- Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. TNF alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a κB-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna BP, Estes ML, Jacobs BS, Hudson S, Ransohoff RM. Human astrocytes proliferate in response to tumor necrosis factor alpha. J Neuroimmunol. 1990;30:239–243. doi: 10.1016/0165-5728(90)90108-y. [DOI] [PubMed] [Google Scholar]
- Barone FC, Parsons AA. Therapeutic potential of anti-inflammatory drugs in focal stroke. Expert Opin Invest Drugs. 2000;9:2281–2306. doi: 10.1517/13543784.9.10.2281. [DOI] [PubMed] [Google Scholar]
- Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocytes glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 sub-groups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-α gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Geddes JW, Knapp PE, McFall RW, Keller JN, Holtsberg FW, Parthasarathy S, Steiner SM, Mattson MP. Anti-death properties of TNF against metabolic poisoning: mitochondrial stabilization by MnSOD. J Neuroimmunol. 1999;93:53–71. doi: 10.1016/s0165-5728(98)00190-8. [DOI] [PubMed] [Google Scholar]
- Canki M, Potash MJ, Bentsman G, Chao W, Flynn T, Heinemann M, Gelbard H, Volsky DJ. Isolation and long term culture of primary ocular human immunodeficiency virus type 1 isolates in primary astrocytes. J Neurovirol. 1997;3:10–15. doi: 10.3109/13550289709015788. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of NMDA receptors. Brain Behav Immun. 1995;9:355–365. doi: 10.1006/brbi.1995.1033. [DOI] [PubMed] [Google Scholar]
- Chaparro-Huerta V, Rivera-Cervantes MC, Torres-Mendoza BM, Beas-Zarate C. Neuronal death and tumor necrosis factor-alpha response to glutamate induced excitotoxicity in cerebral cortex of neonatal rats. Neurosci Lett. 2002;333:95–98. doi: 10.1016/s0304-3940(02)01006-6. [DOI] [PubMed] [Google Scholar]
- Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- Chen ZL, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91:917–925. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- Combs CK, Carlo CJ, Kao SC, Landret GE. β-Amyloid stimulation of microglia and monocytes results in TNF-α dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21:1179–1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter R, Williams C, Ryan L, Erichsen D, Lopez A, Peng H, Zheng J. Fractalkine (CX3CL1) and brain inflammation: Implications for HIV-1-associated dementia. J Neurovirol. 2002;8:1–14. doi: 10.1080/13550280290100950. [DOI] [PubMed] [Google Scholar]
- Cutler RG, Mattson MP. Sphingomyelin and ceramide as regulators of development and lifespan. Mech Ageing Dev. 2001;122:895–908. doi: 10.1016/s0047-6374(01)00246-9. [DOI] [PubMed] [Google Scholar]
- Diem R, Meyer R, Weishaupt JH, Bahr M. Reduction of potassium currents and Phosphoinositol 3-kinase-dependent Akt phosphorylation by tumor necrosis factor-α rescues axotomized retinal ganglion cells from retrograde cell death. J Neurosci. 2001;21:2058–2066. doi: 10.1523/JNEUROSCI.21-06-02058.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diesing TS, Swindells S, Gelbard H, Gendelman HE. HIV-associated dementia: a basic science and clinical perspective. AIDS Read. 2002;12:358–368. [PubMed] [Google Scholar]
- Dreyer EB, Kaiser PK, Offermann JT, Lipton SA. HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science. 1990;248:364–367. doi: 10.1126/science.2326646. [DOI] [PubMed] [Google Scholar]
- Dugas N, Lacroix C, Kilchherr E, Delfraissy JF, Tardieu M. Role of CD23 in astrocytes inflammatory reaction during HIV-1 related encephalitis. Cytokine. 2001;15:96–107. doi: 10.1006/cyto.2001.0896. [DOI] [PubMed] [Google Scholar]
