

Abstract
The presence of neuroantigen-primed T cells recognizing self-myelin antigens within the CNS is necessary for the development of demyelinating autoimmune disease like multiple sclerosis. This study was undertaken to investigate the role of myelin basic protein (MBP)-primed T cells in the expression of proinflammatory cytokines in microglial cells. MBP-primed T cells alone induced specifically the microglial expression of interleukin (IL)-1β, IL-1α tumor necrosis factor α, and IL-6, proinflammatory cytokines that are primarily involved in the pathogenesis of MS. This induction was primarily dependent on the contact between MBP-primed T cells and microglia. The activation of microglial NF-κB and CCAAT/enhancer-binding protein β (C/EBPβ) by MBP-primed T cell contact and inhibition of contact-mediated microglial expression of proinflammatory cytokines by dominant-negative mutants of p65 and C/EBPβ suggest that MBP-primed T cells induce microglial expression of cytokines through the activation of NF-κB and C/EBPβ. In addition, we show that MBP-primed T cells express very late antigen-4 (VLA-4), and functional blocking antibodies to α4 chain of VLA-4 (CD49d) inhibited the ability of MBP-primed T cells to induce microglial proinflammatory cytokines. Interestingly, the blocking of VLA-4 impaired the ability of MBP-primed T cells to induce microglial activation of only C/EBPβ but not that of NF-κB. This study illustrates a novel role of VLA-4 in regulating neuroantigen-primed T cell-induced activation of microglia through C/EBPβ.
Multiple sclerosis (MS)1 is the most common human demyelinating disease of the central nervous system (CNS). Although the etiology of MS is not completely understood, studies of MS patients suggest that the observed demyelination in the CNS is a result of a T cell-mediated autoimmune response (1). According to one hypothesis (2), T cells may be activated in spleen and lymph nodes, organs that have been found to contain myelin basic protein (MBP) mRNA and protein in mouse, rat, and human. In contrast, according to the other view (3), T cells may be activated in the CNS periphery by the first encounter with myelin-related autoantigens such as MBP, proteolipid protein, and myelin oligodendrocyte glycoprotein. However, after activation, these potentially damaging T cells may cross the blood-brain barrier and accumulate in the CNS where they recognize autoantigens, proliferate, and trigger a broad-spectrum inflammatory cascade, leading to myelin degradation and clinical symptoms (1, 4-6). Although the infiltration of neuroantigen-specific T cells into the CNS is a key event in the pathogenesis of MS or experimental allergic encephalomyelitis (EAE), the animal model of MS (4, 5), the mechanisms through which neuroantigen-specific T cells play an etiologic role in MS remain unclear.
The hallmark of brain inflammation in MS is the activation of glial cells, especially that of microglia that produce a variety of proinflammatory and neurotoxic factors including proinflammatory cytokines. Among proinflammatory cytokines, primary inflammatory cytokines such as interleukin (IL)-1β/α, tumor necrosis factor (TNF) α/β, and IL-6 play a predominant role because they are involved at multiple levels of neuroimmune regulation (6-8). An analysis of cerebrospinal fluid from MS patients has shown increased levels of proinflammatory cytokines compared with those of normal control, and the levels of those cytokines in the cerebrospinal fluid of MS patients also correlate with disease severity (6, 9, 10). Consistently, the blockade of proinflammatory cytokine synthesis or function by signaling inhibitors or neutralizing antibodies or gene knock-out can also prevent the development of EAE (11-13). However, the mechanisms by which these proinflammatory cytokines are produced in the CNS of MS patients are poorly understood.
Here we report that neuroantigen-specific T cells induce specifically microglial expression of proinflammatory cytokines (IL-1β, IL-1α, TNF-α, and IL-6) through cell-cell contact. Very-late antigen-4 (VLA-4) molecules on T cell surface play an important role in contact-mediated microglial expression of proinflammatory cytokines through the regulation of microglial activation of CCAAT/enhancer-binding protein β (C/EBPβ) but not that of NF-κB.
MATERIALS AND METHODS
Reagents
Fetal bovine serum (FBS), Hanks’ balanced salt solution (HBSS), DMEM/F-12, RPMI 1640, l-glutamine, and β-mercaptoethanol were from Mediatech. Mouse recombinant interferon-γ was obtained from R&D. Assay systems for IL-1β and TNF-α were purchased from BD Biosciences. Bovine myelin basic protein was purchased from Invitrogen. [α-32P]dCTP and [α-32P]ATP were obtained from PerkinElmer Life Sciences. The dominant-negative mutant of C/EBPβ (ΔC/EBPβ) was kindly provided by Dr. Steve Smale of the University of California (Los Angeles, CA).
Isolation and Purification of Antigen-primed T Cells
Female SJL/J mice were immunized subcutaneously with 400 μg of bovine MBP and 60 μg of Mycobacterium tuberculosis (H37RA, Invitrogen) in Incomplete Freund’s Adjuvant (Calbiochem). Lymph nodes were collected from these mice, and single cell suspension was prepared in RPMI 1640 medium containing 10% FBS, 2 mm l-glutamine, 50 μm β-mercaptoethanol, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells were cultured at a concentration of 4–5 × 106 cells/ml in six-well plates. Cells isolated from MBP-immunized mice were incubated with 50 μg/ml MBP for 4 days. The non-adherent cells were collected and passed through the nylon wool column preincubated for a period of 30 min with RPMI 1640 supplemented with 10% FBS at 37 °C, 5% CO2. The first 15–20-ml eluant was collected, centrifuged at 500 × g, and resuspended in RPMI 1640 medium-FBS. Viability and purity of the cells were checked by trypan blue exclusion and FACS analysis, respectively. Approximately 98% cells were found as CD3-positive T cells (14). These T cell populations were used to stimulate microglial cells.
Flow Cytometry
Surface expression of VLA-4 on MBP-primed T cells was checked by flow cytometry as described previously (14). 1 × 106 cells suspended in RPMI 1640 medium-FBS were incubated in the dark with appropriately diluted FITC-labeled antibodies to CD49d (integrin α4 chain) (BD Biosciences) at 4 °C for 30 min. Following incubation, cell suspension was centrifuged, washed three times, and resuspended in 500 μl of RPMI 1640 medium-FBS. The cells were then analyzed through FACS (BD Biosciences). A minimum of 10,000 cells was accepted for FACS analysis. Cells were gated based on morphological characteristics. Apoptotic and necrotic cells were not accepted for FACS analysis.
