

Abstract
The presence of autoreactive T cells recognizing self myelin antigens is necessary for the development of central nervous system autoimmune diseases such as multiple sclerosis (MS). The present study was undertaken to investigate the role of myelin basic protein (MBP)-primed T cells in the expression of inducible nitric oxide synthase (iNOS) in microglial cells. MBP-primed T cells alone markedly induced the production of NO and the expression of iNOS protein and mRNA in mouse BV-2 microglial cells. Similarly, MBP-primed T cells also induced the production of NO in mouse primary microglia. This induction of NO production was primarily dependent on the contact between MBP-primed T cells and microglia. The expression of very late antigen-4 (VLA-4) on the surface of MBP-primed T cells and inhibition of MBP-primed T cell-induced microglial NO production by functional blocking of antibodies to the α4 chain of VLA-4 (CD49d) suggest that VLA-4 integrin on MBP-primed T cells plays an important role in contact-mediated induction of iNOS. Since IFN-β has been used to treat MS patients, we examined the effect of IFN-β on MBP-primed T cell-induced the production of NO. Surprisingly, IFN-β alone induced the production of NO in microglial cells. However, the pretreatment of MBP-primed T cells with IFN-β inhibited the expression of VLA-4 integrin on the surface of MBP-primed T cells and thereby inhibited the ability of those T cells to induce the production of NO in microglial cells. This study illustrates a novel role of neuroantigen-primed T cells in inducing contact-mediated expression of iNOS in microglial cells that may participate in the pathogenesis of MS.
Nitric oxide (NO),1 a short-lived and bioactive free radical, has been recognized to have many functions in normal physiological and pathophysiological conditions (1, 2). At low concentrations, NO has been shown to play a unique role in neurotransmission and vasodilation; however, at higher concentrations, it is a potent neurotoxin (1, 2). Consistently, NO, derived in excessive amounts from the activation of inducible nitric oxide synthase (iNOS) in glial cells (microglia and astrocytes), is assumed to contribute to oligodendrocyte degeneration in demyelinating diseases and neuronal death during neurodegenerative diseases (3-7). Evidence from several laboratories emphasizes the involvement of NO in the pathophysiology of multiple sclerosis (MS) and experimental allergic encephalomyelitis (EAE), the animal model of MS (8-10). Analysis of cerebrospinal fluid from MS patients has shown increased levels of nitrite and nitrate as compared with normal control (11). The reaction of NO with O2− forms peroxynitrite, ONOO−, a strong nitrosating agent capable of nitrosating tyrosine residues of a protein to nitrotyrosine. Increased levels of nitrotyrosine have been found in demyelinating lesions of MS brains as well as in spinal cords of mice with EAE (12, 13). Consistently, uric acid, a scavenger of peroxynitrite, markedly inhibits the appearance of EAE in mice, and the incidence of MS has been found very rare among gout patients having higher levels of uric acid, suggesting the critical involvement of peroxynitrite in the disease process of EAE and MS. Subsequently, semiquantitative reverse transcription-PCR for iNOS mRNA in MS brains also shows markedly higher expression of iNOS mRNA in MS brains than in normal brains (14, 15).
However, the mechanism by which NO is generated in the MS brain is unclear, but it is known that the resident CNS macrophage, the microglia, is an important source. MS is the most common human autoimmune disease of the CNS of unknown etiology. The infiltration of neuroantigen-specific T cells into the CNS is considered a key event in the pathogenesis of MS or EAE (16, 17). However, the mechanisms through which neuroantigen-specific T cells play an etiologic role in MS remain unclear. Since NO produced in excessive amount from the activation of iNOS participates in the pathogenesis of MS, we investigated the role of neuroantigen-specific T cells in the expression of iNOS in microglial cells.
We herein report that MBP-primed T cells induce the expression of iNOS in mouse microglial cells through cell-cell contact and that very late antigen-4 (VLA-4) on the T cell surface plays an important role in this process. Interestingly, interferon-β (IFN-β) inhibited the expression of VLA-4 and thereby blocked the ability of MBP-primed T cells to induce microglial iNOS.
MATERIALS AND METHODS
Reagents
Fetal bovine serum (FBS), Hanks’ balanced salt solution, Dulbecco’s modified Eagle’s medium/F-12, RPMI 1640, l-glutamine, and β-mercaptoethanol were from Mediatech. l-NG-Monomethylarginine (l-NMA) and d-NG-monomethylarginine (D-NMA) were purchased from Biomol. Arginase was purchased from Sigma. Antibodies against mouse macrophage iNOS were obtained from Calbiochem. 125I-labeled protein A and [α-32P]dCTP were obtained from PerkinElmer Life Sciences. Functional blocking antibodies and FITC-labeled antibodies to CD49d (the α4 chain of VLA-4) were obtained from Pharmingen.
Isolation and Purification of T Cells from Lymph Nodes
Female SJL/J mice were immunized subcutaneously with 400 μg of bovine MBP and 60 μg of Mycobacterium tuberculosis (H37RA; Difco Laboratories) in immunofluorescent assay (Calbiochem). On 10–12 days of immunization, lymph nodes were collected from these mice, and a single cell suspension was prepared in RPMI 1640 medium containing 10% FBS, 2 mm l-glutamine, 50 μm β-mercaptoethanol, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells cultured at a concentration of 4–5 × 106 cells/ml in six-well plates were incubated with 50 μg/ml MBP for 4 days. The nonadherent cells were collected and passed through the nylon wool column preincubated for a period of 30 min with RPMI 1640 supplemented with 10% FBS at 37 ºC, 5% CO2. The first 15–20 ml eluant was collected, centrifuged at 500 × g, and resuspended in RPMI 1640-FBS. Viability of the cells was checked by trypan blue exclusion.
Flow Cytometry
The purity of lymph node cells, obtained after nylon wool column separation, was checked by flow cytometry using FITC-labeled anti-CD3 (BIOSOURCE International). Briefly, 1 × 106 cells suspended in RPMI 1640-FBS were incubated in the dark with appropriately diluted FITC-labeled anti-CD3 at 4 ºC for 30 min. Following incubation, the cell suspension was centrifuged, washed thrice, and resuspended in 500 μl of RPMI-FBS. The cells were then analyzed through FACS (BD Biosciences). A minimum of 10,000 cells were accepted for FACS analysis. Cells were gated based on morphological characteristics. Apoptotic and necrotic cells were not accepted for FACS analysis. More than 98% cells were found as CD3-positive T cells. These T cell populations were used to stimulate microglial cells. Similarly, FITC-labeled antibodies to CD49d (integrin α4 chain) (Pharmingen) was used to examine the surface expression of VLA-4 on T cells.