- Fiala M, Looney DJ, Stins M, Way DD, Zhang L, Gan X, Chiappelli F, Schweitzer ES, Shapshak P, Weinand M, Graves MC, Witte M, Kim KS. TNF-alpha opens a paracellular route for HIV invasion across blood brain barrier. Mol Med. 1997;3:553–564. [PMC free article] [PubMed] [Google Scholar]
- Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, Gelbard HA. Tumor necrosis factor-α inhibits glutamate uptake by primary human astrocytes: implications for pathogenesis of HIV-1 dementia. J Biol Chem. 1996;271:15303–15306. doi: 10.1074/jbc.271.26.15303. [DOI] [PubMed] [Google Scholar]
- Fontana G, Valenti L, Raiteri M. gp120 can revert antagonism at the glycine site of NMDA receptors mediating GABA release from cultured hippocampal neurons. J Neurosci Res. 1997;49:732–738. doi: 10.1002/(SICI)1097-4547(19970915)49:6<732::AID-JNR7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Foos TM, Wu JY. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochem Res. 2002;27:21–26. doi: 10.1023/a:1014890219513. [DOI] [PubMed] [Google Scholar]
- Fotin-Mleczek M, Henkler F, Samel D, Reichwein M, Hausser A, Parmryd I, Scheurich P, Schmid JA, Wajant H. Apoptotic crosstalk of TNF receptors: TNF-R2 induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1 dependent activation of caspase-8. J Cell Sci. 2002;115:2757–2770. doi: 10.1242/jcs.115.13.2757. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Mattson MP. The transcription factor NF-κB mediates increases in calcium currents and decreases in NMDA- and AMPA/kainate-induced currents induced by tumor necrosis factor-α in hippocampal neurons. J Neurochem. 1998;70:1876–1886. doi: 10.1046/j.1471-4159.1998.70051876.x. [DOI] [PubMed] [Google Scholar]
- Garden GA. Microglia in human immunodeficiency virus-associated neurodegeneration. Glia. 2002;40:240–251. doi: 10.1002/glia.10155. [DOI] [PubMed] [Google Scholar]
- Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D’Emilia DM, Friedlander RM, Yuan J, Masliah E, Lipton SA. Caspase cascade in human immunodeficiency virus associated neurodegeneration. J Neurosci. 2002;22:4015–4024. doi: 10.1523/JNEUROSCI.22-10-04015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner S. HIV infection and dementia. Science. 2000;287:602–604. doi: 10.1126/science.287.5453.602. [DOI] [PubMed] [Google Scholar]
- Gary DS, Bruce-Keller A, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab. 1998;18:1283–1287. doi: 10.1097/00004647-199812000-00001. [DOI] [PubMed] [Google Scholar]
- Gelbard HA. Neuroprotective strategies for HIV-1-associated neurologic disease. Ann New York Acad Sciences. 1999;890:312–313. doi: 10.1111/j.1749-6632.1999.tb08008.x. [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Zheng J, Coulter CL, Ghorpade A, Che M, Thylin M, Rubocki R, Persidsky Y, Hahn F, Reinhard J, Jr, Swindells S. Suppression of inflammatory neurotoxins by highly active antiretroviral therapy in human immunodeficiency virus-associated dementia. J Infect Dis. 1998;178:1000–1007. doi: 10.1086/515693. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Ginis I, Jaiswal R, Klimanis D, Liu J, Greenspon J, Hallenbeck JM. TNF-α-induced tolerance to ischemic injury involves differential control of NF-κB transactivation: role of NF-κB association with p300 adapter. J Cereb Blood Flow Metab. 2002;22:142–152. doi: 10.1097/00004647-200202000-00002. [DOI] [PubMed] [Google Scholar]
