

Abstract
Human immunodeficiency virus type 1 (HIV-1) infection is known to cause disorders of the CNS, including HIV-associated dementia (HAD). HIV-1 coat protein gp120 (glycoprotein 120) induces neuronal apoptosis and has been implicated in the pathogenesis of HAD. However, the mechanism by which gp120 causes neuronal apoptosis is poorly understood. The present study underlines the importance of gp120 in inducing the production of ceramide, an important inducer of apoptosis, in human primary neurons. gp120 induced the activation of sphingomyelinases (primarily the neutral one) and the production of ceramide in primary neurons. Antisense knockdown of neutral (NSMase) but not acidic (ASMase) sphingomyelinase markedly inhibited gp120-mediated apoptosis and cell death of primary neurons, suggesting that the activation of NSMase but not ASMase plays an important role in gp120-mediated neuronal apoptosis. Similarly, the HIV-1 regulatory protein Tat also induced neuronal cell death via NSMase. Furthermore, gp120-induced production of ceramide was redox sensitive, because reactive oxygen species were involved in the activation of NSMase but not ASMase. gp120 coupled CXCR4 (CXC chemokine receptor 4) to induce NADPH oxidase-mediated production of superoxide radicals in neurons, which was involved in the activation of NSMase but not ASMase. These studies suggest that gp120 may induce neuronal apoptosis in the CNS of HAD patients through the CXCR4-NADPH oxidase-superoxide-NSMase-ceramide pathway.
Keywords: HIV-1 gp120, ceramide, sphingomyelinases, NADPH oxidase, neurons, apoptosis
Introduction
Human immunodeficiency virus type 1 (HIV-1)-associated dementia (HAD) refers to the severe form of neurological disabilities that is observed in 20-30% of patients in the late stages of acquired immunodeficiency syndrome (AIDS) (Kolson and Gonzalez-Scarano, 2000). From a clinical standpoint, HAD is characterized by neurologic impairments manifested by cognitive, behavioral, and motor dysfunction (Wiley et al., 1986; Bagasra et al., 1996; Saha and Pahan, 2003). This steadily (or, in some cases, abruptly) proceeds to severe global dementia and muteness. Histopathologically, it is identified by infiltration of inflammatory cells into the CNS, gliosis, abnormalities of dendritic processes, and neuronal apoptosis (Wiley et al., 1986; Bagasra et al., 1996; Saha and Pahan, 2003). Without treatment, AIDS patients usually die within 4 months of the onset of HAD, which is much less than one-half of the survival expectancy of untreated AIDS patients without HAD (Bindels et al., 1991).
Several studies have established HAD as a consequence of HIV-1 infection of the brain rather than of an opportunistic pathogen. However, the exact mechanism by which the virus causes this disorder is not completely understood. A number of HIV-1 proteins have been shown to be released from HIV-1-infected cells and/or to be present in the extracellular milieu in the HIV-1-infected brain (Brew et al., 1995; Saha and Pahan, 2003). Some of these proteins have been shown to possess neurotoxic properties (Lipton et al., 1991; Dawson et al., 1993; Bennett et al., 1995; Liu et al., 2002). One such protein is gp120 (glycoprotein 120), the viral envelope glycoprotein, which is shed from HIV-infected macrophages during viral replication. It has been suggested that gp120 may be the etiological agent for the observed neuronal loss in postmortem brains of HIV-positive patients. Indeed, gp120 causes cell death in various neuronal populations in vitro, including midbrain dopaminergic neurons, hippocampal, cortical, and cerebellar granule neurons (Lipton et al., 1991; Dawson et al., 1993; Bennett et al., 1995), as well as in vivo in rodent brains (Bansal et al., 2000). However, the mechanism by which gp120 leads to apoptosis and cell death in neurons is essentially unknown.
Because ceramide, the lipid second-messenger molecule produced from the degradation of sphingomyelin by sphingomyelinases (SMases), induces apoptosis and cell death in various cell types, including glial and neuronal cells (Brugg et al., 1996; Hannun, 1996; Wiesner and Dawson, 1996; Singh et al., 1998; Ruvolo, 2003), we decided to investigate the effect of gp120 on the induction of ceramide production in neuronal cells. Here, we present the evidence that gp120 induces the activation of sphingomyelinases and the production of ceramide in human primary neurons. We also show that the activation of neutral (NSMase) but not acidic (ASMase) sphingomyelinase plays the vital role in gp120-mediated apoptosis of neurons and that CXCR4 (CXC chemokine receptor 4)-coupled NADPH oxidase-mediated superoxide production in neurons is responsible for gp120-induced activation of neutral sphingomyelinase.
Materials and Methods
Reagents. Neurobasal media and B27 supplement were purchased from Invitrogen (San Diego, CA). HIV-1 coat protein gp120 (expressed in Chinese hamster ovary cells; strain, HIV-1 MN) was obtained from US Biological (Swampscott, MA). Recombinant human stromal-derived factor-1α (SDF-1α) was purchased from R & D Systems (Minneapolis, MN). HIV-1 gp120 monoclonal antibodies (mAbs) (2G12) capable of neutralizing HIV-1 MN, HIV-1 Tat (86 amino acid protein produced in Escherichia coli), HIV-1 Tat monoclonal antibodies (1D9), and AMD3100 were obtained from National Institutes of Health AIDS Research and Reference Program. Antibodies against p22phox, glial fibrillary acidic protein (GFAP), and MAP-2 (microtubule-associated protein-2) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Phosphorothioate-labeled antisense and scrambled oligodeoxynucleotides were synthesized in the DNA-synthesizing facility of Invitrogen.
Isolation of human primary neurons. Human primary neurons were prepared as described previously (Zhang et al., 2002) with some modifications. All of the experimental protocols were reviewed and approved by the Institutional Review Board (IRB number 224-01-FB) of the University of Nebraska Medical Center. Briefly, 11- to 17-week-old fetal brains obtained from the Human Embryology Laboratory (University of Washington, Seattle, WA) were dissociated by trituration and trypsinization (0.25% trypsin in PBS at 37°C for 15 min). The trypsin was inactivated with 10% heat-inactivated FBS (Mediatech, Washington, DC). The dissociated cells were filtered successively through 380 and 140 μm meshes (Sigma, St. Louis, MO) and pelleted by centrifugation. The cell pellet was washed once with PBS and once with Neurobasal media containing 2% B27 and 1% antibiotic-antimycotic mixture (Sigma). In the first step, neurons were enriched by allowing the cells (3 × 106/ml) to adhere to poly-d-lysine-coated plates or coverslips for 5 min. Nonadherent cells were removed, and adherent cells (mostly neurons) were further treated with 10 μm cytosine arabinoside (AraC) for 10 d to prevent proliferation of dividing cells. More than 98% of this preparation was positive for MAP-2, a marker for neurons.
Treatment of primary neurons. During treatment with gp120, cells were incubated in Neurobasal medium containing 2% B27 supplement without antioxidant (Invitrogen).
Assay of neutral and acidic sphingomyelinases. Activities of SMase(s) were assayed as described previously (Liu et al., 1998). Briefly, after stimulation, the cells were washed with PBS, harvested, and resuspended in buffer A (100 mm Tris-HCl, 0.1% Triton X-100, 1 mm EDTA, and protease inhibitors, pH 7.4). The cell suspension was sonicated and centrifuged at 500 × g at 4°C for 5 min. The supernatant was used as the enzyme source. For NSMase, the reaction mixture contained enzyme preparation in 100 mm Tris-HCl, 5 nmol of [14C]sphingomyelin, 5 nmol of phosphatidylserine, 5 mm DTT, 0.1% Triton X-100 (1.54 mm), and 5 mm MgCl2, pH 7.4, in a final volume of 100 μl. ASMase activity was measured in a 100 μl reaction mixture consisting of enzyme preparation in 100 mm sodium acetate, 5 nmol of [14C]sphingomyelin, and 0.1% Triton X-100, pH 5.0. The enzyme reaction was initiated by the addition of 50 μl of substrate and stopped by the addition of 1.5 ml of chloroform/methanol (2:1, v/v) and 0.2 ml of water. After vortexing and phase separation, aqueous phase was removed for counting.