T Cell Proliferation Assay
T cells isolated from MBP-immunized mice were cultured in 96-well U-bottomed microtiter plate (Coaster) in 200 μl of RPMI 1640 containing 10% FBS, 2 mm l-glutamine, 50 μm β-mercaptoethanol, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells (2 × 105 cells) in the wells were stimulated with different concentrations of MBP. Unstimulated cells were kept as controls. After 72 h of stimulation, cells were pulsed with [3H]thymidine (0.5 μCi/well) for another 24 h. Cells were then harvested on GF/C glass fiber filter (Whatman), and the level of [3H]thymidine incorporation in cells was assessed by scintillation counter (Beckman Instruments).
Treatment of MBP-primed T Cells with IFN-β before Their Co-culture with Microglial Cells
T cells isolated from MBP-immunized mice were cultured in RPMI 1640 (supplemented with 10% FBS, 2 mm l-glutamine, 50 μm β-mercaptoethanol, 100 units/ml penicillin, 100 μg/ml streptomycin) containing 50 μg/ml MBP in the presence or absence of different concentrations of IFN-β for 4 days before their co-culture with microglia. One day before this co-culture, another dose of IFN-β was administered. Cells were then centrifuged, washed, and used in co-culture.
Isolation of Mouse Primary Microglia
Microglial cells were isolated from mixed glial cultures according to the procedure of Giulian and Baker (18). Briefly, on days 7–9, the mixed glial cultures were washed three times with Dulbecco’s modified Eagle’s medium/F-12 and subjected to a shake at 240 rpm for 2 h at 37 ºC on a rotary shaker. The floating cells were washed and seeded onto plastic tissue culture flasks and incubated at 37 ºC for 2 h. The attached cells were removed by trypsinization and seeded on to new plates for further studies. From 90 to 95% of this preparation was found to be positive for Mac-1 surface antigen. For the induction of NO production, cells were stimulated with MBP-primed T cells in serum-free Dulbecco’s modified Eagle’s medium/ F-12. Mouse BV-2 microglial cells (a kind gift from the Virginia Bocchini of University of Perugia) were also maintained and induced with different stimuli as indicated above.
Assay for NO Synthesis
Synthesis of NO was determined by assay of culture supernatants for nitrite, a stable reaction product of NO with molecular oxygen. Briefly, supernatants were centrifuged to remove cells, and 400 μl of each supernatant was allowed to react with 200 μl of Griess reagent (19-22) and incubated at room temperature for 15 min. The optical density of the assay samples was measured spectrophotometrically at 570 nm. Fresh culture medium served as the blank. Nitrite concentrations were calculated from a standard curve derived from the reaction of NaNO2 in the assay. Protein was measured by the procedure of Bradford (23).
Immunoblot Analysis for iNOS
Immunoblot analysis for iNOS was carried out as described earlier (20-22). Briefly, cells were detached by scraping, washed with Hanks’ buffer, and homogenized in 50 mm Tris-HCl (pH 7.4) containing protease inhibitors (1 mm phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml pepstatin A, and 5 μg/ml leupeptin). After electrophoresis, the proteins were transferred onto a nitrocellulose membrane, and the iNOS band was visualized by immunoblotting with antibodies against mouse macrophage iNOS and 125I-labeled protein A (20-22).
RNA Isolation and Northern Blot Analysis
Cells were taken out of the culture dishes directly by adding Ultraspec-II RNA reagent (Biotecx Laboratories, Inc.), and total RNA was isolated according to the manufacturer’s protocol. For Northern blot analyses, 20 μg of total RNA was electrophoresed on 1.2% denaturing formaldehyde-agarose gels, electrotransferred to Hybond nylon membrane (Amersham Biosciences), and hybridized at 68 ºC with 32P-labeled cDNA probe using Express Hyb hybridization solution (Clontech) as described by the manufacturer. The cDNA probe was made by polymerase chain reaction amplification using two primers (forward primer, 5′-CTC CTT CAA AGA GGC AAA AAT A-3′; reverse primer, 5′-CAC TTC CTC CAG GAT GTT GT-3′) (24-27). After hybridization, the filters were washed two or three times in solution I (2× SSC, 0.05% SDS) for 1 h at room temperature followed by solution II (0.1× SSC, 0.1% SDS) at 50 ºC for another hour. The membranes were then dried and exposed to x-ray films (Eastman Kodak Co.). The same amount of RNA was hybridized with a probe for glyceraldehyde 3-phosphate dehydrogenase.
RESULTS
MBP-primed T Cells Induce the Production of NO and the Expression of iNOS in BV-2 Microglial Cells
T cells isolated from lymph nodes of MBP-primed mice proliferated in response to MBP, and maximum proliferation was observed at 50 or 100 μg/ml MBP (Fig. 1). However, these cells did not proliferate in response to bovine serum albumin (BSA) (data not shown), suggesting that these cells were primed specifically against MBP. Therefore, in subsequent studies, these cells were treated with 50 μg/ml MBP for priming. After priming, these T cells were purified by a nylon wool column. As shown in the FACS analysis of Fig. 2, MBP-primed T cells were more than 98% pure after nylon wool column purification. Next, we examined whether these purified MBP-primed T cells can induce the production of NO in mouse BV-2 microglial cells. MBP-primed T cells were washed and added to BV-2 microglial cells in direct contact. There was no induction of NO production when MBP-primed T cells were added to microglial cells at a ratio of 0.1:1 (Fig. 3). However, the induction of NO production started from the ratio of 0.2:1 of T cell:microglia, reached the maximum at the ratio of 0.7:1, and decreased at higher concentrations of T cells (Fig. 3). This decrease in NO production was due to the increase in microglial cell death in the presence of higher concentrations of MBP-primed T cells (data not shown). In contrast to MBP-primed T cells, normal T cells were unable to induce the production of NO in BV-2 microglial cells (Table I).
Fig. 1. Proliferation of T lymphocytes with MBP.
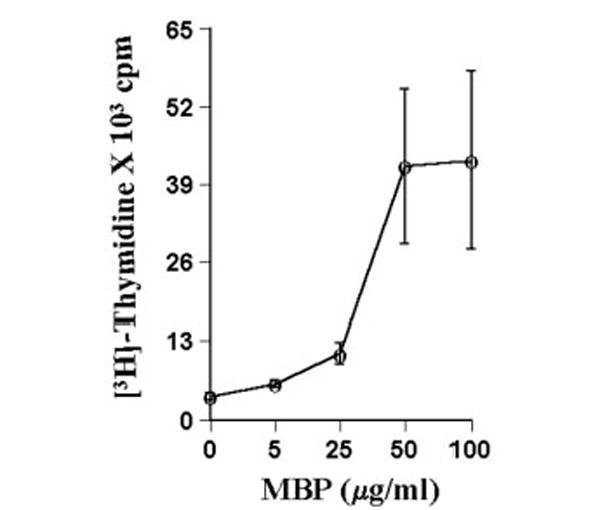
T cells suspended in RPMI containing 10% FBS were treated with different concentrations of MBP. After 4 days of incubation, proliferation was assayed using [3H]thymidine as described under “Materials and Methods.” Data are mean ± S.D. of three different experiments.