- Giulian D, Wendt E, Vaca K, Noonan CA. The envelope glycoprotein of HIV-1 stimulates release of neurotoxins from monocytes. Proc Natl Acad Sci USA. 1993;90:2769–2773. doi: 10.1073/pnas.90.7.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass JD, Johnson RT. Human immunodeficiency virus and the brain. Annu Rev Neurosci. 1996;19:1–26. doi: 10.1146/annurev.ne.19.030196.000245. [DOI] [PubMed] [Google Scholar]
- Gray F, Scaravilli F, Everall I, Chretien F, An S, Boche D, Adle-Biassette H, Wingertsmann L, Durigon M, Hurtrel B, Chiodi F, Bell J, Lantos P. Neuropathology of early HIV-1 infection. Brain Pathol. 1996;6:1–15. doi: 10.1111/j.1750-3639.1996.tb00775.x. [DOI] [PubMed] [Google Scholar]
- Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 KDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- Griffin DE. Cytokines in the brain during viral infection: clues to HIV-associated dementia. J Clin Invest. 1997;100:2948–2951. doi: 10.1172/JCI119847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Jin YX, Ishikawa M, Huang YM, van der Meide PH, Link H, Xiao BG. Regulation of beta-chemokine mRNA expression in adult rat astrocytes by lipopolysaccharide, proinflammatory and immunoregulatory cytokines. Scand J Immunol. 1998;48:502–508. doi: 10.1046/j.1365-3083.1998.00422.x. [DOI] [PubMed] [Google Scholar]
- Gupta S. A decision between life and death during TNFα signaling. J Clin Immunol. 2002;22:185–194. doi: 10.1023/a:1016089607548. [DOI] [PubMed] [Google Scholar]
- Hallenbeck JM. Many faces of tumor necrosis factoring stroke. Nat Med. 2002;8:1363–1368. doi: 10.1038/nm1202-1363. [DOI] [PubMed] [Google Scholar]
- Han Y, He T, Huang DR, Pardo CA, Ransohoff RM. TNF-alpha mediates SDF-1 alpha-induced NF-kappa B activation and cytotoxic effects in primary astrocytes. J Clin Invest. 2001a;108:425–435. doi: 10.1172/JCI12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Wang J, He T, Ransohoff RM. TNFα down regulates CXCR4 expression in primary murine astrocytes. Brain Res. 2001b;888:1–10. doi: 10.1016/s0006-8993(00)02924-3. [DOI] [PubMed] [Google Scholar]
- Haviv R, Stein R. Nerve growth factor inhibits apoptosis induced by tumor necrosis factor in PC12 cells. J Neurosci Res. 1999;55:269–277. doi: 10.1002/(SICI)1097-4547(19990201)55:3<269::AID-JNR1>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Houzen H, Kikuchi S, Kanno M, Shinpo K, Tashiro K. Tumor necrosis factor enhances outward potassium currents in cultured rat cortical neuron. J Neurosci Res. 1997;50:990–999. doi: 10.1002/(SICI)1097-4547(19971215)50:6<990::AID-JNR9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation. 2000;7:153–159. doi: 10.1159/000026433. [DOI] [PubMed] [Google Scholar]
- Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, Agid Y, Dugas B, Hirsch EC. FcepsilonRII/CD23 is expressed in Parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci. 1999;19:3440–3447. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kast RE. Feedback between glial tumor necrosis factor-alpha and gp-120 from HIV-infected cells helps maintain infection and destroy neurons. Neuroimmunomodulation. 2002–2003;10:85–92. doi: 10.1159/000065184. [DOI] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci USA. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kim G-M, Xu J, Xu J, Song S-K, Yan P, Ku G, Xu XM, Hsu CY. Tumor necrosis factor deletion reduces factor-κβ activation, cellular inhibitor of apoptosis protein 2 expression, and functional recovery after traumatic spinal cord injury. J Neurosci. 2001;21:6617–6625. doi: 10.1523/JNEUROSCI.21-17-06617.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolson DL, Gonzalez-Scarano F. HIV and HIV dementia. J Clin Invest. 2000;106:11–13. doi: 10.1172/JCI10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langford D, Masliah E. Crosstalk between components of the blood brain barrier and cells of the CNS in microglial activation in AIDS. Brain Pathol. 