Assay of superoxide production. Superoxide production was detected by LumiMax Superoxide Anion Detection kit (Stratagene, La Jolla, CA) following the protocol of the manufacturer. Briefly, 5 × 105 cells suspended in 100 μl of superoxide anion (SOA) assay medium were added to 100 μl of reagent mixture containing 0.2 mm luminol, 0.25 mm enhancer, and 2 μm of either native gp120 or boiled gp120 in SOA assay medium. Light emission was recorded at regular intervals in a TD-20/20 Luminometer (Turner Designs, Sunnyvale, CA).
Lipid extraction. Approximately 5 × 105 cells were exposed to gp120 for different periods of time, and lipids were extracted according to the methods described previously (Pahan et al., 1998, 2000).
Quantification of ceramide levels by diacylglycerol kinase assay. Ceramide content was quantified using diacylglycerol (DAG) kinase as described previously (Pahan et al., 1998, 2000). Briefly, dried lipids were solubilized in 20 μl of an octyl β-d-glucoside-cardiolipin solution (7.5% octyl β-d-glucoside and 5 mm cardiolipin in 1 mm diethylenetriaminepentaacetic acid) by sonication in a sonicator bath. The reaction was then performed in a final volume of 100 μl containing the 20 μl sample solution, 50 mm imidazole HCl, 50 mm NaCl, 12.5 mm MgCl2, 1 mm EGTA, 2 mm dithiothreitol, 6.6 μg of DAG kinase, and 1 mm [γ-32P]ATP (specific activity of 1-5 × 105 cpm/nmol), pH 6.6, for 30 min at room temperature. The labeled ceramide-1-phosphate was resolved with a solvent system consisting of methyl acetate/n-propanol/chloroform/methanol/0.25% KCl in water/acetic acid (100:100:100:40:36:2). A standard sample of ceramide was phosphorylated under identical conditions and developed in parallel. Both standard and experimental samples had identical RF values (0.46). Quantification of ceramide-1-phosphate was performed by autoradiography and densitometric scanning by a FluorChem 8800 Imaging System (Alpha Innotech, San Leandro, CA). Values are expressed as either arbitrary units (absorbance) or percentage change.
Immunostaining of MAP-2, GFAP, and p22phox. Briefly, coverslips containing 200-300 cells/mm2 were fixed with 4% paraformaldehyde for 20 min, followed by treatment with cold ethanol (-20°C) for 5 min and two rinses in PBS. Samples were blocked with 3% BSA in PBS containing Tween 20 (PBST) for 30 min and incubated in PBST containing 1% BSA and goat anti-p22phox (1:50), rabbit anti-GFAP (1:50), or goat anti-MAP-2 (1:50). After three washes in PBST (15 min each), slides were further incubated with Cy5 (Jackson ImmunoResearch, West Grove, PA). For negative controls, a set of culture slides was incubated under similar conditions without the primary antibodies. The samples were mounted and observed under a Bio-Rad (Hercules, CA) MRC1024ES confocal laser-scanning microscope.
Fragment end labeling of DNA. Fragmented DNA was detected in situ by the terminal deoxynucleotidyl transferase (TdT)-mediated binding of 3′-OH ends of DNA fragments generated in response to apoptotic signals, using a commercially available kit (TdT FragEL) from Calbiochem (La Jolla, CA). Briefly, coverslips were treated with 20 μg/ml proteinase K for 15 min at room temperature and washed before TdT staining.
Cell viability measurement. Mitochondrial activity was measured with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma).
Lactate dehydrogenase measurement. The activity of lactate dehydrogenase (LDH) was measured using the direct spectrophotometric assay using an assay kit from Sigma.
Results
HIV-1 gp120 leads to cell death and apoptosis in human primary neurons
gp120 has been shown to induce apoptosis in different types of neuronal cells (Lipton et al., 1991; Dawson et al., 1993; Bennett et al., 1995). It can also induce neuronal apoptosis in the absence of glia (Bachis et al., 2003). At first, we examined whether gp120 was able to induce apoptosis and cell death in human primary neurons. Cells were treated with 200 pm gp120 for 6 hr, followed by analysis of apoptotic bodies by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL). It is apparent from Figure 1A that gp120 treatment markedly induced TdT-mediated labeling of DNA fragments. In the other set of experiments, cells were treated with gp120 for 18 hr, followed by immunostaining with antibodies against MAP-2. We observed loss of neuronal processing and marked loss of MAP-2 immunoreactivity after gp120 treatment (Fig. 1B). To understand whether the effect of gp120 is specific, gp120 was boiled for 5 min, and cells were treated with denatured gp120 for 18 hr. In contrast to the effect of native gp120 on MAP-2 immunoreactivity, denatured gp120 was unable to induce the loss of neuronal processing and MAP-2 immunoreactivity (data not shown).
Figure 1.

gp120 induces apoptosis and cell death in human primary neurons. A, Coverslips containing human primary neurons were incubated with 200 pm gp120 for 6 hr, followed by TdT-mediated end labeling of 3′-OH ends of DNA fragments. B, After 18 hr of treatment with 200 pm gp120, cells were immunostained with MAP-2.
HIV-1 gp120 induces the production of ceramide in human primary neurons
Cells were treated with gp120, and the level of ceramide was measured at different time periods using the DAG kinase assay. We found that the production of ceramide in response to gp120 increased steadily from 30 min to 6 hr of incubation in human primary neurons (Fig. 2A). Within 30 min of treatment, gp120 was able to induce an increase in the level of ceramide by approximately threefold, and, with additional increase in duration of treatment, the level of ceramide increased markedly (Fig. 2A). Because DAG kinase phosphorylates both DAG and ceramide using [γ-32P]ATP as substrate, both lipids can be quantified in the same assay. Although the DAG content was much higher than the ceramide content, the level of DAG was unchanged at different time points of stimulation, in contrast to a time-dependent increase in the production of ceramide (Fig. 2B).
Figure 2.