Fig. 2. FACS analysis of T lymphocytes with FITC-labeled anti-CD3.
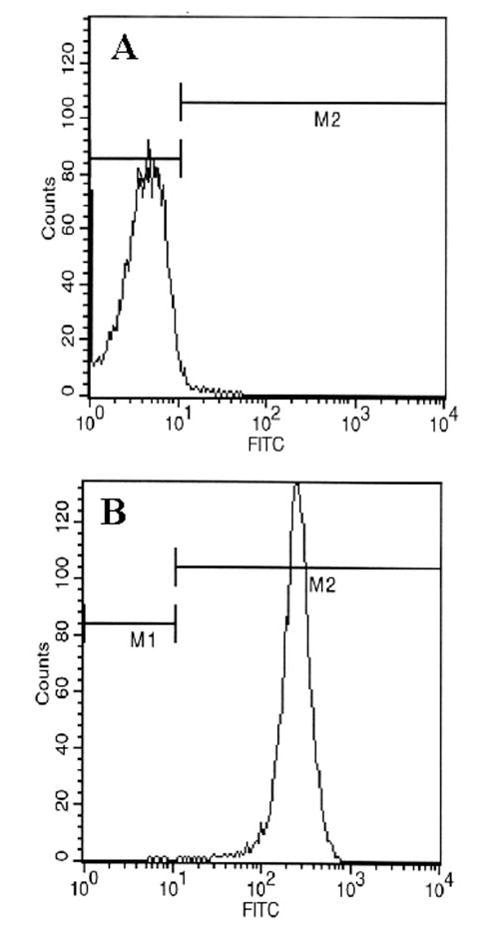
Purified T cells were treated with 0.5 ml of appropriately diluted FITC-labeled anti-CD3 for 30 min followed by FACS analysis in a FACScan as described under “Materials and Methods.” A, FITC-unconjugated; B, FITC-conjugated.
Fig. 3. MBP-primed T lymphocytes induce the production of NO in mouse BV-2 microglial cells.
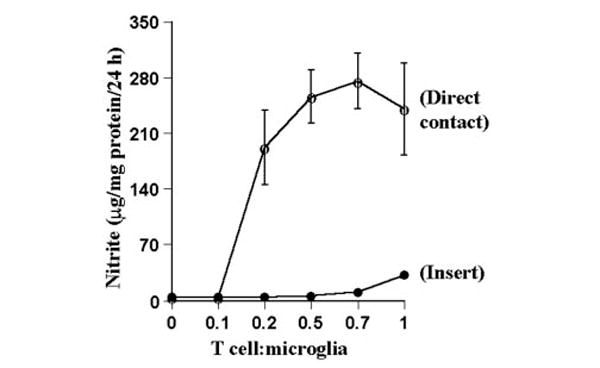
BV-2 cells received different concentrations of MBP-primed T cells in direct contact or within insert under serum-free conditions. After 24 h, supernatants were used for nitrite assay as described under “Materials and Methods.” Data are mean ± S.D. of three different experiments.
Table I. Induction of NO production in mouse BV-2 microglial cells by MBP-primed T cells.
BV-2 microglial cells received MBP-primed T cells at 1 to 0.5 ratio under serum-free conditions in the presence or absence of l-NMA (0.1 mm), d-NMA (0.1 mm), and arginase (100 units/ml). After 24 h of incubation, concentrations of nitrite were measured in the supernatants as described under “Materials and Methods.” Data are mean ± S.D. of three different experiments.
| Treatments | Nitrite |
|---|---|
| μg/24 h/mg of protein | |
| Control BV-2 cells | 3.6 ± 0.5 |
| BV-2 + normal T cells (1:0.5) | 3.6 ± 0.3 |
| BV-2 + normal T cells (1.1) | 3.8 ± 0.5 |
| BV-2 + MBP-primed T cells (1:0.5) | 256 ± 34 |
| BV-2 + MBP-primed T cells (1:0.5) + l-NMA | 6.3 ± 1.7 |
| BV-2 + MBP-primed T cells (1:0.5) + d-NMA | 255 ± 43 |
| BV-2 + MBP-primed T cells (1:0.5) + arginase | 4.6 ± 1.5 |
Next, we examined whether the contact between MBP-primed T cells and microglial cells is necessary to induce the production of NO. First, MBP-primed T cells were placed in a culture insert, where they were in close proximity to, but not contacting, microglia. In contrast to marked induction of NO production by MBP-primed T cell:microglia contact (Fig. 3 and Table I), very little induction of NO production was observed when MBP-primed T cells were placed within culture inserts (Fig. 3). Second, the conditioned supernatant of MBP-primed T cells was added to microglial cells. Fifty μl of supernatant is equivalent to T cells sufficient for the ratio of 0.5:1 of T cell: microglia. The induction of NO production by 50 μl of that supernatant (7.7 ± 2.4 μg/24 h/mg of protein) was very low as compared with that (256 ± 34 μg/24 h/mg of protein) of 0.5:1 of T cell:microglia (Tables I and II). These observations suggest that the induction of NO production by MBP-primed T cells primarily depend on the contact between T cells and microglia. However, higher volumes of supernatants of MBP-primed T cells did induce the production of NO in BV-2 glial cells by 5–9-fold (Table II).
Table II. Induction of NO production in BV-2 microglial cells by supernatants of MBP-primed T cells.
BV-2 microglial cells received different amount of supernatants of MBP-primed T cells under serum-free conditions. The total volume of supernatant was adjusted to 1 ml. After 24 h of incubation, concentrations of nitrite were measured in the supernatants as described under “Materials and Methods.” Data are mean ± S.D. of three different experiments.