2001;11:306–312. doi: 10.1111/j.1750-3639.2001.tb00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence DM, Major EO. HIV-1 and the brain: connection between HIV-1-associated dementia, neuropathology and neuroimmunology. Microbes Infect. 2002;4:301–308. doi: 10.1016/s1286-4579(02)01542-3. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Hou J, Benveniste EN. Transcriptional regulation of intercellular adhesion molecule-1 in astrocytes involves NF-kappaB and C/EBP isoforms. J Immunol. 1998;92:196–207. doi: 10.1016/s0165-5728(98)00209-4. [DOI] [PubMed] [Google Scholar]
- Letendre SL, Lanier ER, McCutchan JA. Cerebrospinal fluid beta chemokine concentration in neurocognitivity impaired individuals infected with human immunodeficiency virus type 1. J Infect Dis. 1999;180:310–319. doi: 10.1086/314866. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Neuronal injury associated with HIV-1: approaches to treatment. Annu Rev Pharmacol Toxicol. 1998;38:159–177. doi: 10.1146/annurev.pharmtox.38.1.159. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Gendelman HE. Dementia associated with the acquired immunodeficiency syndrome. N Engl J Med. 1995;332:934–940. doi: 10.1056/NEJM199504063321407. [DOI] [PubMed] [Google Scholar]
- Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K. Human immunodeficiency virus type-1 (HIV-1) tat induces nitric oxide synthase in human astroglia. J Biol Chem. 2002;277:39312–39319. doi: 10.1074/jbc.M205107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lung HL, Leung KN, Stadlin A, Ma CM, Tsang D. Induction of tumor necrosis factor receptor type 2 gene expression by tumor necrosis factor-α in rat primary astrocytes. Life Sci. 2001;68:2081–2091. doi: 10.1016/s0024-3205(01)00997-3. [DOI] [PubMed] [Google Scholar]
- MacEwan DJ. TNF receptor subtype signaling: differences and cellular consequences. Cell Signal. 2002;14:477–492. doi: 10.1016/s0898-6568(01)00262-5. [DOI] [PubMed] [Google Scholar]
- Magnuson DS, Knudsen BE, Geiger JD, Brownstone RM, Nath A. Human immunodeficiency virus type 1 tat activates non-N-methyl-d-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann Neurol. 1995;37:373–380. doi: 10.1002/ana.410370314. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Camandola S. NF-κB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001;107:247–254. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Culmsee C, Yu Z, Camandola S. Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem. 2000;74:443–456. doi: 10.1046/j.1471-4159.2000.740443.x. [DOI] [PubMed] [Google Scholar]
- McManus CM, Brosnan CF, Berman JW. Cytokine induction of MIP-1 alpha and MIP-1 beta in human fetal microglia. J Immunol. 1998;160:1449–1455. [PubMed] [Google Scholar]
- Medvedev AE, Espevik T, Ranges G, Sundan A. Distinct roles of the two tumor necrosis factor (TNF) receptors in modulating TNF and lymphotoxin alpha effects. J Biol Chem. 1996;271:9778–9784. doi: 10.1074/jbc.271.16.9778. [DOI] [PubMed] [Google Scholar]
- Moosmayer D, Dinkel A, Gerlach E, Hessabi B, Grell M, Pfizenmaier K, Scheurich P. Coexpression of the human TNF receptors TR60 and TR80 in insect cells: analysis of receptor complex formation. Lymphokine Cytokine Res. 1994;13:295–301. [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood–brain barrier. Neuroscience. 1999;93:1449–1464. doi: 10.1016/s0306-4522(99)00225-0. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Mogi M, Ichinose H, Togari A. Cytokines in Parkinson’s disease. J Neural Transm Suppl. 2000;58:143–151. [PubMed] [Google Scholar]
- Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, Geiger JD. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol. 1996;70:1475–1480. doi: 10.1128/jvi.70.3.1475-1480.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas RS, Compston A, Brown DR. Inhibition of tumour necrosis factor-α- induced NF-κB p52 converts the metabolic effects of microglial-derived TNF- α on mouse cerebellar neurons to neurotoxicity. J Neurochem. 2001;76:1431–1438. doi: 10.1046/j.1471-4159.2001.00141.x. [DOI] [PubMed] [Google Scholar]
- Pahan K, Dobashi K, Ghosh B, Singh I. Induction of the manganese superoxide dismutase gene by sphingomyelinase and ceramide. J Neurochem. 1999;73:513–520. doi: 10.1046/j.1471-4159.1999.0730513.x. [DOI] [PubMed] [Google Scholar]
- Pahan K, Liu X, McKinney MJ, Wood C, Sheikh FG, Raymond JR. Expression of a dominant-negative mutant of p21ras inhibits induction of nitric oxide synthase and activation of nuclear factor-κB in primary astrocytes. J Neurochem. 2000;74:2288–2295. doi: 10.1046/j.1471-4159.2000.0742288.x. [DOI] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Liu X, Hilger S, McKinney M, Petro TM. Induction of nitric-oxide synthase and activation of NF-kappa B by interleukin-12 p40 in microglial cells. J Biol Chem. 2001;276:7899–7905. doi: 10.1074/jbc.M008262200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel CA, Mukhtar M, Pomerantz RJ. Human immunodeficiency virus type 1 Vpr induces apoptosis in human neuronal cells. J Virol. 2000;74:9717–9726. doi: 10.1128/jvi.74.20.9717-9726.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RT, Collins JS, Wiener H, Acton R, Go RC. The role of TNF and its receptors in Alzheimer’s disease. Neurobiol Aging. 2000;22:873–883. doi: 10.1016/s0197-4580(01)00291-3. [DOI] [PubMed] [Google Scholar]
- Perry SW, Dewhurst S, Bellizzi MJ, Gelbard HA. Conflicting effects of TNFα in normal and diseased brain: intraneural receptor crosstalk as one of the mechanism for explaining the paradox. J Neurovirol. 2002;8:611–624. doi: 10.1080/13550280290101021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Gendelman HE. Murine models for human immunodeficiency virus type 1-associated dementia: the development of new treatment testing paradigms. J Neurovirol. 2002;8:49–52. doi: 10.1080/13550280290167993. [DOI] [PubMed] [Google Scholar]
- Petito CK, Vecchio D, Chen Y-T. HIV antigen and DNA in AIDS spinal cords correlate with macrophage infiltration but not with vacuolar myelopathy. J Neuropathol Exp Neurol. 1994;53:86–94. doi: 10.1097/00005072-199401000-00011. [DOI] [PubMed] [Google Scholar]
- Pimentel-Muinos FX, Seed B. Regulated commitment of TNF receptor signaling: a molecular switch for death or activation. Immunity. 1999;11:783–793. doi: 10.1016/s1074-7613(00)80152-1. [DOI] [PubMed] [Google Scholar]
- Pulliam L, Zhou M, Stubblebine M, Bitler CM. Differential modulation of cell death proteins in human brain cells by TNF alpha and platelet activating factor. J Neurosci Res. 1998;54:530–538. doi: 10.1002/(SICI)1097-4547(19981115)54:4<530::AID-JNR10>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Quasney MW, Zhang Q, Sargent S, Mynatt M, Glass J, McArthur J. Increased frequency of the TNFα-308A allele in adults with HIV dementia. Ann Neurol. 2001;50:157–162. [PubMed] [Google Scholar]
- Reimann-Philipp U, Ovase R, Weigel PH, Grammas P. Mechanism of cell death in primary cortical neurons and PC12 cells. J Neurosci Res. 2001;64:654–660. doi: 10.1002/jnr.1119. [DOI] [PubMed] [Google Scholar]
- Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem J. 1994;300:281–290. doi: 10.1042/bj3000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousselet E, Callebert J, Parain K, Joubert C, Hunot S, Hartmann A, Jacque C, Perez-Diaz F, Cohen-Salmon C, Launay JM, Hirsch EC. Role of TNF-α receptors in mice intoxicated with the parkinsonian toxin MPTP. Exp Neurol. 2002;177:183–192. doi: 10.1006/exnr.2002.7960. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Cellular defense against excitotoxic insults. J Neurochem. 2001;76:1601–1611. doi: 10.1046/j.1471-4159.2001.00203.x. [DOI] [PubMed] [Google Scholar]
- Schlomann U, Rathke-Hartlieb S, Yamamoto S, Jockusch H, Bartsch JW. Tumor necrosis factor α induces a metalloprotease-disintegrin, ADAM8 (CD 156): implications for neuron–glia interactions during neurodegeneration. J Neurosci. 2000;20:7964–7971. doi: 10.1523/JNEUROSCI.20-21-07964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- Segui B, Cuvillier O, Adam-Klages S, Garcia V, Malagarie-Cazenave S, Leveque S, Caspar-Bauguil S, Coudert J, Salvayre R, Kronke M, Levade T. Involvement of FAN in TNF-induced apoptosis. J Clin Invest. 2001;108:143–151. doi: 10.1172/JCI11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi B, De Girolami U, He J, Wang S, Lorenzo A, Busciglio J, Gabuzda D. Apoptosis induced by HIV-1 infection of central nervous system. J Clin Invest. 1996;98:1979–1990. doi: 10.1172/JCI119002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi B, Raina J, Lorenzo A, Busciglio J, Gabuzda D. Neuronal apoptosis induced by Hiv-1 Tat protein and TNF-alpha: potentiation of neurotoxicity mediated by oxidative stress and implications for HIV dementia. J Neurovirol. 1998;4:281–290. doi: 10.3109/13550289809114529. [DOI] [PubMed] [Google Scholar]
- Sippy BD, Hofman FM, Wallach D, Hinton DR. Increased expression of TNF-alpha receptors in the brain of patients with AIDS. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;10:511–521. [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O’Callaghan JP. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson’s disease. FASEB J. 2002;16:1474–1476. doi: 10.1096/fj.02-0216fje. [DOI] [PubMed] [Google Scholar]
- Talley AK, Dewhurst S, Perry SW, Dollard SC, Gummuluru S, Fine SM, New D, Epstein LG, Gendelman HE, Gelbard HA. Tumor necrosis factor alpha-induced apoptosis in human neuronal cells: protection by antioxidant N-acetylcysteine and genes bcl-2 and crmA. Mol Cell Biol. 1995;15:2359–2366. doi: 10.1128/mcb.15.5.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamatani M, Che YH, Matsuzaki H, Ogawa S, Okado H, Miyake S, Mizuno T, Tohyama M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFκB activation in primary hippocampal neurons. J Biol Chem. 1999;274:8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- Tarkowski E, Blennow K, Wallin A, Tarkowski A. Intracerebral production of tumor necrosis factor-alpha, a local neuro-protective agent, in Alzheimer’s disease and vascular dementia. J Clin Immunol. 1999;19:223–230. doi: 10.1023/a:1020568013953. [DOI] [PubMed] [Google Scholar]
- Torres-Munoz J, Stockton P, Tacoronte N, Roberts B, Maronpot RR, Petito CK. Detection of HIV-1 gene sequences in hippocampal neurons isolated from postmortem AIDS brains by laser capture microdissection. J Neuropathol Exp Neurol. 2001;60:885–892. doi: 10.1093/jnen/60.9.885. [DOI] [PubMed] [Google Scholar]
- Tyor WR, Glass JD, Griffin JW, Becker PS, McArthur JC, Bezman L, Griffin DE. Cytokine expression in the brain during acquired immunodeficiency syndrome. Ann Neurol. 1992;31:349–360. doi: 10.1002/ana.410310402. [DOI] [PubMed] [Google Scholar]