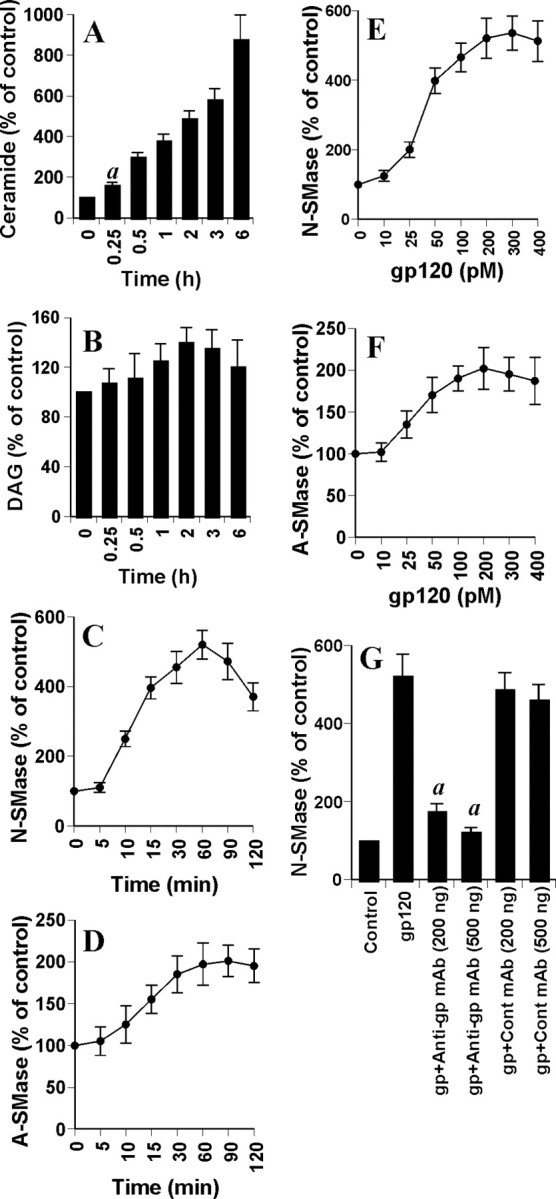
Effect of gp120 on the induction of ceramide production and the activation of NSMase and ASMase in human primary neurons. Cells were incubated with 200 pm gp120 for different time intervals. Lipids were extracted, and ceramide (A) and DAG (B) contents were determined. Activities of NSMase (C) and ASMase (D) were assayed in total-cell extract. Cells were treated with different concentrations of gp120 for 1 hr. Activities of NSMase (E) and ASMase (F) were assayed. G, Cells preincubated with anti-gp120 mAb (2G12) and control IgG1 for 30 min were treated with 200 pm gp120. Activity of NSMase was assayed after 1 hr of treatment. Control group served as 100%, and data from other groups were expressed as percentage of control. Results are mean ± SD of three different experiments. ap < 0.001 versus 0 hr.
HIV-1 gp120 induces the activation of sphingomyelinases in human primary neurons
Five distinct sphingomyelinases have been identified based on their pH optima, cellular localization, and cation dependence (Hannun, 1996; Ruvolo, 2003). However, the neutral membrane-bound Mg2+-independent sphingomyelinase and the lysosomal acid pH-optimum sphingomyelinase have been the best studied for their roles in ceramide generation (Hannun, 1996; Ruvolo, 2003). Because gp120 induced the generation of ceramide, we investigated whether gp120 was capable of inducing the activation of NSMase and ASMase. Marked induction of NSMase activity was observed at 10 min of treatment with gp120, with the maximum induction (approximately fivefold) observed at 1 hr (Fig. 2C). Although gp120 also induced the activation of ASMase (Fig. 2D), the level of its induction was much lower than that of NSMase (Fig. 2C). To understand the optimum dose of gp120 for the activation of sphingomyelinases, cells were treated with different doses of gp120, and, after 1 hr of stimulation, activities of NSMase and ASMase were measured. It is evident from Figure 2, E and F, that gp120 was able to induce the activation of NSMase and ASMase, even at 25 pm. However, maximum induction of NSMase was observed at 200 pm or higher concentration of gp120 (Fig. 2E). On the other hand, maximum induction of ASMase was observed at 100 pm or higher concentration of gp120 (Fig. 2 F). To prove that gp120 but not any contaminant in the gp120 preparation is involved in the activation of SMase, we investigated the effect of HIV-1 gp120 monoclonal antibodies (2G12) on gp120-induced activation of NSMase. Marked inhibition of gp120-induced activation of NSMase in neurons by different doses of gp120 monoclonal antibody 2G12 but not control (IgG1) mAb (Fig. 2G) suggests that the effect is caused by gp120.
Recently, glial activation has been attributed to be one of the major causes of several neurodegenerative disorders, including HAD (Bezzi et al., 2001; Saha and Pahan, 2003). Our human fetal neuronal preparation contains essentially neurons, because these cells were first selected based on differential attachment property of neurons and glia on poly-d-lysine-coated surface and then grown in the presence of AraC. However, a few glial cells (∼2%) are present as a contaminant. Although challenged in several studies (Dawson et al., 1993; Bachis et al., 2003), it has been proposed that gp120 toxicity depends predominantly on the activation of microglia (Bezzi et al., 2001). After activation, microglia is known to produce several proinflammatory cytokines, including tumor necrosis factor-α (TNF-α) (Saha and Pahan, 2003). Because TNF-α is a prototype inducer of NSMase (Hannun, 1996), we were prompted to investigate whether TNF-α had any role in gp120-induced neuronal activation of NSMase. However, neutralizing antibodies against human TNF-α (clone 28401;R&D Systems) capable of blocking TNF-α-induced activation of NSMase had no effect on gp120-induced activation of NSMase in human primary neurons (data not shown), suggesting that, in our experimental setup, glial activation does not play a role in gp120-induced activation of NSMase in neurons. Furthermore, these results also suggest that gp120 induces activation of NSMase in neurons independent of TNF-α.
HIV-1 gp120 induces apoptosis and cell death in human primary neurons through the activation of NSMase
Next, we decided to investigate the role of NSMase and ASMase in gp120-mediated neuronal damage. Antisense oligonucleotides provide an effective tool with which to investigate the in vivo functions of different proteins in primary neurons. Using the method of Bloomfield and Giles (1992) and based on the translational initiation site, we selected different antisense (ASO) and scrambled (ScO) oligonucleotide sequences against human NSMase and ASMase and screened their efficacy to inhibit gp120-induced activation of respective SMase in primary neurons. After screening of several antisenses, we found the following specific ASO and ScO against NSMase and ASMase: NSMase, ASO, 5′-CAGCGAGCCCGTCCACCAGCC-3′; ScO, 5′-CACGCGTCCGACGCCGCACGA-3′; ASMase, ASO, 5′-GACATCTCGGAGCCGGGGCA-3′; ScO, 5′-GGAAACCCGGTTAGGCCCGG-3′.
As observed in Figure 3, A and B, ASO against NSMase markedly inhibited gp120-induced activation of NSMase but not ASMase. Similarly, ASO against ASMase inhibited gp120-induced activation of ASMase but not NSMase. On the other hand, respective scrambled oligonucleotides did not influence the activation of either NSMase or ASMase in gp120-treated primary neurons (Fig. 3A,B). Next, we investigated the effect of antisense knockdown of NSMase and/or ASMase on gp120-induced neuronal apoptosis. Under the experimental condition of ASO-ScO treatment, gp120 treatment resulted in a marked increase in apoptosis (Fig. 3C,F). However, ASO but not ScO against NSMase markedly blocked gp120-induced apoptosis of primary neurons (Fig. 3C-F). On the other hand, ASO against ASMase did not inhibit gp120-induced apoptosis (Fig. 3F). To further confirm this observation, we examined gp120-induced cell death in ASO- and ScO-treated neurons. Cell viability was determined by the MTT metabolism assay. After 18 hr of treatment, gp120 reduced cell viability, as evidenced by a decrease in MTT metabolism (Fig. 3G). Interestingly, ASO against NSMase but not ASMase effectively prevented gp120-induced loss of MTT metabolism (Fig. 3G). Similarly, gp120 also induced an increase in LDH release, and treatment of cells with ASO against NSMase but not ASMase resulted in significant reduction in LDH release (Fig. 3H). On the other hand, ScO against NSMase had no effect on gp120-induced loss of MTT metabolism (Fig. 3G) and increase in LDH release (Fig. 3H). These studies suggest that activation of NSMase but not ASMase plays the key role in gp120-mediated neuronal apoptosis and cell death.
Figure 3.