| Treatments | Nitrite |
|---|---|
| μg/24 h/mg of protein | |
| Control BV-2 cells | 3.6 ± 0.5 |
| BV-2 + normal T cell supernatant (200 μl) | 3.6 ± 0.4 |
| BV-2 + MBP-primed T cell supernatant (25 μl) | 3.6 ± 0.4 |
| BV-2 + MBP-primed T cell supernatant (50 μl) | 7.7 ± 2.4 |
| BV-2 + MBP-primed T cell supernatant (100 μl) | 18.9 ± 2.9 |
| BV-2 + MBP-primed T cell supernatant (200 μl) | 39.6 ± 3.2 |
The inhibition of MBP-primed T cell-induced NO production by arginase, an enzyme that degrades the substrate (l-arginine) of NOS and l-NMA, a competitive inhibitor of NOS, but not by d-NMA, a negative control of l-NMA, suggests that MBP-primed T cells induce the production of NO in BV-2 microglial cells through NOS-dependent arginine metabolism (Table I). To understand further the mechanism of NO production, we examined the effect of MBP-primed T cells on protein and mRNA levels of iNOS. Western blot analysis with antibodies against murine macrophage iNOS and Northern blot analysis for iNOS mRNA clearly showed that MBP-primed T cells alone are capable of inducing the expression of iNOS protein (Fig. 4A) and iNOS mRNA (Fig. 4B). Consistent with the induction of NO production, MBP-primed T cells did not induce the expression of iNOS protein and mRNA in BV-2 microglial cells when added at a ratio of 0.1:1. The induction of iNOS protein and mRNA started at the ratio of 0.2:1 of T cell:microglia, peaked at the ratio of 0.7:1, and decreased at higher concentrations of T cells (Fig. 4). Consistent with a role of NFκB activation in the expression of iNOS (24-31), we have also found that MBP-primed T cell:microglia contact induced the activation of NFκB in BV-2 microglial cells and that SN50, a specific inhibitor of NFκB activation (32), but not that SN50M, a nonfunctional mutant of SN50, inhibited MBP-primed T cell-induced the production of microglial NO (data not shown).
Fig. 4. MBP-primed T lymphocytes induce the expression of iNOS in mouse BV-2 microglial cells.
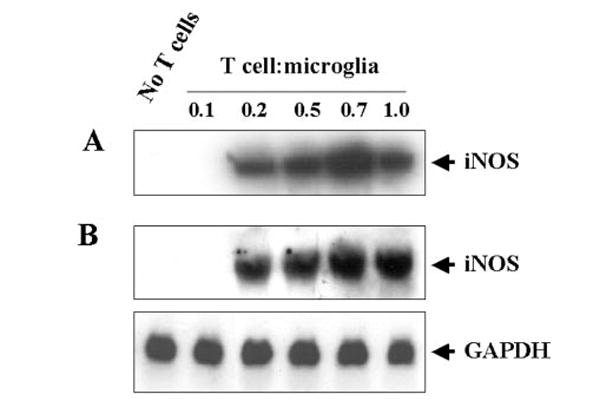
BV-2 cells were stimulated with different concentrations of MBP-primed T cells under serum-free conditions. In A, cell homogenates were electrophoresed, transferred on nitrocellulose membrane, and immunoblotted with antibodies against mouse macrophage iNOS as described under “Materials and Methods.” In B, after 6 h of incubation, Northern blot analysis for iNOS mRNA was carried out as described under “Materials and Methods.” GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
MBP-primed T Cells Induce the Production of NO in Mouse Primary Microglia
To understand whether MBP-primed T cells were able to induce the production of NO in primary cells, we examined the effect of MBP-primed T cells on the induction of NO production in mouse primary microglia (Table III). Consistent with the induction of NO production in BV-2 microglial cells, MBP-primed T cells dose-dependently induced the production of NO in mouse primary microglia. Similar to BV-2 cells, MBP-primed T cells were unable to induce the production of NO in primary microglia when added at a ratio of 0.1:1 of T cells and microglia (Table III). However, the induction of NO production started from the ratio of 0.2:1 of T cell:microglia, reached the maximum at the ratio of 0.5:1, and decreased at higher concentrations of T cells (Table III).
Table III. MBP-primed T cells induce the production of NO in mouse primary microglia.
Mouse primary microglia received MBP-primed T cells at different ratios under serum-free conditions. After 24 h of incubation, concentrations of nitrite were measured in the supernatants as described under “Materials and Methods.” Data are mean ± S.D. of three different experiments.
| Treatments | Nitrite |
|---|---|
| μg/24 h/mg protein | |
| Control microglia | 3.4 ± 0.5 |
| Microglia + normal T cells (1.0.5) | 3.45 ± 0.4 |
| Microglia + normal T cells (1.1) | 3.6 ± 0.4 |
| Microglia + MBP-primed T cells (1.0.1) | 3.4 ± 0.4 |
| Microglia + MBP-primed T cells (1.0.2) | 156 ± 11 |
| Microglia + MBP-primed T cells (1.0.5) | 363 ± 46 |
| Microglia + MBP-primed T cells (1.0.7) | 349 ± 35 |
| Microglia + MBP-primed T cells (1.1) | 310 ± 23 |
Role of VLA-4 in MBP-primed T Cell-induced Production of NO in BV-2 Microglial Cells
Integrins being present on the cell surface are mainly involved in integrating cell-cell interaction (33). It has been shown that T cells activated by anti-CD3 express more VLA-4 integrin and that VLA-4 plays an important role in T cell contact-mediated activation of microglial cells (34). Therefore, we investigated whether neuroantigen-primed T cells require VLA-4 to induce contact-mediated production of NO in microglial cells. At first, we analyzed the expression of VLA-4 on the surface of MBP-primed T cells by FACS analysis using FITC-labeled antibodies against the α4 chain of VLA-4 antigen. Fig. 5A represents autofluorescence as this was observed in unconjugated normal T cells. As areas under M1 and M2 in Fig. 5 represent autofluorescence and fluorescence due to VLA-4, respectively, there was very little expression of VLA-4 on the surface of normal T cells (Fig. 5B) in contrast to marked expression of VLA-4 on the surface of MBP-primed T cells (Fig. 5C). Analysis of three separate experiments shows that MBP priming induced the expression of VLA-4 by 16.85 ± 2.15-fold. These results suggest that neuroantigen priming induces the expression of VLA-4 on the surface of T cells.
Fig. 5. Expression of VLA4 on the surface of MBP-primed T cells.
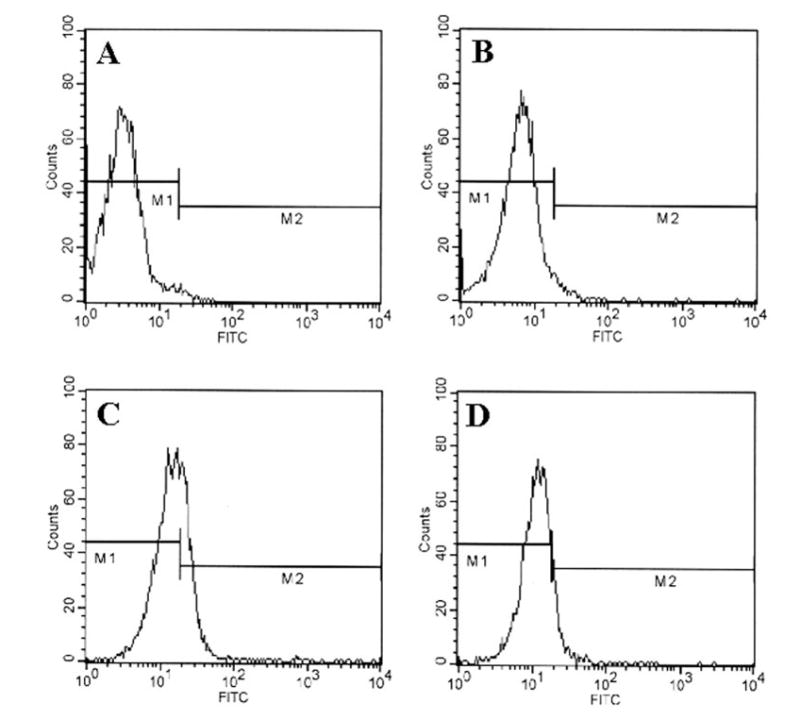
Normal or MBP-primed T cells were treated with 0.5 ml of appropriately diluted FITC-labeled anti-VLA α4 for 30 min followed by FACS analysis. A, FITC-unconjugated normal T cells; B, FITC-conjugated normal T cells; C, FITC-conjugated MBP-primed T cells. In D, similarly MBP-primed T cells that were treated with 50 units/ml IFN-β during priming with 50 μg/ml MBP for 4 days were analyzed by FACS.