Isolation of Mouse Primary Microglia
Microglial cells were isolated from mixed glial cultures according to the procedure of Guilian and Baker (15). On days 7–9, the mixed glial cultures were washed three times with DMEM/F-12 and subjected to a shake at 240 rpm for 2 h at 37 °C on a rotary shaker. The floating cells were washed and seeded onto plastic tissue culture flasks and incubated at 37 °C for 2 h. The attached cells were removed by trypsinization and seeded onto new plates for further studies. 90–95% of this preparation was found to be positive for Mac-1 surface antigen. For the induction of TNF-α production, cells were stimulated with MBP-primed T cells in serum-free DMEM/F-12. Mouse BV-2 microglial cells (a kind gift from Virginia Bocchini of University of Perugia) were also maintained and induced with different stimuli as indicated above.
Stimulation of Mouse BV-2 Microglial Cells and Primary Microglia by MBP-primed T Cells
Microglial cells were stimulated with different concentrations of MBP-primed T cells under serum-free condition. After 1 h of incubation, culture dishes were shaken and washed three times with HBSS to lower the concentration of T cells. We performed FACS analysis to quantify the amount of T cells removed from microglia by this procedure. Areas under M1 and M2 in Fig. 1 represent autofluorescence and fluorescence attributed to CD3, respectively. Scales for M1 and M2 have been determined from nonconjugated cells. FACS analysis of adherent microglial cells using FITC-labeled anti-CD3 antibodies showed that >80% T cells were removed from microglial cells by this procedure (Fig. 1). Microglial cells then were incubated in serum-free medium for different period time.
FIG. 1. FACS analysis of BV-2 microglia and MBP-primed T cell coculture with FITC-labeled anti-CD3.

Purified MBP-primed T cells were added to BV-2 glial cells (0.5:1 of T cell:glia) under serum-free condition. A, after 1 h of incubation, cells (adherent and non-adherent) were taken out for FACS analysis. B, after 1 h of incubation, culture dishes were shaken and washed three times with HBSS to remove non-adherent cells. Adherent cells were taken out for FACS analysis. Cells were treated with 0.5 ml of appropriately diluted FITC-labeled anti-CD3 for 30 min followed by FACS analysis in a FACScan as described under “Materials and Methods.”
Assay for IL-1β and TNF-α Synthesis
Concentrations of IL-1β and TNF-α were measured in culture supernatants by a high sensitivity enzyme-linked immunosorbent assay (BD Biosciences) according to manufacturer’s instruction as described previously (16-18). Protein was measured by the procedure of Bradford (19).
RNA Isolation and Analysis of Different Proinflammatory and Anti-inflammatory Cytokines by Gene Array
BV-2 microglial cells were stimulated with MBP-primed T cells (0.5:1 of T cell:microglia), and after 1 h of stimulation, culture dishes were shaken and washed three times with HBSS to lower T cell concentration. BV-2 cells then received serum-free medium, and after 3 h of incubation, the expression of different proinflammatory and anti-inflammatory cytokines was analyzed in adherent BV-2 cells by a gene array kit (GEArray™, Super-Array Inc.) following manufacturer’s protocol. Total RNA was isolated from stimulated or unstimulated BV-2 microglial cells by using Ultra-spec-II RNA reagent (Biotecx Laboratories Inc.). This RNA was used as a template for [32P]cDNA probe synthesis using [α-32P]dCTP and Moloney murine leukemia virus reverse transcriptase. The GEArray membrane was prehybridized, hybridized with [32P]cDNA probe, and washed two or three times in solution I (2× SSC, 0.05% SDS) for 1 h at room temperature followed by solution II (0.1× SSC, 0.1% SDS) at 50 °C for another hour. The membranes were then dried and exposed to x-ray films (Eastman Kodak Co.).
Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assay (EMSA)
After different minutes of stimulation of BV-2 microglial cells by MBP-primed T cells, culture dishes were shaken and washed three times with HBSS to lower T cell concentration. Nuclear extracts then were prepared from adherent BV-2 cells using the method of Dignam et al. (20) with slight modifications. Cells were harvested, washed twice with ice-cold phosphate-buffered saline, and lysed in 400 μl of buffer A (10 mm HEPES, pH 7.9, 10 mm KCl, 2 mm MgCl2, 0.5 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml pepstatin A, and 5 μg/ml leupeptin) containing 0.1% Nonidet P-40 for 15 min on ice, vortexed vigorously for 15 s, and centrifuged at 14,000 rpm for 30 s. The pelleted nuclei were resuspended in 40 μl of buffer B (20 mm HEPES, pH 7.9, 25% (v/v) glycerol, 0.42 m NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 0.5 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml pepstatin A, and 5 μg/ml leupeptin). After 30 min on ice, lysates were centrifuged at 14,000 rpm for 10 min. Supernatants containing the nuclear proteins were diluted with 20 μl of modified buffer C (20 mm HEPES, pH 7.9, 20% (v/v) glycerol, 0.05 m KCl, 0.2 mm EDTA, 0.5 mm dithiothreitol, and 0.5 mm phenylmethylsulfonyl fluoride) and stored at 70 °C until use. Nuclear extracts were used for EMSA using 32P-end-labeled double-stranded (NF-κB, 5′ -AGT TGA GGG GAC TTT CCC AGG C-3′ (Promega), and C/EBPβ, 5′-TGC AGA TTG CGC AAT CTG CA-3′ (Santa Cruz Biotechnology, Inc.)) oligonucleotides. Double-stranded mutated (NF-κB, 5′-AGT TGA GGC GAC TTT CCC AGG C-3′, and C/EBPβ, 5′-TGC AGA GAC TAG TCT CTG CA-3′ (Santa Cruz Biotechnology, Inc.)) oligonucleotides were used to verify the specificity of NF-κB and C/EBPβ binding to DNA.
Assay of Transcriptional Activities of NF-κB and C/EBPβ
To assay the transcriptional activity of NF-κB and C/EBPβ, cells at 50–60% confluence in 12-well plates were transfected with 0.5 μg of either PBIIX-Luc, an NF-κB-dependent reporter construct (21), or pC/EBPβ-Luc, an C/EBPβ-dependent reporter construct (21), using the LipofectAMINE Plus method (Invitrogen) (22-23). All of the transfections included 50 ng/μg total DNA of pRL-TK (a plasmid encoding Renilla luciferase used as transfection efficiency control, Promega). After 24 h of transfection, cells were stimulated with MBP-primed T cells (0.5:1 of T cell:microglia). After 1 h of stimulation, culture dishes were shaken and washed three times with HBSS to lower T cell concentration. BV-2 cells then were incubated with serum-free medium for 5 h. Adherent BV-2 cells were analyzed for firefly and Renilla luciferase activities according to standard instructions provided in the Dual luciferase kit (Promega) in a TD-20/20 luminometer (Turner Designs). Relative luciferase activity of cell extracts was typically represented as (firefly luciferase value/Renilla luciferase value) × 10−3.