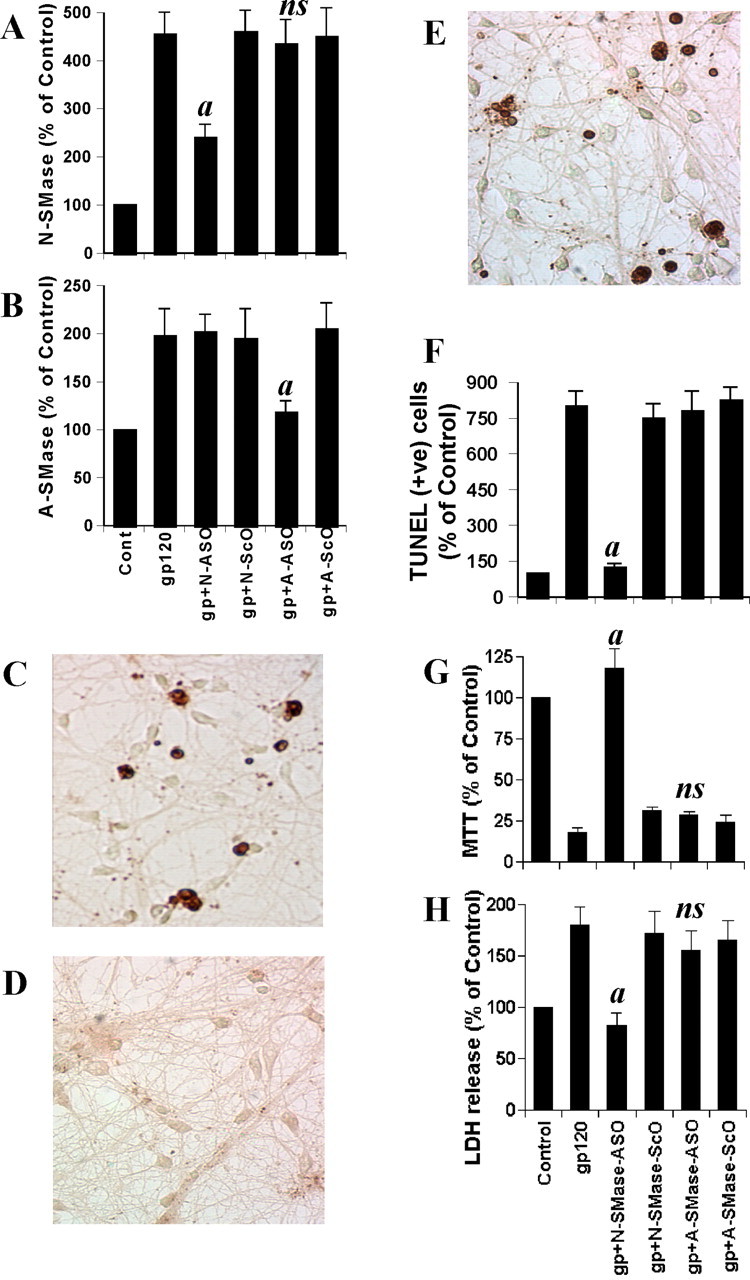
Effect of antisense oligonucleotides against NSMase and ASMase on gp120-mediated cell death in human primary neurons. Cells received 1 μm of either ASO or ScO against NSMase and ASMase. After 48 hr of incubation, cells were treated with 200 pm gp120, and, after 1 hr of stimulation, activities of NSMase (A) and ASMase (B) were measured. Coverslips containing primary neurons were preincubated with 1 μm ASO or ScO against NSMase and ASMase for 40 hr, followed by stimulation with 200 pm gp120. Control cells received only vehicle (PBS). After 6 hr of stimulation, TUNEL (C, control; D, ASO; E, ScO) was performed. TUNEL-positive (+ve) cells were counted manually in four different images of each of three coverslips by three blinded individuals (F). Cells preincubated with 1 μm ASO or ScO against NSMase and ASMase for 40 hr received 200 pm gp120. After 18 hr of stimulation, cells were subjected to MTT reduction activity assay (G), and culture medium was collected for LDH release assay (H). Values obtained from the control group served as 100%, and data obtained in other groups were calculated as percentage of control accordingly. Results are mean ± SD of three different experiments. ap < 0.001 versus gp120. ns, Not significant.
Does HIV-1 regulatory protein Tat also induce neuronal cell death via NSMase?
We investigated whether, in a manner similar to gp120, another neurotoxic HIV-1 protein such as Tat (Kolson and Gonzalez-Scarano, 2000; Saha and Pahan, 2003) also induces cell death in human primary neurons via NSMase. At first, we examined whether recombinant Tat is capable of activating NSMase and/or ASMase. As evident in Figure 4A, Tat induced the activation of NSMase at a different time period of stimulation. Inhibition of Tat-induced activation of NSMase in neurons by HIV-1 Tat monoclonal antibody 1D9 (data not shown) suggests that the effect is caused by Tat but not any contaminant. In contrast to gp120 (Fig. 2C), Tat was unable to induce activation of NSMase at minute intervals; however, with an increase in the time of incubation, Tat markedly induced the activation of NSMase (Fig. 4A). On the other hand, Tat was unable to induce the activation of ASMase in neurons under the same condition (data not shown). Next, we examined whether antisense knockdown of NSMase was capable of preventing Tat-induced neuronal death. After 18 hr of treatment, Tat reduced cell viability, as evidenced by a decrease in MTT metabolism (Fig. 4B). However, ASO but not ScO against NSMase markedly prevented gp120-induced loss of MTT metabolism (Fig. 4B), suggesting that Tat also induces neuronal cell death via NSMase.
Figure 4.

Role of NSMase in Tat-mediated cell death in human primary neurons. A, Cells were incubated with 100 ng/ml Tat for different time intervals. Activity of NSMase was assayed in total-cell extract. B, Cells received 1 μm of either ASO or ScO against NSMase. After 40 hr of incubation, cells were treated with 100 ng/ml Tat, and, after 18 hr of treatment, cells were subjected to MTT reduction activity assay. Values obtained from the control group (Cont) served as 100%, and data obtained in other groups were calculated as percentage of control accordingly. Results are mean ± SD of three different experiments. ap < 0.001 versus Tat.
Involvement of reactive oxygen species in gp120-induced production of ceramide and activation of NSMase in human primary neurons
Next, we investigated mechanisms by which gp120 induced the activation of NSMase in neurons. Previous observations (Liu et al., 1998; Singh et al., 1998) that reactive oxygen species (ROS) are involved in cytokine-induced production of ceramide and activation of NSMase prompted us to investigate whether ROS play a role in gp120-induced production of ceramide and activation of NSMase in neurons. We examined the effect of antioxidants [N-acetylcysteine (NAC) and diphenyliodonium (DPI)] on gp120-induced production of ceramide and activation of SMases. Interestingly, both NAC and DPI dose dependently prevented gp120-induced generation of ceramide (Fig. 5A), suggesting that gp120-induced ceramide generation in neurons is redox sensitive. Next, we investigated whether gp120-induced activation of NSMase and/or ASMase is redox sensitive. Although both NAC and DPI inhibited gp120-induced activation of NSMase (Fig. 5B), these two antioxidants had no effect on the activation of ASMase (Fig. 5C), suggesting that gp120-induced activation of NSMase but not ASMase is redox sensitive.
Figure 5.