Next, to examine whether this VLA-4 is involved in contact-mediated production of NO, we used functional blocking antibodies against CD49d (the α4 chain of VLA-4). It is apparent from Fig. 6 that incubation of MBP-primed T cells with different concentrations of antibodies against CD49d inhibited its ability to induce the production of NO in microglial cells. Although antibodies against CD49d at a dose of 10 or 20 μg/ml were not effective in blocking the production of NO (data not shown), marked inhibition was observed when antibodies were used at a concentration of 50 or 100 μg/ml. In contrast, control IgG did not block the ability of MBP-primed T cells to induce microglial production of NO (Fig. 6). These studies suggest that VLA-4 on the surface of MBP-primed T cells is involved in contact-mediated induction of NO production in microglial cells.
Fig. 6. Functional blocking antibodies against the α4 chain of VLA4 inhibit the ability of MBP-primed T cells to induce the production of NO in BV-2 microglial cells.
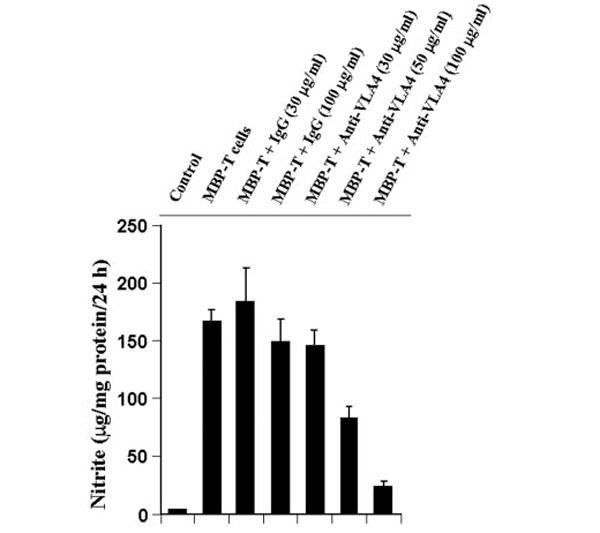
MBP-primed T cells were mixed with either different concentrations of antibodies against the α4 chain of VLA4 or control IgG and rocked gently for 1 h at room temperature. Cells were centrifuged, washed twice, and added to BV-2 microglial cells at a ratio of 0.7:1 T cell:microglia. After 24 h of stimulation, supernatants were used for nitrite assay. Data are mean ± S.D. of three different experiments.
Effect of IFN-β on MBP-primed T Cell-induced Production of NO in BV-2 Microglial Cells
Recent clinical trials have demonstrated that IFN-β or betaseron decreases the number of relapses in relapsing-remitting MS (35, 36). However, the mode of action of IFN-β in MS remains unclear. Since MS is a T cell-mediated disease and MBP-primed T cells induce iNOS in microglial cells, we investigated the effect of IFN-β on MBP-primed T cell-induced production of NO in BV-2 microglial cells. MBP-primed T-cells, with or without preconditioning with different doses of IFN-β, were co-cultured with BV-2 microglial cells for 24 h. It is evident from Fig. 7A that the production of NO in BV-2 glial cells was markedly inhibited by more than 90% when they were co-cultured with MBP-primed T cells preconditioned with IFN-β. On the other hand, addition of IFN-β to the co-culture of BV-2 glial cells and MBP-primed T cells did not result in inhibition of NO production (Fig. 7B), suggesting that pretreatment of MBP-primed T cells with IFN-β is necessary to inhibit the ability of T cells to induce the production of NO. Surprisingly, IFN-β alone induced the production of NO in BV-2 microglial cells (Fig. 7C). These observations suggest that despite the ability of IFN-β itself to induce the production of NO in microglial cells, pretreatment of MBP-primed T cells with IFN-β inhibits the ability of MBP-primed T cells to induce contact-mediated production of NO in microglial cells.
Fig. 7. Effect of IFN-β on MBP-primed T cell-induced production of NO in BV-2 microglial cells.
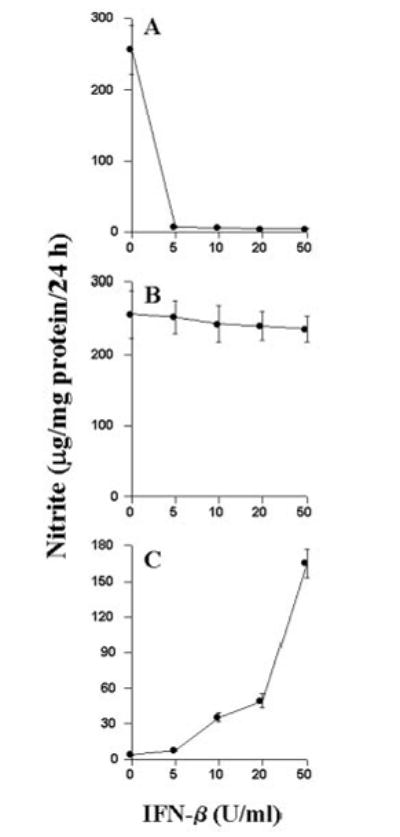
BV-2 cells were stimulated under serum-free conditions with MBP-primed T cells, which were pretreated with different concentrations of IFN-β (A), MBP-primed T cells followed by the treatment of different concentrations of IFN-β (B), and different concentrations of IFN-β (C). After 24 h of stimulation, supernatants were used for nitrite assay. Data are mean ± S.D. of three different experiments.