RESULTS
MBP-primed T Cells Induced the Production of IL-1β and TNF-α in BV-2 Microglial Cells
T cells isolated from MBP-immunized mice proliferated in response to MBP, and the maximum proliferation was observed at 50 or 100 μg/ml MBP (14). Therefore, these cells were stimulated with 50 μg/ml MBP. After nylon-wool column purification, MBP-primed T cells were ~98% pure as obtained by the FACS analysis (14). We next examined whether these purified MBP-primed T cells can induce the production of proinflammatory cytokines like IL-1β and TNF-α in mouse BV-2 microglial cells. MBP-primed T cells were washed and added to BV-2 microglial cells in direct contact. After 1 h of contact, culture dishes were shaken and washed three times to remove MBP-primed T cells. Normal T cells (isolated from normal mice) were unable to induce the production of IL-1β and TNF-α in BV-2 microglial cells (Fig. 2A). There was also very little or no induction of IL-1β and TNF-α production when MBP-primed T cells were added to microglial cells at a ratio of 0.1:1 (Fig. 2B). However, the marked induction of IL-1β and TNF-α production was observed at the ratio of 0.2:1 of T cell:microglia (Fig. 2B). The induction of TNF-α production did not further significantly increase at 0.5:1 or 0.7:1 of T cell:microglia (Fig. 2B). However, the induction of IL-1β production increased further at the ratio of 0.5:1 of T cell:glia (Fig. 2B). On the other hand, a decrease in IL-1β and TNF-α production was observed at a higher concentration of T cells (1:1). This decrease in cytokine production was the result of the increase in microglial cell death in the presence of a higher concentration of MBP-primed T cells (data not shown). The time-dependent experiment using MBP-primed T cells in the ratio of 0.5:1 of T cell:microglia shows that the microglial induction of IL-1β and TNF-α production peaked at 24 h of incubation and decreased at a longer hour of incubation (Fig. 2C).
FIG. 2. MBP-primed T cells induce the production of IL-1β and TNF-α in mouse BV-2 microglial cells and primary microglia.

BV-2 cells received different concentrations of normal (A) and MBP-primed T cells (B) in direct contact under serum-free condition. After 1 h of incubation, culture dishes were shaken and washed three times with HBSS to remove the burden of T cells. Adherent BV-2 cells then were incubated in serum-free medium for 23 h, and supernatants were used to assay IL-1β and TNF-α as mentioned under “Materials and Methods.” Data are mean ± S.D. of three different experiments. C, BV-2 cells were stimulated with MBP-primed T cells (0.5:1 of T cell:glia) under serum-free condition. After 1 h of incubation, culture dishes were shaken and washed three times with HBSS to remove the burden of T cells. At different hours of incubation in serum-free medium, supernatants were used to assay IL-1β and TNF-α. Data are mean ± S.D. of three different experiments. D, BV-2 cells received different concentrations of MBP-primed T cells within insert under serum-free condition. After 24 h, supernatants were used to assay IL-1β and TNF-α. Data are mean ± S.D. of three different experiments. E, BV-2 cells received different concentrations of conditioned supernatant of MBP-primed T cells under serum-free condition. After 24 h, supernatants were used to assay IL-1β and TNF-α. Data are mean ± S.D. of three different experiments. F, mouse primary microglia received different concentrations of MBP-primed T cells in direct contact under serum-free condition. After 1 h of incubation, culture dishes were shaken and washed three times with HBSS to remove the burden of T cells. Adherent cells were incubated with serum-free medium for another 23 h, and supernatants were used to assay IL-1β and TNF-α. Data are mean ± S.D. of three different experiments.
We next examined whether the contact between MBP-primed T cells and microglial cells or the soluble factors produced from activated T cells is necessary to induce the production of IL-1β and TNF-α. First, MBP-primed T cells were placed in a culture insert where they were in close proximity to but not contacting microglia. In contrast to marked induction of IL-1β and TNF-α production by MBP-primed T cell:microglia contact (Fig. 2B), very little cytokine production was observed when MBP-primed T cells were placed within culture inserts (Fig. 2D). Second, a different amount of the conditioned supernatant of MBP-primed T cells was added to microglial cells. 50 μl of supernatant is equivalent to T cells sufficient for the ratio of 0.5:1 of T cell:microglia. The induction of both IL-1β and TNF-α production by 50 μl of that of supernatant (Fig. 2E) was very low (<5%) as compared with that by 0.5:1 of T cell:microglia (Fig. 2B). However, higher volumes of supernatants of MBP-primed T cells induced higher amounts of IL-1β and TNF-α in BV-2 microglial cells (Fig. 2E). These observations strongly suggest that the induction of IL-1β and TNF-α production by MBP-primed T cells primarily depends on the contact between T cells and microglia.
To understand whether MBP-primed T cells were able to induce the production of IL-1β and TNF-α in primary cells, we examined the effect of MBP-primed T cells on the induction of IL-1β and TNF-α production in mouse primary microglia (Fig. 2F). Similar to BV-2 cells, MBP-primed T cells were unable to induce the production of IL-1β and TNF-α in primary microglia when added at a ratio of 0.1:1 of T cells and microglia (Fig. 2F). However, the induction of IL-1β and TNF-α production started from the ratio of 0.2:1 of T cell:microglia, reached to the maximum at the ratio of 0.5:1, and decreased at higher concentrations of T cells (Fig. 2F).
Effect of MBP-primed T Cells on the Expression of Different Cytokines in BV-2 Microglial Cells
To understand further the mechanism of cytokine production, we examined the effect of MBP-primed T cells on the expression of different cytokines by cytokine gene array analysis using the GEArray kit. After 1 h of stimulation of BV-2 microglial cells by MBP-primed T cells, culture dishes were shaken and washed three times with HBSS to remove any extra or loosely bound T cells. Following 3 h of incubation in serum-free medium, total RNA was isolated from adherent BV-2 microglial cells to perform gene array analysis. This gene array analysis allowed us to study the expression of 23 different proinflammatory and anti-inflammatory cytokines such as granulocyte colony-stimulating factor, interferon-γ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-11, IL-12p35, IL-12p40, IL-13, IL-15, IL-16, IL-17, IL-18, lymphotoxin-β, TNF-α, and TNF-β. Normal T cells were unable to induce any of these cytokines in BV-2 microglial cells (Fig. 3A). On the other hand, MBP-primed T cells in the ratio of 0.5:1 of T cell:microglia strongly induced contact-mediated expression of IL-1β, IL-1α, TNF-α, and IL-6 (Fig. 3, B and C), cytokines that are involved in the pathogenesis of MS (6, 9, 13). This induction of IL-1β, IL-1α, TNF-α, and IL-6 by MBP-primed T cell contact is specific because under the same condition, MBP-primed T cells did not induce other proinflammatory cytokines in microglial cells.