- Tyor WR, Wesselingh SL, Griffin JW, McArthur JC, Griffin DE. Unifying hypothesis for the pathogenesis of HIV-associated dementia complex, vacuolar myelopathy, and sensory neuropathy. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;9:379–388. [PubMed] [Google Scholar]
- Vandenabeele P, Declercq W, Vanhaesebroeck B, Grooten J, Fiers W. Both TNF receptors are required for TNF-mediated induction of apoptosis in PC60 cells. J Immunol. 1995;154:2904–2913. [PubMed] [Google Scholar]
- Veerhuis R, Hoozemans JJ, Janssen I, Boshuizen RS, Langeveld JP, Eikelenboom P. Adult human microglia secrete cytokines when exposed to neurotoxic prion protein peptide: no intermediary role for prostaglandin E2. Brain Res. 2002;925:195–203. doi: 10.1016/s0006-8993(01)03273-5. [DOI] [PubMed] [Google Scholar]
- Venters HD, Tang Q, Liu Q, VanHoy RW, Dantzer R, Kelley KW. A new mechanism of neurodegeneration: a proinflammatory cytokine inhibits receptor signaling by survival peptide. Proc Natl Acad Sci USA. 1999;96:9879–9884. doi: 10.1073/pnas.96.17.9879. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Wang CX, Shuaib A. Involvement of inflammatory cytokines in central nervous system injury. Prog Neurobiol. 2002;67:161–172. doi: 10.1016/s0301-0082(02)00010-2. [DOI] [PubMed] [Google Scholar]
- Wesselingh SL, Power C, Glass JD, Tyor WR, McArthur JC, Farber JM, Griffin JW, Griffin DE. Intracerebral cytokine mRNA expression in acquired immunodeficiency syndrome dementia. Ann Neurol. 1993;33:576–582. doi: 10.1002/ana.410330604. [DOI] [PubMed] [Google Scholar]
- Wilde GJ, Pringle AK, Sundstrom LE, Mann DA, Iannotti F. Attenuation and augmentation of ischaemia-related neuronal death by tumor necrosis factor-alpha in vitro. Eur J Neurosci. 2000;12:3863–3870. doi: 10.1046/j.1460-9568.2000.00273.x. [DOI] [PubMed] [Google Scholar]
- Xu L, Zhan Y, Wang Y, Feuerstein GZ, Wang X. Recombinant adenoviral expression of dominant negative IkappaB-alpha protects brain from cerebral ischemic injury. Biochem Biophys Res Commun. 2002;299:14–17. doi: 10.1016/s0006-291x(02)02573-1. [DOI] [PubMed] [Google Scholar]
- Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. 2002;22:3025–3032. doi: 10.1523/JNEUROSCI.22-08-03025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh MW, Kaul M, Zheng J, Nottet HS, Thylin M, Gendelman HE, Lipton SA. Cytokine stimulated, but not HIV-infected human monocyte derived macrophages produce neurotoxic levels of 1-cysteine. J Neuroimmunol. 2000;164:4265–4270. doi: 10.4049/jimmunol.164.8.4265. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Imaizumi T, Fujimoto K, Matsuo N, Kimura K, Cui X, Matsumiya T, Tanji K, Shibata T, Tamo W. Synergistic stimulation by tumor necrosis factor-alpha and interferon-gamma, of fractalkine expression in human astrocytes. Neurosci Lett. 2001;303:132–136. doi: 10.1016/s0304-3940(01)01699-8. [DOI] [PubMed] [Google Scholar]
- Zheng J, Thylin MR, Ghorpade A, Xiong H, Persidsky Y, Cotter R, Niemann D, Che M, Zeng YC, Gelbard HA, Shepard RB, Swartz JM, Gendelman HE. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-associated dementia. J Neuroimmunol. 1999;98:185–200. doi: 10.1016/s0165-5728(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP. Lack of p50 subunit of nuclear factor-κB increases vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci. 1999;19:8856–8865. doi: 10.1523/JNEUROSCI.19-20-08856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Zhou D, Cheng G, Mattson MP. Neuroprotective role for the p50 subunit of NF-kappaB in an experimental model of Huntington’s disease. J Mol Neurosci. 2000;15:31–44. doi: 10.1385/JMN:15:1:31. [DOI] [PubMed] [Google Scholar]