Effect of IFN-β on the Surface Expression of VLA-4 in MBP-primed T Cells
Since IFN-β inhibited the ability of MBP-primed T cells to induce contact-mediated production of NO in microglia, we investigated the effect of IFN-β on the expression of VLA-4 on the surface of MBP-primed T cells. MBP-primed T cells were treated with 50 units/ml IFN-β during priming with 50μg/ml MBP for 4 days before the FACS analysis. One day before FACS analysis, another dose of IFN-β (50 units/ml) was administered. Cells were then centrifuged, washed, and used in co-culture. Consistent with the inhibitory effect of IFN-β on MBP-primed T cell-induced production of NO (Fig. 7A), pretreatment of MBP-primed T cells with IFN-β led to the inhibition of VLA-4 expression (compare Fig. 5D with 5C). Analysis and quantitation of FACS data obtained from four separate experiments showed that IFN-β treatment of MBP-primed T cells resulted in 50.8 ± 12.1% inhibition of VLA-4 expression. These results suggest that IFN-β inhibits the ability of MBP-primed T cells to induce microglial production of NO by inhibiting the expression of VLA-4 on the surface of MBP-primed T cells.
DISCUSSION
Activation of microglia, CNS-resident professional macrophages, has been implicated in the pathogenesis of a variety of neurodegenerative diseases, including multiple sclerosis (MS), Alzheimer’s disease, Creutzfeld-Jacob disease, and human immunodeficiency virus dementia (37). Upon activation, microglia produce and secrete potentially neuroinflammatory molecules including NO that play a pivotal role in demyelination of MS patients (7, 24, 25, 28). Bagasra et al. (38) have shown that iNOS-expressing cells in MS brains were predominantly, but not exclusively, microglia/macrophages. Therefore, understanding the mechanism by which iNOS is induced in CNS microglia of MS patients can thus impact upon the rational treatment of the disease. However, the mechanism by which microglia are activated to induce the expression of iNOS for the production of NO in MS brain remains unclear. Although lipopolysaccharide is a potent inducer of iNOS in microglia, it has not been demonstrated to have a physiological relevance in MS (39). IFN-γ, alone or in combinations with other proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and IL-1β, can also induce iNOS in microglia (7, 24, 25, 28); however, microglia isolated from adult human brain tend to be a poor source of iNOS-derived NO in response to these proinflammatory cytokines (40, 41). These observations suggest that apart from proinflammatory cytokines, microglia can be activated by some other mechanism(s) for the induction of iNOS in the CNS of MS patients.
The infiltration of neuroantigen-specific T cells into the CNS is considered a key event in the pathogenesis of MS or EAE. However, the biological role of such T cells within the CNS is poorly understood. Several lines of evidence presented in this manuscript clearly support the conclusion that MBP-primed T cells potently induce the expression of iNOS in microglial cells through cell-cell contact. This conclusion is based on the following observations. First, MBP-primed T cells induce the production of NO and the expression of iNOS through the activation of NFκB in mouse microglial cells. This effect was dose-dependent, and it peaked at 0.7:1 or 0.5:1 of T cell:microglia. In contrast, normal T cells were unable to induce the production of NO and the expression of iNOS. Second, the placement of MBP-primed T cells in a culture insert, where they were in close proximity to but not contacting microglia, was unable to induce the production of NO in microglial cells. Third, soluble factors of MBP-primed T cells equivalent to T cells of 0.5:1 or 0.7:1 of T cell:microglia were very poor inducers of NO.
Integrins and adhesion molecules are involved in cell-cell interaction. It has been shown that an increased number of lymphocytes express VLA-4 in active MS lesions (42) and that blocking VLA-4 function with a monoclonal antibody prevents experimental allergic encephalomyelitis, an animal model of MS, in mice (43). Consistently, short term treatment of MS patients with anti-α4 integrin antibody significantly reduces BBB dysfunction as assessed by contrast-enhanced MRI (44). Earlier, Yong and colleagues (34) have reported that this VLA-4-VCAM-1 interaction plays a crucial role when anti-CD3-stimulated T cells activate microglia through cell-cell contact, prompting us to investigate the role of VLA-4 in neuroantigen-primed T cell-induced microglial production of NO. The observations presented in this manuscript that MBP-primed T cells expressed VLA-4 on their surface and that blocking of that VLA-4 by neutralizing antibodies against the α4 chain of VLA-4 inhibited the ability of MBP-primed T cells to induce the production of NO clearly suggest that VLA-4 integrin on the surface of neuroantigen-primed T cells plays an important role in contact-mediated induction of iNOS in microglia. NO, being a major mediator in immune and autoimmune functions, has also been shown to increase the permeability of the BBB, allowing substances to enter into the brain passively, leading to vasogenic edema and secondary brain damage (45). Although the precise molecular mechanisms for NO-induced breakdown of the BBB are not completely understood, in a cell culture model of the BBB, NO leads to a rapid breakdown in model barrier integrity and resulted in a reduction in cellular ATP content and glyceraldehyde 3-phosphate dehydrogenase activity (46). If these cell culture studies have significance in vivo in the CNS of EAE and MS, it is possible that neuroantigen-primed T cells may increase the permeability of BBB through contact-mediated induction of NO production in microglia and astroglia2 present near the BBB junction.
Sedgwick et al. (42) have shown that microglia and T cells do interact in vivo. In the graft-versus-host disease model, activated microglia were found to cluster around T cell infiltrates and to be associated with single or clustered microglia (42). Furthermore, microglia isolated from animals of graft-versus-host disease proliferated and exhibited functions of activated microglia, such as phagocytosis and motility (42). In light of these in vivo observations and the proximity of activated T cells and microglia in the perivascular space or CNS parenchyma of MS lesions (48), the induction of iNOS by the contact between MBP-primed T cells and microglia described in this manuscript is relevant to MS pathogenesis. The in vitro model of our study used a 0.5:1 or 0.7:1 ratio of neuroantigen-primed T cells to microglia, and at present, it is unclear whether this is a relevant ratio. Depending on the fact that microglia constitute only 2–5% of total CNS cells in normal brain (37), the present study suggests that infiltration of very few neuroantigen-primed T cells would be sufficient to activate CNS microglia for contact-mediated induction of iNOS.
Several studies have shown that IFN-β decreases the number of relapses and MRI-detected lesions in relapsing/remitting MS patients (35, 36, 49). However, the precise mechanism(s) by which IFN-β is effective in the treatment of MS have remained unclear. It has been suggested that IFN-β attenuates the disease process of MS probably through mediating systemic immunity (50), decreasing T cell reactivity to produce IFN-γ (51), stimulating the production of IL-10, an anti-inflammatory cytokine (17), inhibiting antigen presentation to T cells (52), or modifying humoral immune response (53). Our current results have underlined another important mechanism by which IFN-β may be efficient in MS. In this regard, IFN-β blocks the ability of neuroantigen-primed T cells to induce contact-mediated production of the oligodendrocyte-toxic molecule, NO, in microglia by inhibiting the expression of VLA-4 on the surface of neuroantigen-primed T cells. It is not known whether IFN-β can penetrate the blood-brain barrier. However, we have found that IFN-β alone can induce the production of NO and the activation of NFκB (data not shown) in microglial cells, suggesting that apart from inhibiting the functions of activated T cells, if IFN-β itself can enter into the CNS, it may augment the inflammatory response. Consistent with our observation, it has been found that IFN-β is beneficial in relapsing/remitting MS but not in acute MS attacks (54, 55) when the integrity of the blood-brain barrier is questionable.