FIG. 3. MBP-primed T cells induce the expression of different proinflammatory cytokines in mouse BV-2 microglial cells.
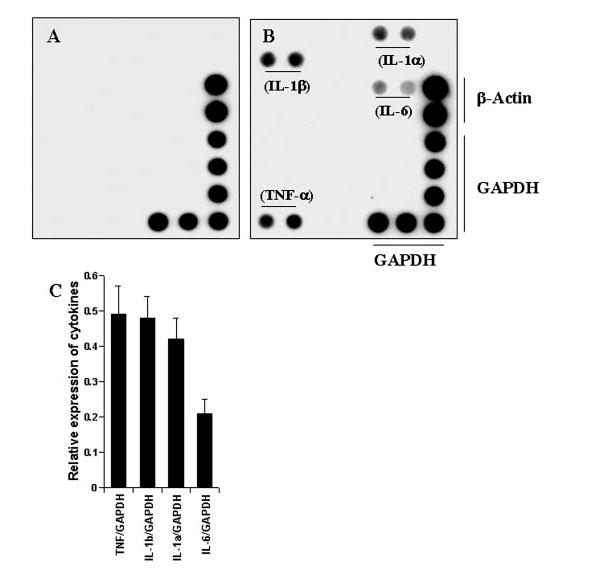
BV-2 cells were stimulated with either normal (A) or MBP-primed T cells (B) (0.5:1 of T cell:glia) under serum-free condition. After 1 h of stimulation, culture dishes were shaken and washed three times with HBSS to lower T cell concentration. BV-2 cells then received serum-free medium, and after 3 h of incubation, total RNA was isolated from adherent BV-2 cells for the analysis of different proinflammatory and anti-inflammatory cytokines by a gene array kit (GEArray) following manufacturer’s protocol as described under “Materials and Methods.” C, the relative expression of different cytokines (cytokines/glyceraldehyde-3-phosphate dehydrogenase (GAPDH)) was measured after scanning the bands with a Fluor Chem 8800 Imaging System (Alpha Innotech Corporation). Data are expressed as the mean ± S.D. of three different experiments.
Role of VLA-4 in MBP-primed T Cell-induced Expression of Proinflammatory Cytokines in BV-2 Microglial Cells
Integrins being present on cell surface are mainly involved in integrating cell-cell interaction (24). It has been shown that T cells activated by anti-CD3 express more VLA-4 integrin and that VLA-4 plays an important role in T cell contact-mediated activation of microglial cells (25). Therefore, we investigated whether neuroantigen-primed T cells require VLA-4 to induce contact-mediated expression of proinflammatory cytokines in microglial cells. At first, we analyzed the expression of VLA-4 on the surface of MBP-primed T cells by FACS analysis using FITC-labeled antibodies against the α4 chain of VLA-4 antigen. Fig. 4A represents autofluorescence as this was observed in unconjugated normal T cells. As areas under M1 and M2 in Fig. 4 represent autofluorescence and fluorescence because of VLA-4, respectively, there was very little expression of VLA-4 on the surface of normal T cells (Fig. 4B) in contrast to marked expression of VLA-4 on the surface of MBP-primed T cells (Fig. 4C). An analysis of three separate experiments shows that MBP priming induced the expression of VLA-4 by 20.4 ± 3.6-fold. These results suggest that neuroantigen priming induces the expression of VLA-4 on the surface of T cells.
FIG. 4. Expression of VLA-4 on the surface of MBP-primed T cells.
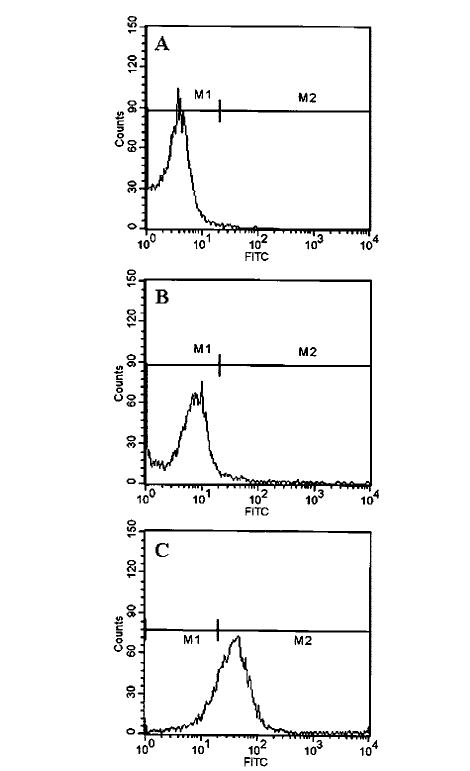
Normal or MBP-primed T cells were treated with 0.5 ml of appropriately diluted FITC-labeled anti-VLA α4 for 30 min followed by FACS analysis. A, FITC-nonconjugated normal T cells. B, FITC-conjugated normal T cells. C, FITC-conjugated MBP-primed T cells.
To examine whether this VLA-4 is involved in contact-mediated expression of proinflammatory cytokines, we used functional blocking antibodies against CD49d (the α4 chain of VLA-4). It is apparent from Fig. 5A that incubation of MBP-primed T cells with different concentration of antibodies against CD49d inhibited its ability to induce the production of IL-1β and TNF-α in BV-2 microglial cells. Although antibodies against CD49d at a dose of 10 or 20 μg/ml were not effective in blocking the production of IL-1β and TNF-α (data not shown), marked inhibition was observed when antibodies were used at a concentration of 50 or 100 μg/ml. In contrast, control IgG did not block the ability of MBP-primed T cells to induce microglial production of IL-1β and TNF-α (Fig. 5A). Consistently, gene array analysis also showed that antibodies against CD49d but not control IgG at a concentration of 50 μg/ml markedly inhibited the ability of MBP-primed T cells to induce the expression of IL-1β, IL-1α, TNF-α, and IL-6 in microglial cells (Fig. 5B). These studies suggest that VLA-4 being present on the surface of MBP-primed T cells is involved in contact-mediated induction of proinflammatory cytokines in microglial cells.
FIG. 5. Functional blocking antibodies against the α4 chain of VLA-4 inhibit the ability of MBP-primed T cells to induce the expression of proinflammatory cytokines in BV-2 microglial cells.