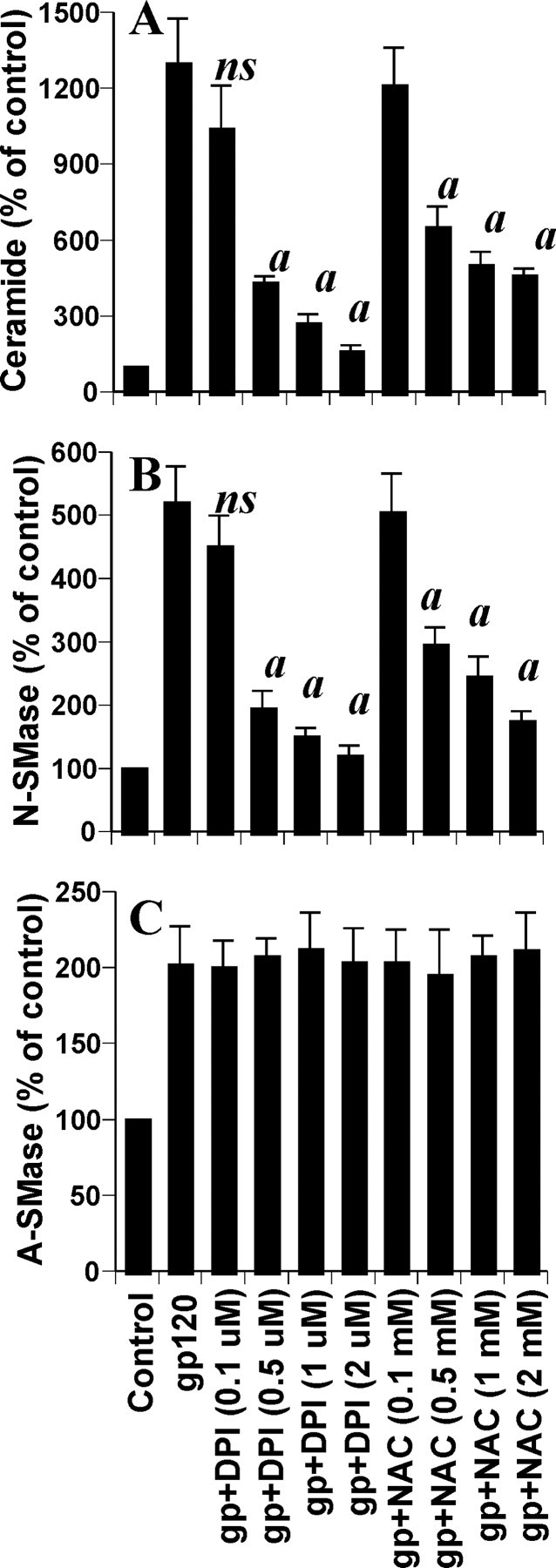
Effects of NAC and DPI on gp120-induced production of ceramide and activation of sphingomyelinases in human primary neurons. Cells preincubated with different concentrations of either NAC or DPI for 1 hr received 200 pm gp120. A, After 12 hr of incubation, level of ceramide was measured. After 1 hr of incubation, activities of NSMase (B) and ASMase (C) were assayed. Values obtained from the control group served as 100%, and data obtained in other groups were calculated as percentage of control accordingly. Results are mean ± SD of three different experiments. ap < 0.001 versus gp120. ns, Not significant.
Involvement of NADPH oxidase in gp120-induced activation of NSMase in human primary neurons
Next, we attempted to identify the ROS-producing molecule that couples gp120 to NSMase in neurons. Because NADPH oxidase is capable of producing ROS (superoxide radicals) in response to different stimuli (Bokoch and Knaus, 2003), we investigated whether human primary neurons expressed NADPH oxidase. Immunofluorescence analysis in Figure 6A as well as Western blot analysis in Figure 6B showed that control neurons abundantly expressed p22phox, a subunit of NADPH oxidase. Next, we were prompted to investigate whether human primary neurons produce superoxide radicals in response to gp120. Interestingly, treatment of primary neurons with gp120 resulted in time-dependent release of superoxide (Fig. 6C). The production of superoxide was observed as early as 60 sec of stimulation, peaked at 90 sec of stimulation, and decreased afterward. To confirm that p22phox is responsible for gp120-induced production of superoxide in neurons, we used ASO against human p22phox. We selected several antisenses and screened for their efficacy to inhibit the protein expression of p22phox by Western blot. Following ASO but not ScO against p22phox effectively inhibited the expression of p22phox in neurons (Fig. 6B): p22phox, ASO; 5′-CGCCAGCGCCTGCTCGTTGGC-3′; ScO, 5′-GGCCTTCCCGGTTAGCCCCGG-3′.
Figure 6.
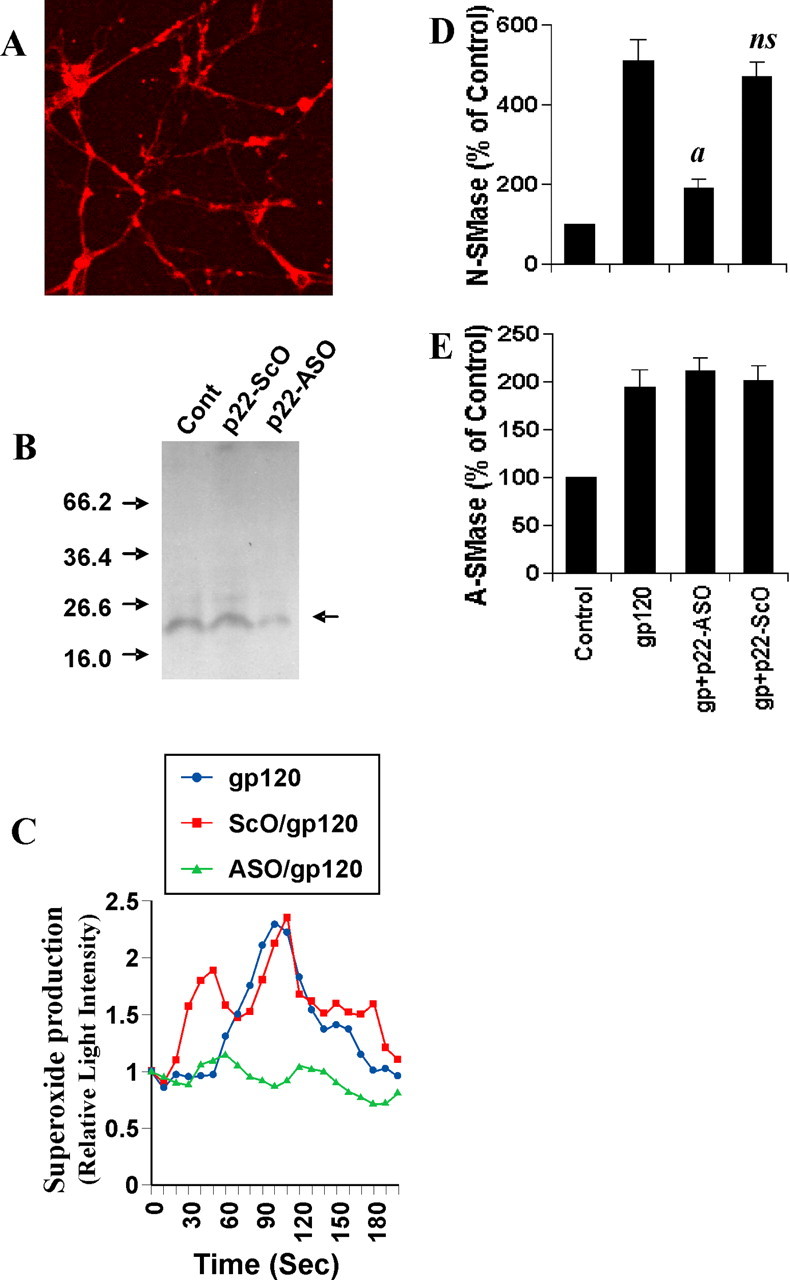
Role of NADPH oxidase in gp120-induced activation of NSMase but not ASMase in human primary neurons. A, Cells were immunostained with antibodies against p22phox. B, Cells were incubated with 1 μm of either ASO or ScO against p22phox. Control cells (Cont) received only vehicle (PBS). After 48 hr, the expression of p22phox was examined by Western blot. Briefly, total-cell homogenates containing protease inhibitor mixture (Sigma) were electrophoresed in 14% SDS-polyacrylamide gel, followed by transfer onto a nitrocellulose membrane and immunoblotting with polyclonal antibodies against human p22phox. C, Cells were incubated with 1 μm either ASO or ScO against p22phox. Control cells received only vehicle (PBS). After 40 hr, cells were stimulated with 200 pm gp120. At different second intervals, superoxide production was assayed in whole cells using the LumiMax Superoxide Anion Detection kit (Stratagene). Results are the mean of two separate experiments. After 1 hr of stimulation of gp120, activities of NSMase (D) and ASMase (E) were assayed. Values obtained from the control group served as 100%, and data obtained in other groups were calculated as percentage of control accordingly. Results are mean ± SD of three different experiments. ap < 0.001 versus gp120. ns, Not significant.