Consistent with the inhibitory effect of IFN-β on the surface expression of VLA-4 in neuroantigen-primed T cells (Fig. 5), after analyzing the cell surface expression of VLA-4 by flow cytometry in 10 MS patients before and during IFN-β treatment, Calabresi et al. (56) have shown that the mean VLA-4 fluorescence of the lymphocytes of MS patients decreases on treatment as compared with untreated controls. However, this inhibitory effect of IFN-β on surface expression of VLA-4 was absent in in vitro studies with peripheral blood mononuclear cells (56). By Western blot analysis, Chabot et al. (34) have reported that the total cellular level of the 80-kDa CD49d is lower (about 40%) in T lymphocytes treated with IFN-β than in untreated cells. In contrast, under the same condition, the cell surface levels of α4 integrin as assessed by flow cytometry were not affected (34), leading to the speculation that IFN-β treatment might affect the affinity of integrin molecules without altering their expression on the cell surface. However, in our experiment, IFN-β treatment has been found to inhibit the mean VLA-4 fluorescence on the surface of neuroantigen (MBP)-primed T cells as compared with untreated cells (Fig. 5). The only difference between these two studies is that activated T cells were incubated with IFN-β for 3 days in the former study by Chabot et al. (34) but for 4 days in the present one (Fig. 5). Therefore, this discrepancy about the effect of IFN-β on surface expression of VLA-4 in T cells could be explained by the possible complexity involved in the turnover of cell surface integrins.
NO and peroxynitrite (a reaction product of NO and O2−) are potentially toxic molecules to neurons and oligodendrocytes that may mediate toxicity through the formation of iron-NO complexes of iron containing enzyme systems, oxidation of protein sulfhydryl groups, nitration of proteins, and nitrosylation of nucleic acids and DNA strand breaks (2). NO derived from microglia has also been implicated in the damage of myelin-producing oligodendrocytes in demyelinating disorders such as MS (7). Because neuroantigen-primed T cells infiltrate into the CNS of MS patients, the induction of iNOS expression by MBP-primed T cells in microglia suggests that neuroantigen-primed T cells may induce the neural injury in the inflamed CNS through the induction of NO production.
Acknowledgments
We thank Tom Dunn and associates of University of Nebraska Medical Center (UNMC) College of Dentistry for help in preparing this manuscript.
The abbreviations used are
- NO
nitric oxide
- NOS
NO synthase
- iNOS
inducible NOS
- ONOO−
peroxynitrite
- MBP
myelin basic protein
- CNS
central nervous system
- MS
multiple sclerosis
- VLA-4
very late antigen-4
- IFN
interferon
- EAE
experimental allergic encephalomyelitis
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- FACS
fluorescence-activated cell sorter
- BBB
blood brain barrier
Footnotes
This work was supported by Grant NS39940 from the National Institutes of Health.
S. Dasgupta and K. Pahan, unpublished observation.
References
- 1.Nathan C. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- 2.Jaffrey SR, Snyder SH. Annu Rev Cell Dev Biol. 1995;11:417–440. doi: 10.1146/annurev.cb.11.110195.002221. [DOI] [PubMed] [Google Scholar]
- 3.Galea E, Feinstein DL, Reis DJ. Proc Natl Acad Sci U S A. 1992;89:10945–10949. doi: 10.1073/pnas.89.22.10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koprowski H, Zheng YM, Heber-Katz E, Fraser N, Rorke L, Fu ZF, Hanlon C, Dietzshold B. Proc Natl Acad Sci U S A. 1993;90:3024–3027. doi: 10.1073/pnas.90.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitrovic B, Ignarro LJ, Montestruque S, Smoll A, Merril JE. Neuroscience. 1994;61:575–585. doi: 10.1016/0306-4522(94)90435-9. [DOI] [PubMed] [Google Scholar]
- 6.Murphy GM, Yang L, Cordell B. J Biol Chem. 1998;273:20967–20971. doi: 10.1074/jbc.273.33.20967. [DOI] [PubMed] [Google Scholar]
- 7.Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE. J Immunol. 1993;151:2132–2141. [PubMed] [Google Scholar]
- 8.Kolb H, Kolb-Bachofen V. Immunol Today. 1992;13:157–160. doi: 10.1016/0167-5699(92)90118-Q. [DOI] [PubMed] [Google Scholar]
- 9.McCatney-Francis N, Allen JB, Mizel DE, Albina JE, Xie Q-W, Nathan CF, Wahl SM. J Exp Med. 1993;178:749–754. doi: 10.1084/jem.178.2.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding M, Zhang M, Wong JL, Rogers NE, Ignarro LJ, Voskuhl RR. J Immunol. 1998;160:2560–2564. [PubMed] [Google Scholar]
- 11.Johnson AW, Land JM, Thompson EJ, Bolanos JP, Clark JB, Heales SJR. J Neurol Neurosurg Psychiatry. 1995;58:107–115. doi: 10.1136/jnnp.58.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenner T, Brocke S, Szafer F, Sobel RA, Parkinson JF, Perez DH, Steinman L. J Immunol. 1997;158:2940–2946. [PubMed] [Google Scholar]
- 13.Hooper DC, Scott GS, Zborek A, Mikheeva T, Kean RB, Koprowski H, Spitsin SV. FASEB J. 2000;14:691–698. doi: 10.1096/fasebj.14.5.691. [DOI] [PubMed] [Google Scholar]
- 14.Brosan CF, Battistini L, Raine CS, Dickson DW, Casadevall A, Lee SC. Dev Neurosci. 1994;16:152–161. doi: 10.1159/000112102. [DOI] [PubMed] [Google Scholar]
- 15.Bo L, Dawson TM, Wesselingh S, Mork S, Choi S, Kong PA, Hanley D, Trapp BD. Ann Neurol. 1994;36:778–786. doi: 10.1002/ana.410360515. [DOI] [PubMed] [Google Scholar]
- 16.Utz U, McFarland HF. J Neuropathol Exp Neurol. 1994;53:351–358. doi: 10.1097/00005072-199407000-00005. [DOI] [PubMed] [Google Scholar]