MBP-primed T cells were mixed with either different concentrations of antibodies against the α4 chain of VLA-4 or control IgG and rocked gently for 1 h at room temperature. Cells were centrifuged, washed twice, and added to BV-2 microglial cells at a ratio of 0.5:1 T cell:microglia. After 1 h of stimulation, culture dishes were shaken and washed to lower T cell concentration. A, adherent cells were incubated with serum-free medium for another 23 h. Supernatants were used to assay IL-1β and TNF-α. Data are mean ± S.D. of three different experiments. B, adherent cells were incubated with serum-free medium for another 3 h. The expression of different cytokines was analyzed by a gene array kit (GEArray). The relative expression of different cytokines (cytokines/glyceraldehyde-3-phosphate dehydrogenase (GAPDH)) was measured after scanning the bands with a Fluor Chem 8800 Imaging System. Data are mean ± S.D. of three different experiments.
MBP-primed T Cells Induce the Activation of NF-κB and C/EBPβ in BV-2 Microglial Cells
It has been shown that activation of both NF-κB and C/EBPβ is involved in the expression of different proinflammatory cytokines in monocytes and macrophages following inflammatory insult (18, 26-28). These findings prompted us to ask whether activation of NF-κB and C/EBPβ may be responsible for the proinflammatory cytokine release following MBP-primed T cell contact in BV-2 microglial cells. Activation of NF-κB and C/EBPβ was monitored by both DNA-binding and transcriptional activities. DNA-binding activities of NF-κB and C/EBPβ were evaluated by the formation of a distinct and specific complex in a gel shift DNA-binding assay. Treatment of BV-2 glial cells with MBP-primed T cells at a ratio of 0.5:1 of T cell:glia resulted in the induction of DNA binding activity of NF-κB at different minutes of stimulation (Fig. 6A). This gel shift assay detected a specific band in response to MBP-primed T cells that was not found in the case of mutated double-stranded oligonucleotide, suggesting that MBP-primed T cell microglial contact induces the DNA binding activity of NF-κB. We then tested the effect of MBP-primed T cell on NF-κB -dependent transcription of luciferase in BV-2 glial cells using the expression of luciferase from a reporter construct, PBIIX-Luc, as an assay. Consistently, MBP-primed T cells also induced NF-κB -dependent transcription of luciferase in BV-2 microglial cells (Fig. 6B). MBP-primed T cells did not induce the activation of NF-κB in BV-2 microglial cells when added at a ratio of 0.1:1. However, the activation of NF-κB started at the ratio of 0.2:1 of T cell:microglia, peaked at the ratio of 0.5:1, and decreased at a higher concentration of T cells (Fig. 6B).
FIG. 6. MBP-primed T cells induce the activation of NF-κB in BV-2 microglial cells.

A, cells incubated in serum-free DMEM/F-12 were stimulated with MBP-primed T cells (0.5:1 of T cell:glia). After different minutes of stimulation, culture dishes were shaken and washed three times with HBSS. Adherent cells were taken out to prepare nuclear extracts, and nuclear proteins were used for EMSA as described under “Materials and Methods.” The upper, middle, and lower arrows indicate the induced NF-κB band, nonspecific band, and the unbound probe, respectively. B, cells plated at 50–60% confluence in 12-well plates were cotransfected with 0.5 μg of PBIIX-Luc and 25 ng of pRL-TK using LipofectAMINE Plus as described under “Materials and Methods.” After 24 h of transfection, BV-2 cells were stimulated with different concentrations of MBP-primed T cells. After 1 h of stimulation, T cells were removed followed by the incubation of adherent BV-2 cells in serum-free medium for another 5 h. Firefly and Renilla luciferase activities were obtained by analyzing total cell extract of BV-2 cells as described under “Materials and Methods.” Data are mean ± S.D. of three different experiments.
Similarly, MBP-primed T cells also induced the DNA binding activity of C/EBPβ in different minutes of stimulation (Fig. 7A). The specific band in response to MBP-primed T cells (upper arrow) was observed only in the case of the double-stranded wild type oligonucleotides but not in the case of the mutated ones (Fig. 7A). It is evident from C/EBPβ-dependent luciferase assay in Fig. 7B that MBP-primed T cells markedly induced the transcriptional activity of C/EBPβ. Similar to NF-κB, there was no activation of C/EBPβ in BV-2 microglial cells when MBP-primed T cells were added at a ratio of 0.1:1. However, the activation of C/EBPβ started at the ratio of 0.2:1 of T cell:microglia and peaked at the ratio of 0.7:1 (Fig. 7B).
FIG. 7. MBP-primed T cells induce the activation of C/EBPβ in BV-2 microglial cells.

A, cells incubated in serum-free DMEM/F-12 were stimulated with MBP-primed T cells (0.5:1 of T cell:glia). After different minutes of stimulation, culture dishes were shaken and washed three times with HBSS and EMSA was carried out as described earlier. The upper and lower arrows indicate the induced C/EBPβ band and the unbound probe, respectively. B, BV-2 cells plated at 50–60% confluence in 12-well plates were cotransfected with 0.5 μg of pC/EBPβ -Luc (an C/EBPβ -dependent reporter construct) and 25 ng of pRL-TK. After 24 h of transfection, BV-2 cells were stimulated with different concentrations of MBP-primed T cells. After 1 h of stimulation, T cells were removed followed by the incubation of adherent BV-2 cells in serum-free medium for another 5 h. Firefly and Renilla luciferase activities were obtained by analyzing total cell extract of BV-2 cells. Data are mean ± S.D. of three different experiments.
Role of NF-κB and C/EBPβ in MBP-primed T Cell-induced Expression of Proinflammatory Cytokines in BV-2 Microglial Cells
We next examined whether the activation of both NF-κB and C/EBPβ is important for the expression of proinflammatory cytokines in cells stimulated with MBP-primed T cells. Overexpression of dominant-negative molecules provides an effective tool with which to investigate the in vivo functions of different transcription factors or signaling molecules. Therefore, microglial NF-κB was inhibited by a dominant-negative mutant of p65 (Δp65) (29). Similarly, we used the dominant-negative mutant of C/EBPβ (ΔC/EBPβ) (30) to inhibit microglial activation of C/EBPβ. It is apparent from Fig. 8A that the expression of Δp65 and ΔC/EBPβ but not that of the empty vector inhibited MBP-primed T cell-induced production of IL-1β and TNF-α in BV-2 microglial cells. Consistently, gene array analysis also showed that both Δp65 and ΔC/EBPβ but not the empty vector were able to inhibit the expression of IL-1β, IL-1α, TNF-α, and IL-6 (Fig. 8B) in BV-2 microglial cells stimulated with MBP-primed T cells. These studies suggest that MBP-primed T cells induce the expression of proinflammatory cytokines in microglial cells through the activation of NF-κB and C/EBPβ.
FIG. 8. Dominant-negative mutants of p65 (Δp65) and C/EBPβ (ΔC/EBPβ) inhibit MBP-primed T cell-induced expression of proinflammatory cytokines in BV-2 microglial cells.