It is apparent from Figure 6C that ASO but not ScO against human p22phox markedly inhibited gp120-induced production of superoxide radicals, suggesting that gp120 induces the production of superoxide through NADPH oxidase. Because gp120-induced activation of NSMase is redox sensitive, we next examined whether p22phox is required for gp120-induced activation of NSMase in neurons. Consistent with the inhibition of gp120-induced activation of NSMase but not ASMase by DPI, ASO against p22phox markedly inhibited gp120-induced activation of NSMase (Fig. 6D) but not ASMase (Fig. 6E), suggesting that gp120 induces the activation of NSMase through NADPH oxidase.
Is NADPH oxidase involved in gp120-induced apoptosis and cell death of human primary neurons?
Because gp120 induced neuronal cell death via NSMase (Fig. 3) and p22phox was required for gp120-induced activation of NSMase (Fig. 6), we were prompted to investigate the effect of p22phox knockdown on gp120-induced apoptosis and cell death in human primary neurons. Treatment of neurons with gp120 led to apoptosis (Fig. 7A) and cell death, as evidenced by loss of MTT metabolism (Fig. 7B) and increase in LDH release (Fig. 7C). However, ASO but not ScO against p22phox effectively prevented gp120-induced apoptosis, loss of MTT metabolism, and release of LDH (Fig. 7). Similarly, DPI at 1 μm concentration also protected neurons against gp120-induced apoptosis and cell death (data not shown). These results suggest that NADPH oxidase is involved in gp120-induced neuronal cell death and that gp120 leads to neuronal apoptosis and cell death via NADPH oxidase-mediated activation of NSMase.
Figure 7.

Role of NADPH oxidase in gp120-induced apoptosis and cell death in human primary neurons. Cells were incubated with 1 μm of either ASO or ScO against p22phox. Control cells received only vehicle (PBS). After 40 hr, cells were stimulated with 200 pm gp120. After 6 hr of stimulation, TUNEL-positive (+ve) cells were counted manually in four different images of each of three coverslips by three blinded individuals (A). After 18 hr of stimulation, cells were subjected to MTT reduction activity assay (B), and culture medium was collected for LDH release assay (C). Values obtained from the control group served as 100%, and data obtained in other groups were calculated as percentage of control accordingly. Results are mean ± SD of three different experiments. ap < 0.001 versus gp120.
Do superoxide radicals induce the activation of NSMase in human primary neurons?
Because NADPH oxidase is involved in gp120-mediated activation of NSMase, we investigated whether superoxide radicals alone were capable of activating NSMase in neurons. Superoxide radicals were generated by exogenous addition of hypoxanthine and xanthine oxidase. Although addition of either hypoxanthine or xanthine oxidase alone to neurons was unable to induce the activation of NSMase (data not shown), addition of both markedly induced the activation of NSMase (Fig. 8). An approximately fivefold increase in NSMase activity was observed within 15 min of treatment with hypoxanthine and xanthine oxidase (Fig. 8). However, the activity of NSMase decreased afterward. On the other hand, exogenous production of superoxide radicals had no effect on the activation of ASMase (data not shown). These studies suggest that superoxide radicals alone are sufficient to induce the activation of NSMase but not ASMase. Next, we investigated whether superoxide radicals stimulate gp120-induced activation of NSMase. As expected, marked stimulation of gp120-induced activation of NSMase was observed after 15 min of treatment with hypoxanthine and xanthine oxidase (Fig. 8), suggesting that superoxide radicals are also capable of stimulating the induction of NSMase.
Figure 8.

Superoxide radicals alone induce the activation of NSMase in human primary neurons. Cells were treated with 200 pm gp120 in the presence or absence of hypoxanthine (5 μm) and xanthine oxidase (0.5 mU/ml) (X plus XO). At different minute intervals, activity of NSMase was assayed. Values obtained from the control group served as 100%, and data obtained in other groups were calculated as percentage of control accordingly. Results are mean ± SD of three different experiments.
Does gp120 couple CXCR4 for the activation of NSMase in human primary neurons?
Because gp120 of different strains of HIV-1, including MN, is known to transduce intracellular signals via CXCR4, and because neurons also express CXCR4 (Meucci et al., 1998; Doranz et al., 1999; Su et al., 1999; van der Meer et al., 2000), we investigated whether gp120 induced the activation of NSMase via CXCR4. We used AMD3100 to antagonize functions of CXCR4. As observed in Figure 9A, AMD3100 dose dependently inhibited gp120-induced activation of NSMase. More than 85% inhibition of gp120-induced activation of NSMase was observed at a concentration of 2 or 5 nm AMD3100 (Fig. 9A), suggesting that gp120 uses CXCR4 for the activation of NSMase. Alternatively, we also used SDF-1α as an agonist of CXCR4 (Bezzi et al., 2001) to investigate whether ligation of CXCR4 alone induces the activation of NSMase. Interestingly, SDF-1α alone time dependently induced the activation of NSMase, exhibiting maximum activation at 60 min (Fig. 9B). Next, we examined the effect of NSMase knockdown on SDF-1α-induced neuronal cell death. Treatment of human primary neurons with SDF-1α led to cell death, as evidenced by loss of MTT metabolism (Fig. 9C) and increase in LDH release (Fig. 9D). However, ASO but not ScO against NSMase effectively prevented SDF-1α-induced loss of MTT metabolism and release of LDH (Fig. 9C,D), suggesting that coupling of CXCR4 alone also induces neuronal cell death via NSMase. Because NADPH oxidase-mediated superoxide release was involved in the activation of NSMase, we were prompted to examine further whether SDF-1α alone produces superoxide radicals in human primary neurons. Interestingly, SDF-1α alone induced the production of superoxide radicals in different minute intervals (Fig. 9E), and DPI, a specific inhibitor of NADPH oxidase, blocked SDF-1α-induced production of superoxide (Fig. 9E), suggesting that coupling of CXCR4 alone produces superoxide radicals via NADPH oxidase. Consistently, ASO but not ScO against p22phox also saved neurons from SDF-1α-mediated loss of MTT (Fig. 9C) and release of LDH (Fig. 9D), suggesting the involvement of NADPH oxidase in SDF-1α-induced neuronal cell death. Furthermore, gp120 was also unable to induce the production of superoxide in AMD3100-treated neurons (Fig. 9F), suggesting that gp120 produces superoxide radicals via CXCR4. Together, these studies suggest that gp120 induces neuronal cell death via the CXCR4-NADPH oxidase-NSMase pathway.
Figure 9.