- 17.Chabot S, Yong VW. Neurology. 2000;55:1497–1505. doi: 10.1212/wnl.55.10.1497. [DOI] [PubMed] [Google Scholar]
- 18.Giulian D, Baker TJ. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feinstein DL, Galea E, Roberts S, Berquist H, Wang H, Reis DJ. J Neurochem. 1994;62:315–321. doi: 10.1046/j.1471-4159.1994.62010315.x. [DOI] [PubMed] [Google Scholar]
- 20.Pahan K, Namboodiri AMS, Sheikh FG, Smith BT, Singh I. J Biol Chem. 1997;272:7786–7791. doi: 10.1074/jbc.272.12.7786. [DOI] [PubMed] [Google Scholar]
- 21.Pahan K, Sheikh FG, Khan M, Namboodiri AMS, Singh I. J Biol Chem. 1998;273:2591–2600. doi: 10.1074/jbc.273.5.2591. [DOI] [PubMed] [Google Scholar]
- 22.Pahan K, Raymond JR, Singh I. J Biol Chem. 1999;274:7528–7536. doi: 10.1074/jbc.274.11.7528. [DOI] [PubMed] [Google Scholar]
- 23.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 24.Jana M, Liu X, Koka S, Ghosh S, Petro TM, Pahan K. J Biol Chem. 2001;276:44527–44533. doi: 10.1074/jbc.M106771200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pahan K, Sheikh FG, Liu X, Hilger S, McKinney M, Petro TM. J Biol Chem. 2001;276:7899–7905. doi: 10.1074/jbc.M008262200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pahan K, Liu X, McKinney MJ, Wood C, Sheikh FG, Raymond JR. J Neurochem. 2000;74:2288–2295. doi: 10.1046/j.1471-4159.2000.0742288.x. [DOI] [PubMed] [Google Scholar]
- 27.Pahan K, Liu X, Wood C, Raymond JR. FEBS Lett. 2000;472:203–207. doi: 10.1016/s0014-5793(00)01465-4. [DOI] [PubMed] [Google Scholar]
- 28.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. J Clin Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie Q, Kashiwabara Y, Nathan C. J Biol Chem. 1994;269:4705–4708. [PubMed] [Google Scholar]
- 30.Taylor BS, de Vera ME, Ganster RW, Wang Q, Shapiro RA, Morris SM, Billiar TR, Geller DA. J Biol Chem. 1998;273:15148–15156. doi: 10.1074/jbc.273.24.15148. [DOI] [PubMed] [Google Scholar]
- 31.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. J Biol Chem. 1998;273:12219–12226. doi: 10.1074/jbc.273.20.12219. [DOI] [PubMed] [Google Scholar]
- 32.Lin Y-Z, Yao SY, Veach RA, Torgerson TR, Hawiger J. J Biol Chem. 1995;270:14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- 33.Hynes RO. Cell. 1987;48:549–554. doi: 10.1016/0092-8674(87)90233-9. [DOI] [PubMed] [Google Scholar]
- 34.Chabot S, Williams G, Yong VW. J Clin Invest. 1997;100:604–612. doi: 10.1172/JCI119571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yong VW, Chabot S, Stuve O, Williams G. Neurology. 1998;51:682–689. doi: 10.1212/wnl.51.3.682. [DOI] [PubMed] [Google Scholar]
- 36.Paty DW, Li DKB the UBC MS/MRI Study Group, and the IFN Multiple Sclerosis Study Group. Neurology. 1993;43:662–667. doi: 10.1212/wnl.43.4.662. [DOI] [PubMed] [Google Scholar]
- 37.Gonzalez-Scarano F, Baltuch G. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 38.Bagasra O, Michaels FH, Zheng YM, Bobroski LE, Spitsin SV, Fu ZF, Tawadros R, Koprowski H. Proc Natl Acad Sci U S A. 1995;92:12041–12045. doi: 10.1073/pnas.92.26.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin R, McFarland HF, McFarlin DE. Annu Rev Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 40.Zhao ML, Liu JS, He D, Dickson DW, Lee SC. Brain Res. 1998;813:402–405. doi: 10.1016/s0006-8993(98)01023-3. [DOI] [PubMed] [Google Scholar]
- 41.Peterson PK, Hu S, Anderson WR, Chao CC. J Infect Dis. 1994;170:457–460. doi: 10.1093/infdis/170.2.457. [DOI] [PubMed] [Google Scholar]
- 42.Sedgwick JD, Ford AL, Foulcher E, Airriess R. J Immunol. 1998;160:5320–5330. [PubMed] [Google Scholar]
- 43.Traugott U, Reinherz EL, Raine CS. Science. 1983;218:308–310. doi: 10.1126/science.6217550. [DOI] [PubMed] [Google Scholar]
- 44.Cannella B, Raine CS. Ann Neurol. 1995;37:424–435. doi: 10.1002/ana.410370404. [DOI] [PubMed] [Google Scholar]
- 45.Yednock T, Cannon C, Fritz L, Sanchez-Madrid R, Steinman L, Karin N. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- 46.Tubridy N, Behan PO, Capildeo R, Chaudhuri A, Forbes R, Hawkins CP, Hughes RAC, Palace J, Sharrack B, Swingler R, Young C, Moseley IF, MacManus DG, Donoghue S, Miller DH. Neurology. 1999;53:466–472. doi: 10.1212/wnl.53.3.466. [DOI] [PubMed] [Google Scholar]
- 47.Thiel VE, Audus KL. Antioxid Redox Signal. 2001;3:273–278. doi: 10.1089/152308601300185223. [DOI] [PubMed] [Google Scholar]
- 48.Hurst RD, Azam S, Hurst A, Clark JB. Brain Res. 2001;894:181–188. doi: 10.1016/s0006-8993(01)01992-8. [DOI] [PubMed] [Google Scholar]
- 49.Rudick RA, Carpenter CS, Cookfair DL, Tuohy VK, Ransohoff RM. Neurology. 1996;43:2080–2087. doi: 10.1212/wnl.43.10.2080. [DOI] [PubMed] [Google Scholar]
- 50.IFN Multiple Sclerosis Study Group. Neurology. 1993;43:655–661. doi: 10.1212/wnl.43.4.655. [DOI] [PubMed] [Google Scholar]
- 51.Noronha A, Toscas A, Jensen MA. J Neuroimmunol. 1993;46:145–154. doi: 10.1016/0165-5728(93)90244-s. [DOI] [PubMed] [Google Scholar]
- 52.Noronha A, Toscas A, Jensen MA. Ann Neurol. 1995;37:7–15. [Google Scholar]
- 53.O’Gorman MRG, Oger J, Kastrukoff LF. Clin Exp Immunol. 1987;67:66–67. [PMC free article] [PubMed] [Google Scholar]
- 54.Whitaker JN. Ann Neurol. 1994;36:S103–S107. doi: 10.1002/ana.410360724. [DOI] [PubMed] [Google Scholar]
- 55.Connelly JF. Ann Pharmacother. 1994;28:610–616. doi: 10.1177/106002809402800511. [DOI] [PubMed] [Google Scholar]
- 56.Calabresi PA, Pelfrey CM, Tranquill LR, Maloni H, McFarland HF. Neurology. 1997;49:1111–1116. doi: 10.1212/wnl.49.4.1111. [DOI] [PubMed] [Google Scholar]