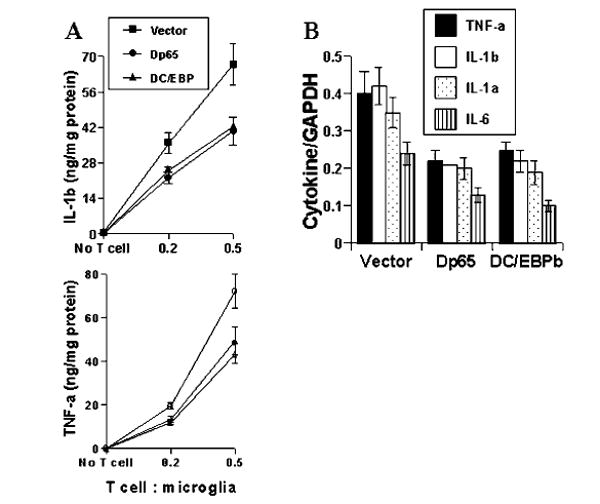
Cells were transfected with 0.5 μg of either Δp65 or ΔC/EBPβ using LipofectAMINE-plus. After 24 h of transfection, cells were stimulated with MBP-primed T cells (0.5:1 of T cell:glia) under serum-free condition. A, after 1 h of stimulation, T cells were removed followed by the incubation of adherent BV-2 cells in serum-free medium for another 23 h. Supernatants were used to assay IL-1β and TNF-α. Data are mean ± S.D. of three different experiments. B, after 1 h of stimulation, T cells were removed followed by the incubation of adherent BV-2 cells in serum-free media for another 3 h. The expression of different cytokines was analyzed by a gene array kit (GEArray). The relative expression of different cytokines (cytokines/glyceraldehyde-3-phosphate dehydrogenase (GAPDH)) was measured after scanning the bands with a Fluor Chem 8800 Imaging System. Data are mean ± S.D. of three different experiments.
Role of VLA-4 in MBP-primed T Cell-induced Activation of NF-κB and C/EBPβ in BV-2 Microglial Cells
Because VLA-4 is also involved in the induction of proinflammatory cytokines in microglial cells, we decided to investigate whether VLA-4 is coupled to the induction of cytokines through the activation of NF-κB and/or C/EBPβ. Function of VLA-4 on MBP-primed T cells was inhibited by functional blocking antibodies against CD49d. These antibodies at a concentration of 50 or 100 μg/ml markedly inhibited MBP-primed T cell-induced microglial expression of proinflammatory cytokines (Fig. 5). However, EMSA studies have shown that these antibodies, at same concentrations, did not inhibit MBP-primed T cell-induced DNA binding activity of NF-κB in BV-2 microglial cells (Fig. 9A). In contrast, MBP-primed T cell-induced DNA binding activity of C/EBPβ in microglial cells was markedly inhibited by anti-CD49d (Fig. 9A). To confirm this observation further, we assayed transcriptional activities of NF-κB and C/EBPβ. It is apparent from Fig. 9B that the blocking of VLA-4 by anti-CD49d inhibited the ability of MBP-primed T cells to induce transcriptional activation of C/EBPβ but not that of NF-κB (Fig. 9B) in BV-2 microglial cells. These studies suggest that VLA-4 is coupled to microglial induction of proinflammatory cytokines through microglial activation of C/EBPβ but not that of NF-κB.
FIG. 9. Functional blocking antibodies against the α4 chain of VLA-4 inhibit the ability of MBP-primed T cells to induce the activation of C/EBPβ but not that of NF-κB in BV-2 microglial cells.

A, MBP-primed T cells were mixed with either different concentrations of antibodies against the α4 chain of VLA-4 or control IgG and rocked gently for 1 h at room temperature. Cells were centrifuged, washed three times, and added to BV-2 microglial cells at a ratio of 0.5:1 T cell:microglia. After 1 h of incubation, culture dishes were shaken and washed three times with HBSS and EMSA was carried out to study the DNA binding activity of NF-κB (upper panel) and C/EBPβ (lower panel) in adherent microglia. B, BV-2 microglial cells plated at 50–60% confluence in 12-well plates were cotransfected with 0.5 μg of either PBIIX-Luc (upper panel) or p C/EBPβ -Luc (lower panel) and 25 ng of pRL-TK. After 24 h of transfection, BV-2 cells were stimulated with either anti-α4-preincubated or control IgG-preincubated MBP-primed T cells (0.5:1 of T cell:glia). After 1 h of stimulation, T cells were removed by shaking and washing followed by the incubation of adherent BV-2 cells in serum-free media for another 5 h. Firefly and Renilla luciferase activities were obtained by analyzing total cell extract of BV-2 cells. Data are mean ± S.D. of three different experiments.
DISCUSSION
Microglia are considered as CNS-resident professional macrophages. The activation of microglia has been implicated in the pathogenesis of a variety of neurodegenerative diseases including Alzheimer’s disease, Creutzfeld-Jacob disease, human immunodeficiency virus dementia, and multiple sclerosis (MS) (31). Upon activation, microglia produce and secrete potentially neurotoxic proinflammatory cytokines including IL-1β, IL-1α, TNF-α, and IL-6 (31) that play a pivotal role in demyelination of MS patients (10-13, 31). However, the mechanism by which proinflammatory cytokines are produced in MS brain is unclear. Although lipopolysaccharide is a potent inducer of microglial activation, it has not been demonstrated to have a physiological relevance in MS (32). The infiltration of neuroantigen-specific T cells into the CNS is considered a key event in the pathogenesis of MS or EAE. However, the biological role of such T cells within the CNS is poorly understood. Several lines of evidence presented in this paper clearly support the conclusion that MBP-primed T cells potently induce the expression of IL-1β, IL-1α, TNF-α, and IL-6 in microglial cells through cell-cell contact. This conclusion is based on the following observations. First, MBP-primed T cells induced the production of IL-1β and TNF-α in mouse BV-2 microglial cells and primary microglia. This effect was dose-dependent, and it peaked at 0.7:1 or 0.5:1 of T cell:microglia. Gene array analysis showed that apart from the induction of IL-1β and TNF-α, MBP-primed T cells were also capable of inducing the expression of IL-1α and IL-6. Second, the placement of MBP-primed T cells in a culture insert where they were in close proximity to, but not contacting microglia, was unable to induce the production of proinflammatory cytokines in microglial cells. Third, soluble factors of MBP-primed T cells equivalent to T cells of 0.5:1 or 0.7:1 of T cell:microglia were very poor inducers of proinflammatory cytokines. However, greater amount of soluble factors induced the production of proinflammatory cytokines to some extent.