Role of CXCR4 in gp120-induced activation of NSMase in human primary neurons. A, Cells incubated with different concentrations of AMD3100 for 1 hr were treated with 200 pm gp120. After 1 hr, activity of NSMase was assayed. B, Cells were incubated with 100 ng/ml SDF-1α for different time intervals, followed by assay of NSMase. C, D, Cells preincubated with 1 μm ASO or ScO against NSMase and p22phox for 40 hr received 100 ng/ml SDF-1α. After 18 hr of stimulation, cells were subjected to MTT reduction activity assay (C), and culture medium was collected for LDH release assay (D). Values obtained from the control group served as 100%, and data obtained in other groups were calculated as percentage of control accordingly. Results are mean ± SD of three different experiments. ap < 0.001 versus SDF-1α. E, Cells preincubated with 2 μm DPI for 1 hr were stimulated with 100 ng/ml SDF-1α. F, Cells preincubated with different concentrations of AMD3100 were stimulated with 200 pm gp120. At different second intervals, superoxide production was assayed in whole cells. Values obtained from the control group served as 100%, and data obtained in other groups were calculated as percentage of control accordingly. Results are the mean of two separate experiments.
Discussion
In HIV-1-infected brain, gp120 has been found in the extracellular milieu. Because of the facts that HIV-1 gp120 has been implicated in the pathogenesis of HAD, that inhibition of gp120 activity could reduce the toxicity of HIV in the CNS, and that gp120 is capable of causing neuronal cell death even in the absence of glia (Lipton et al., 1991; Dawson et al., 1993; Bennett et al., 1995; Bansal et al., 2000; Bachis et al., 2003), it is important to understand the mechanisms by which gp120 induces apoptosis and cell death in neurons. Ceramide, a lipid second-messenger molecule, is a prototype inducer of apoptosis in various cell types, including glial and neuronal cells (Brugg et al., 1996; Wiesner and Dawson, 1996). Once ceramide is generated, different molecules, such as Bcl-2, SAPK (stress-activated protein kinase), PKB (protein kinase B), and KSR (kinase suppressor of ras), become involved downstream of ceramide in the apoptotic pathway (Hannun, 1996; Ruvolo, 2003). Here, we present evidence that gp120 induces the activation of sphingomyelinases and the production of ceramide in human primary neurons. First, gp120 induced the production of ceramide in human primary neurons. Second, gp120 markedly induced the activation of NSMase. On the other hand, activity of ASMase increased marginally in response to gp120. Because gp120 induces neuronal cell death, we investigated whether NSMase and/or ASMase are required for gp120-mediated neuronal cell death. Interestingly, antisense knockdown of NSMase but not ASMase protected neurons from gp120-induced apoptosis and cell death, suggesting a key role of NSMase but not ASMase in gp120-mediated neuronal damage. Furthermore, our studies also suggest that the sphingomyelin cycle may play an important role during neurodegeneration in HAD brain. Consistently, a recent finding by Haughey et al. (2004) has shown that overproduction of ceramide may be involved during neuronal death in HAD patients.
Since the discovery of the sphingomyelin cycle, several inducers have been shown to be coupled to NSMase, including 1 α,25-dihydroxyvitamin D3, radiation, antibody cross-linking, TNF-α, IL-1β (interleukin-1β), Fas, nerve growth factor, brefeldin A, and serum deprivation (Hannun, 1996; Ruvolo, 2003). The mechanisms involved in the activation of NSMase in response to TNF-α are becoming clear. For instance, TNF-α initiates the pathway through TNF receptor 1 (TNFR1) (55 kDa receptor) leading to phospholipase A2 activation, generation of arachidonic acid, and subsequent activation of NSMase (Hannun, 1996). Although binding of Fan to the NSMase-activating domain of TNFR1 is required for the activation of NSMase, binding of TRADD [TNFR1-associated death domain (DD)] to the DD of the same receptor induces the activation of ASMase (Adam-Klages et al., 1996; Ruvolo, 2003). In addition, proteases have also been implicated in the pathway leading from TNF-α to the activation of NSMase (Hannun, 1996; Ruvolo, 2003). Previously, it has been found that the activation of NSMase but not ASMase in TNF-α-stimulated cells is redox sensitive (Liu et al., 1998). Here, we delineate that gp120 induces the activation of NSMase in primary neurons through NADPH oxidase-sensitive superoxide production. First, gp120 induced the production of superoxide in minute intervals. Second, primary neurons expressed p22phox, and antisense knockdown of p22phox strongly inhibited gp120-induced production of superoxide. Third, antisense knockdown of p22phox also inhibited gp120-induced activation of NSMase but not ASMase. Fourth, generation of superoxide radicals by hypoxanthine and xanthine oxidase also induced the activation of NSMase but not ASMase. These studies suggest that superoxide radicals are also produced from NADPH oxidase in neurons in response to gp120, which, in turn, induces the activation of NSMase.
The signaling events for the activation of NADPH oxidase are poorly understood. NADPH oxidase consists of both cytosolic and membrane-bound proteins in the resting state. The two membrane-bound components (p22phox and gp91phox) form the heterodimeric flavocytochrome b558, which contains a putative NADPH binding site, FAD, and two hemes (Bokoch and Knaus, 2003). It possesses all of the electron machinery required to transfer two electrons from NADPH to two molecules of molecular oxygen. The four cytosolic components are Rac-GTP and the phosphoproteins p47phox, p40phox, and p67phox. During the activation of NADPH oxidase, all of the cytosolic components are required for translocation to the membrane and assemble with flavocytochrome b558 to enable the electron flow (Bokoch and Knaus, 2003).
Here, we have delineated a mechanism by which gp120 may activate the NADPH oxidase-NSMase-cell death pathway in neurons. Although virus binding to target cells occurs among the HIV-1 gp120, the CD4 molecule, and the members of the chemokine family of receptors [including CXCR4 (Berger et al., 1999)], gp120 can transduce signals in human neurons in a CD4-independent manner (Hesselgesser et al., 1997). Because different CNS cells (including neurons) express CXCR4 and because gp120 has also been shown to interact with neurons via CXCR4 (Hesselgesser et al., 1997; Meucci et al., 1998; van der Meer et al., 2000), we were prompted to explore the possible involvement of CXCR4 in gp120-induced activation of the NADPH oxidase-NSMase-cell death pathway in human primary neurons. First, gp120-induced activation of NSMase was prevented by AMD3100, an antagonist of CXCR4. Second, SDF-1α (an agonist of CXCR4) alone induced the activation of NSMase. Recent studies suggest that α chemokine SDF-1α may contribute to neuronal cell death during HAD (Rostasy et al., 2003). Therefore, it is important to witness that antisense knockdown of NSMase prevented SDF-1α-induced neuronal cell death. Third, gp120-induced production of superoxide radicals was inhibited by AMD3100, suggesting the involvement of CXCR4 in gp120-induced production of superoxide radicals. Finally, SDF-1α alone induced the production of superoxide radicals in neurons, and DPI (a specific inhibitor of NADPH oxidase) blocked SDF-1α-induced superoxide production. Together, these results demonstrate that gp120 couples CXCR4 to activate NSMase via NADPH oxidase-mediated superoxide release.
In summary, we have demonstrated that gp120 induces neuronal apoptosis through the NSMase-ceramide pathway and that the activation of NSMase is regulated by CXCR4-coupled NADPH oxidase-sensitive production of superoxide radicals. Although the in vitro situation of human fetal neurons in culture does not truly resemble the in vivo situation of neurons in the brain of HAD patients, our results suggest that specific targeting of NSMase may be an important therapeutic avenue to halt neuronal damage in HAD and other neurodegenerative disorders. Alternatively, the use of antioxidants and specific inhibitors of NADPH oxidase may be also beneficial in restoring the cellular redox and thus attenuating gp120-mediated activation of the NSMase-ceramide pathway in the degenerating CNS of HAD brain.
Footnotes
This study was supported by National Institutes of Health Grants NS39940 and AG19487. We thank Dr. Xiaojuan Liu for her help in cell culture and Dr. You Zhou of the University of Nebraska at Lincoln for his help in microscopy.
Correspondence should be addressed to Dr. Kalipada Pahan, Section of Neuroscience, Department of Oral Biology, University of Nebraska Medical Center, 40th and Holdrege, Lincoln, NE 68583-0740. E-mail, kpahan@unmc.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249531-10$15.00/0
References
- Adam-Klages S, Adam D, Wiegmann K, Struve S, Kolanus W, Schneider-Mergener J, Kronke M (1996) FAN, a novel WD-repeat protein, couples the p55 TNF-receptor to neutral sphingomyelinase. Cell 86: 937-947. [DOI] [PubMed] [Google Scholar]
- Bachis A, Major EO, Mocchetti I (2003) Brain-derived neurotrophic factor inhibits human immunodeficiency virus-1/gp120-mediated cerebellar granule cell death by preventing gp120 internalization. J Neurosci 23: 5715-5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagasra O, Lavi E, Bobroski L, Khalili K, Pestaner JB, Pomerantz RJ (1996) Cellular reservoirs of HIV-1 in the central nervous system of infected individuals: identification by the combination of in situ polymerase chain reaction and immunohistochemistry. AIDS 10: 573-585. [DOI] [PubMed] [Google Scholar]
- Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM (2000) Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res 879: 42-49. [DOI] [PubMed] [Google Scholar]
- Bennett BA, Rusyniak DE, Hollingsworth CK (1995) HIV-1 gp120-induced neurotoxicity to midbrain dopamine cultures. Brain Res 705: 168-176. [DOI] [PubMed] [Google Scholar]
- Berger EA, Murphy PM, Farber JM (1999) Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol 17: 657-700. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A (2001) CXCR4-activated astrocyte glutamate release via TNF-α: amplification by microglia triggers neurotoxicity. Nat Neurosci 4: 702-710. [DOI] [PubMed] [Google Scholar]
- Bindels PJ, Poos RMJ, Tong JT, Mulder JW, Jager HJC, Coutinho RA (1991) Trends in mortality among AIDS patients in Amsterdam 1982-1988. AIDS 5: 853-858. [PubMed] [Google Scholar]
- Bloomfield M, Giles IG (1992) FINDPROBE: a computer program to locate potential probe sequences in DNA. Biochem Soc Trans 20: 293S. [DOI] [PubMed] [Google Scholar]
- Bokoch GM, Knaus UG (2003) NADPH oxidases: not just for leukocytes anymore! Trends Biochem Sci 28: 502-508. [DOI] [PubMed] [Google Scholar]
- Brew BJ, Rosenblum M, Cronin K, Price RW (1995) AIDS dementia complex and HIV-1 brain infection: clinical-virological correlations. Ann Neurol 38: 563-570. [DOI] [PubMed] [Google Scholar]
- Brugg B, Mitchel PP, Agid Y, Ruberg M (1996) Ceramide induces apoptosis in cultured mesencephalic neurons. J Neurochem 66: 733-739. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, Uhl GR, Snyder SH (1993) Human immunodeficiency virus type 1 coat protein neurotoxicity mediated by nitric oxide in primary cortical cultures. Proc Natl Acad Sci USA 90: 3256-3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doranz BJ, Orsini MJ, Turner JD, Hoffman TL, Berson JF, Hoxie JA, Peiper SC, Brass LF, Doms RW (1999) Identification of CXCR4 domains that support coreceptor and chemokine receptor functions. J Virol 73: 2752-2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun YA (1996) Functions of ceramide in coordinating cellular responses to stress. Science 274: 1855-1858. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Cutler RG, Tamara A, McArthur JC, Vargas DL, Pardo CA, Turchan J, Nath A, Mattson MP (2004) Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann Neurol 55: 257-267. [DOI] [PubMed] [Google Scholar]
- Hesselgesser J, Halks-Miller M, DelVecchio V, Peiper SC, Hoxie J, Kolson DL, Taub D, Horuk R (1997) CD4-independent association between HIV-1 gp120 and CXCR4: functional chemokine receptors are expressed in human neurons. Curr Biol 7: 112-121. [DOI] [PubMed] [Google Scholar]
- Kolson DL, Gonzalez-Scarano F (2000) HIV and HIV dementia. J Clin Invest 106: 11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Sucher NJ, Kaiser PK, Dreyer EB (1991) Synergistic effects of HIV coat protein and NMDA receptor-mediated neurotoxicity. Neuron 7: 111-118. [DOI] [PubMed] [Google Scholar]
- Liu B, Andrieu-Abadie N, Levade T, Zhang P, Obeid LM, Hannun YA (1998) Glutathione regulation of neutral sphingomyelinase in tumor necrosis factor-alpha-induced cell death. J Biol Chem 273: 11313-11320. [DOI] [PubMed] [Google Scholar]
- Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K (2002) Human immunodeficiency virus type 1 (HIV-1) Tat induces nitric-oxide synthase in human astroglia. J Biol Chem 277: 39312-39319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ (1998) Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci USA 95: 14500-14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Khan M, Namboodiri AM, Singh I (1998) Sphingomyelinase and ceramide stimulate the expression of inducible nitric-oxide synthase in rat primary astrocytes. J Biol Chem 273: 2591-2600. [DOI] [PubMed] [Google Scholar]
- Pahan K, Khan M, Singh I (2000) Interleukin-10 and interleukin-13 inhibit proinflammatory cytokine-induced ceramide production through the activation of phosphatidylinositol 3-kinase. J Neurochem 75: 576-582. [DOI] [PubMed] [Google Scholar]
- Rostasy K, Egles C, Chauhan A, Kneissl M, Bahrani P, Yiannoutsos C, Hunter DD, Nath A, Hedreen JC, Navia BA (2003) SDF-1alpha is expressed in astrocytes and neurons in the AIDS dementia complex: an in vivo and in vitro study. J Neuropathol Exp Neurol 62: 617-626. [DOI] [PubMed] [Google Scholar]
- Ruvolo PP (2003) Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol Res 47: 383-392. [DOI] [PubMed] [Google Scholar]
- Saha RN, Pahan K (2003) Tumor necrosis factor-α at the crossroads of neuronal life and death during HIV-associated dementia. J Neurochem 86: 1057-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh I, Pahan K, Khan M, Singh AK (1998) Cytokine-mediated induction of ceramide production is redox-sensitive: implications to proinflammatory cytokine-mediated apoptosis in demyelinating diseases. J Biol Chem 273: 20354-20362. [DOI] [PubMed] [Google Scholar]
- Su SB, Gong W, Grimm M, Utsunomiya I, Sargeant R, Oppenheim JJ, Ming Wang J (1999) Inhibition of tyrosine kinase activation blocks the down-regulation of CXC chemokine receptor 4 by HIV-1 gp120 in CD4+ T cells. J Immunol 162: 7128-7132. [PubMed] [Google Scholar]
- van der Meer P, Ulrich AM, Gonzalez-Scarano F, Lavi E (2000) Immunohistochemical analysis of CCR2, CCR3, CCR5, and CXCR4 in the human brain: potential mechanisms for HIV dementia. Exp Mol Pathol 69: 192-201. [DOI] [PubMed] [Google Scholar]
- Wiesner DA, Dawson G (1996) Staurosporine induces programmed cell death in embryonic neurons and activation of the ceramide pathway. J Neurochem 66: 1418-1425. [DOI] [PubMed] [Google Scholar]
- Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MBA (1986) Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci USA 83: 7089-7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, McLaughlin R, Goodyer C, LeBlanc A (2002) Selective cytotoxicity of intracellular amyloid beta peptide1-42 through p53 and Bax in cultured primary human neurons. J Cell Biol 156: 519-529. [DOI] [PMC free article] [PubMed] [Google Scholar]