Sedgwick et al. (33) have shown that microglia and T cells do interact in vivo. In the graft-versus-host disease model, activated microglia were found to cluster around T cell infiltrates and to be associated with single or clustered microglia (33). Furthermore, microglia isolated from animals of graft-versus-host disease proliferated and exhibited functions of activated microglia such as phagocytosis and motility (33). In light of these in vivo observations and the proximity of activated T cells and microglia in the perivascular space or CNS parenchyma of MS lesions (34), the induction of proinflammatory cytokines like IL-1α, IL-1β, TNF-α, and IL-6 by the contact between MBP-primed T cells and microglia described in this paper is relevant to MS pathogenesis. Earlier, several reports have shown that non-antigen specific T cells activated by phytohemagglutinin phorbol 12-myristate 13-acetate, IL-2, or anti-CD3 also can induce TNF-α in microglial cells (5, 25, 35). However, the in vitro model of our study shows marked micro-glial induction of IL-1β and TNF-α production by very few neuroantigen-primed T cells used in the ratio of 0.2:1 of T cells to microglia. At present, it is unclear whether this is a relevant ratio. In light of the fact that microglia constitute only 2–5% total CNS cells in normal brain (31), this study suggests that infiltration of a very few neuroantigen-primed T cells would be sufficient to activate CNS microglia for contact-mediated induction of proinflammatory cytokines. In fact, Cross et al. (36) has shown that the antigen-specific T cells in adoptive transfer model of EAE constitute a minority population (only <2% total cell infiltrate into the CNS parenchyma). Although the homing of a huge number of non-antigen-specific T cells and other blood cells into the CNS after the breakdown of the blood-brain barrier multiplies the inflammatory response and aggravates the disease condition in MS and EAE, the minority population (neuroantigen-primed T cells) is instrumental to initiate the disease process and to induce fresh relapse in MS and EAE (1-5). Therefore, our results delineate a potential mechanism by which a few number of neuroantigen-primed T cells may initiate the inflammatory response in the naïve CNS by activating microglia through cell-cell contact to express specifically IL-1β, IL-1α, TNF-α, and IL-6, proinflammatory cytokines each of which plays an important role in the pathophysiology of MS (6, 31).
Integrins and adhesion molecules are involved in cell-cell interaction. It has been shown that an increased number of lymphocytes express VLA-4 in active MS lesions (33) and that blocking VLA-4 function with a monoclonal antibody prevents experimental allergic encephalomyelitis, an animal model of MS, in mice (34). Consistently, short term treatment of MS patients with anti-α4 integrin antibody significantly reduces blood-brain barrier dysfunction as assessed by contrast-enhanced MRI (39). Earlier, Yong and colleagues (25) had reported that this VLA-4-vascular cell adhesion molecule-1 interaction plays a crucial role when anti-CD3-stimulated T cells activate microglia through cell-cell contact, prompting us to investigate the role of VLA-4 in neuroantigen-primed T cell-induced microglial expression of proinflammatory cytokines. The observations presented in this paper that MBP-primed T cells expressed VLA-4 on their surface and that the blocking of VLA-4 by neutralizing antibodies against the α4 chain of VLA-4 inhibited the ability of MBP-primed T cells to induce microglial expression of IL-1β, IL-1α, TNF-α, and IL-6 clearly suggest that VLA-4 integrin on the surface of neuroantigen-primed T cells plays an important role in contact-mediated induction of these proinflammatory cytokines in microglia.
The signaling events in the induction of proinflammatory cytokines are poorly understood. Promoter regions of proinflammatory cytokines (IL-1β, IL-1α, TNF-α, and IL-6) contain consensus sequences for the binding of different transcription factors, especially NF-κB and C/EBPβ (27, 28, 40). The inhibition of NF-κB and C/EBPβ by either inhibitors or dominant-negative mutants also leads to the inhibition of cytokine expression (18, 27, 28, 38), suggesting an essential role of NF-κB and C/EBPβ in the induction of these proinflammatory cytokines. Our results have clearly shown that the activation of NF-κB and C/EBPβ is important for MBP-primed T cell contact-induced expression of proinflammatory cytokines in microglial cells. First, MBP-primed T cells alone induced contact-mediated activation of NF-κB and C/EBPβ in microglial cells. Second, Δp65, a dominant-negative mutant of p65, but not the empty vector inhibited the expression of IL-1β, IL-1α, TNF-α, and IL-6 in BV-2 glial cells stimulated with MBP-primed T cells. Consistently, overexpression of ΔC/EBPβ, a truncated alternate C/EBPβ translation product, LIP, which acts as a dominant-negative inhibitor of C/EBPβ activity (30), inhibited MBP-primed T cell-induced expression of IL-1β, IL-1α, TNF-α, and IL-6 in microglial cells.
VLA-4 being present on the surface of MBP-primed T cells plays an important role in contact-mediated induction of proinflammatory cytokines in microglia. However, the mechanism by which VLA-4 is coupled to microglial induction of proinflammatory cytokines is not known. Therefore, we investigated its role in contact-mediated microglial activation of NF-κB and C/EBPβ. Interestingly, the blocking of VLA-4 function by neutralizing antibodies inhibited the ability of MBP-primed T cells to induce the activation of C/EBPβ but not that of NF-κB. These studies suggest that VLA-4 is coupled to microglial expression of proinflammatory cytokines through the activation of C/EBPβ but not that of NF-κB and probably some other ligand(s) on MBP-primed T cells is/are involved in microglial activation of NF-κB.
Although monocytes/macrophages are the primary source of proinflammatory cytokines in inflammation, CNS microglia manifest a similar response upon activation (17, 18, 31). Proinflammatory cytokines derived from microglia is toxic to myelin-producing oligodendrocytes and can produce demyelination as observed in demyelinating disorders like MS (37). Therefore, understanding the mechanism by which proinflammatory cytokines are generated within the CNS can thus impact upon the rational treatment of inflammatory demyelinating diseases including MS. Because neuroantigen-primed T cells infiltrate into the CNS of MS patients, the induction of proinflammatory cytokine expression by MBP-primed T cells in microglia suggests that neuroantigen-primed T cells may induce the neural injury in the inflamed CNS through the induction of proinflammatory cytokine production.
Acknowledgments
We thank Tom Dunn and associates of University Nebraska Medical College of Dentistry for their help in preparing this paper.
The abbreviations used are
- MS
multiple sclerosis
- CNS
central nervous system
- MBP
myelin basic protein
- EAE
experimental allergic encephalomyelitis
- IL
interleukin
- TNF
tumor necrosis factor
- VLA-4
very-late antigen-4
- FBS
fetal bovine serum
- HBSS
Hanks’ balanced salt solution
- DMEM
Dulbecco’s modified Eagle’s medium
- FACS
fluorescence-activated cell sorter
- FITC
fluorescein isothiocyanate
- EMSA
electrophoretic mobility shift assay
- C/EBPβ
CCAAT/enhancer-binding protein β
- Luc
luciferase
Footnotes
This study was supported by a grant from National Institutes of Health Grant NS39940.
References
- 1.Martin R, McFarland HF, McFarlin DE. Annu Rev Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 2.Liu H, MacKenzie-Graham AJ, Palaszynski K, Liva S, Voskuhl RR. J Neuroimmunol. 2001;116:83–93. doi: 10.1016/s0165-5728(01)00284-3. [DOI] [PubMed] [Google Scholar]
- 3.Iglesias A, Bauer J, Litzenburger T, Schubart A, Linington C. Glia. 2001;36:220–234. doi: 10.1002/glia.1111. [DOI] [PubMed] [Google Scholar]
- 4.Utz U, McFarland HF. J Neuropathol Exp Neurol. 1994;53:351–358. doi: 10.1097/00005072-199407000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Chabot S, Yong VW. Neurology. 2000;55:1497–1505. doi: 10.1212/wnl.55.10.1497. [DOI] [PubMed] [Google Scholar]
- 6.Benveniste EN. J Mol Med. 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- 7.Tyor WR, Glass JD, Baumrind N, McArthur JC, Griffin JW, Becker PS, Griffin DE. Neurology. 1993;43:1002–1009. doi: 10.1212/wnl.43.5.1002. [DOI] [PubMed] [Google Scholar]
- 8.Brosnan CF, Raine CS. Brain Pathol. 1996;6:243–257. doi: 10.1111/j.1750-3639.1996.tb00853.x. [DOI] [PubMed] [Google Scholar]
- 9.Sharief MK, Hentges R. N Engl J Med. 1991;325:467–472. doi: 10.1056/NEJM199108153250704. [DOI] [PubMed] [Google Scholar]
- 10.Maimone D, Gregory S, Arnason BG, Reder AT. J Neuroimmunol. 1991;32:67–74. doi: 10.1016/0165-5728(91)90073-g. [DOI] [PubMed] [Google Scholar]
- 11.Ruddle NH, Bergman CM, McGrath KM, Lingenheld EG, Grunnet ML, Padula SJ, Clark RB. J Exp Med. 1990;172:1193–1200. doi: 10.1084/jem.172.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samoilova EB, Horton JL, Hilliard B, Liu TS, Chen Y. J Immunol. 1998;161:6480–6486. [PubMed] [Google Scholar]
- 13.Esiri MM, Gay D. In: Multiple Sclerosis. Clinical and Pathogenetic Basis. Raine CS, McFarland HF, Tourtellotte WW, editors. Chapman & Hall; London: 1997. pp. 173–186. [Google Scholar]
- 14.Dasgupta S, Jana M, Liu X, Pahan K. J Biol Chem. 2002;277:39327–39333. doi: 10.1074/jbc.M111841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giulian D, Baker TJ. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. Free Rad Biol Med. 1998;24:39–48. doi: 10.1016/s0891-5849(97)00137-8. [DOI] [PubMed] [Google Scholar]
- 17.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. J Clin Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jana M, Dasgupta S, Liu X, Pahan K. J Neurochem. 2002;80:197–203. doi: 10.1046/j.0022-3042.2001.00691.x. [DOI] [PubMed] [Google Scholar]
- 19.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 20.Dignam JD, Lebovitz RM, Roeder RG. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jana M, Liu X, Koka S, Ghosh S, Petro TM, Pahan K. J Biol Chem. 2001;276:44527–44533. doi: 10.1074/jbc.M106771200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K. J Biol Chem. 2002;277:39312–39319. doi: 10.1074/jbc.M205107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pahan K, Jana M, Liu X, Taylor BS, Wood C, Fischer SM. J Biol Chem. 2002;277:45984–45991. doi: 10.1074/jbc.M200250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hynes RO. Cell. 1987;48:549–554. doi: 10.1016/0092-8674(87)90233-9. [DOI] [PubMed] [Google Scholar]
- 25.Chabot S, Williams G, Yong VW. J Clin Invest. 1997;100:604–612. doi: 10.1172/JCI119571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao J, Mackman N, Edgington TS, Fan ST. J Biol Chem. 1997;272:17795–17801. doi: 10.1074/jbc.272.28.17795. [DOI] [PubMed] [Google Scholar]
- 27.Hu HM, Tian Q, Baer M, Spooner CJ, Williams SC, Johnson PF, Schwartz RC. J Biol Chem. 2000;275:16373–16381. doi: 10.1074/jbc.M910269199. [DOI] [PubMed] [Google Scholar]
- 28.Ghosh S, May MJ, Kopp EB. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 29.Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 30.Descombes P, Schibler U. Cell. 1991;67:569–579. doi: 10.1016/0092-8674(91)90531-3. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez-Scarano F, Baltuch G. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 32.Meda L, Cassatella M, Szendrei G, Otvos L, Baron P, Villalba M, Ferrari D, Rossi F. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 33.Sedgwick JD, Ford AL, Foulcher E, Airriess R. J Immunol. 1998;160:5320–5330. [PubMed] [Google Scholar]
- 34.Traugott U, Reinherz EL, Raine CS. Science. 1983;218:308–310. doi: 10.1126/science.6217550. [DOI] [PubMed] [Google Scholar]
- 35.Tan J, Town T, Paris D, Placzek A, Parker T, Crawford F, Yu H, Humphrey J, Mullan M. J Neuroimmunol. 1999;97:77–85. doi: 10.1016/s0165-5728(99)00053-3. [DOI] [PubMed] [Google Scholar]
- 36.Cross AH, O’Mara T, Raine CS. Neurology. 1993;43:1028–1033. doi: 10.1212/wnl.43.5.1028. [DOI] [PubMed] [Google Scholar]
- 37.Heiss E, Herhaus C, Klimo K, Bartsch H, Gerhauser C. J Biol Chem. 2001;276:32008–32015. doi: 10.1074/jbc.M104794200. [DOI] [PubMed] [Google Scholar]
- 38.D’Souza SD, Bonetti B, Balasingam V, Cashman NR, Barker PA, Troutt AB, Raine CS, Antel JP. J Exp Med. 1996;184:2361–2370. doi: 10.1084/jem.184.6.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cannella B, Raine CS. Ann Neurol. 1995;37:424–435. doi: 10.1002/ana.410370404. [DOI] [PubMed] [Google Scholar]
- 40.Udalova IA, Knight JC, Vidal V, Nedospasov SA, Kwiatkowski D. J Biol Chem. 1998;273:21178–21186. doi: 10.1074/jbc.273.33.21178. [DOI] [PubMed] [Google Scholar]