

Abstract
A growing number of organisms have been discovered inhabiting extreme environments, including temperatures in excess of 100 °C. How cellular proteins from such organisms retain their native folds under extreme conditions is still not fully understood. Recent computational and structural studies have identified disulfide bonding as an important mechanism for stabilizing intracellular proteins in certain thermophilic microbes. Here, we present the first proteomic analysis of intracellular disulfide bonding in the hyperthermophilic archaeon Pyrobaculum aerophilum. Our study reveals that the utilization of disulfide bonds extends beyond individual proteins to include many protein-protein complexes. We report the 1.6Å crystal structure of one such complex, a citrate synthase homodimer. The structure contains two intramolecular disulfide bonds, one per subunit, which result in the cyclization of each protein chain in such a way that the two chains are topologically interlinked, rendering them inseparable. This unusual feature emphasizes the variety and sophistication of the molecular mechanisms that can be achieved by evolution.
Keywords: disulfide bond, protein stability, catenane, citrate synthase, thermophile
INTRODUCTION
Proteins from thermophilic organisms are stabilized by a variety of molecular mechanisms1; 2; 3. Among these mechanisms, disulfide bonding has attracted recent attention as a key stabilizing factor in some organisms4; 5; 6. The potentially important role for disulfide bonding in thermophiles was somewhat unexpected in view of classical ideas in biochemistry. In most well-studied organisms, the chemical environment within the cytosol is highly reducing. As a result, the cytosolic proteins of typical organisms do not contain stabilizing disulfide bonds7; 8. However, recent computational, structural and biochemical studies have made it clear that the protein sequences and cellular environments of certain microbes have evolved to utilize widespread disulfide bonding as a defence against thermal denaturation4; 5; 6. Still, the cellular and structural aspects of how thermophiles use disulfide bonding to stabilize their proteins have not yet been fully explored.
Here we extend earlier studies, which focused mainly on the stabilization of individual proteins, to include protein complexes composed of multiple protein chains. Among proteins of known three-dimensional structure, a number of cases had already been noted in which thermophilic protein complexes were held together by a disulfide bond between distinct protein chains (Table 1). However, no genome-wide studies had been conducted, and the frequency of these stabilizing interactions had not been examined. We therefore sought to address whether intermolecular disulfide bonding might be found in abundance in a thermophilic microbe such as Pyrobaculum aerophilum, an organism in which earlier studies had already suggested the prevalence of disulfide bonding within individual protein chains.
Table 1.
Reported protein structures containing intermolecular disulfide bonds.
| Protein | Source | PDB ID | Disulfide | Reference |
|---|---|---|---|---|
| 5′-deoxy-5′-methylthioadenosine phosphorylase | Sulfolobus solfataricus | 1JDS | 125–125′ | Appleby, et al. 200141 |
| Glycosyltrehalose trehalohydrolase | Sulfolobus solfataricus | 1EH9 | 298–298′ | Feese, et al. 200042 |
| Protein-L-Isoaspartate O-Methyltransferase | Sulfolobus tokodaii | 1VBF | 149–149′ | Tanaka, et al. 200443 |
| Sm-Like archaeal protein 1 (Smap1) | Pyrobaculum aerophilum | 1LNX | 8–8′ | Mura, et al. 200344 |
| Aspartate Racemase | Pyrococcus horikoshii | 1JFL | 73–73′ | Liu, et al. 200245 |
| Pyrrolidone Carboxyl Peptidase | Thermococcus litoralis | 1A2Z | 190–190′ | Singleton, et al. 199946 |
| Conserved Hypothetical Protein (Tm0160) | Thermotoga maritima | 1VJL | 38–38′ | Spraggon, et al. 200447 |
| Indole-3-Glycerol Phosphate Synthase | Thermotoga maritima | 1I4N | 102–102′ | Knochel, et al. 200248 |
| Triosephosphate Isomerase | Thermotoga maritima | 1B9B | 541–541′ | Maes, et al. 199949 |
| Elongation Factor Ts | Thermus thermophilus | 1AIP | 190–190′ | Jiang, et al. 199650 |
RESULTS
Quantitation of disulfide bonds by fluorescence
An experimental method was developed to visualize the abundance of intracellular protein disulfide bonds in P. aerophilum cell lysates. First, cysteine residues existing in their free thiol form were blocked immediately upon cell lysis by irreversible alkylation with iodoacetamide. Then, any existing protein disulfide bonds were cleaved by chemical reduction. Finally, the resulting free thiol groups were labeled with the thiol-reactive fluorescent reagent CPM. The result of this procedure was a selective labeling of cysteine residues involved in disulfide bonds.
The reliable measurement of disulfide-bonded cysteines in this assay is dependent on the efficiency and specificity of the reagents utilized, and thus careful consideration was given to their selection. Iodoacetamide is well established in redox studies for alkylating free thiols; this blocks cysteine residues in their reduced form and prevents any subsequent oxidation9; 10; 11. Previous work has demonstrated the high efficiency of this reaction following denaturation of the substrate proteins12; 13. The fluorescent reagent CPM has also been used extensively in the quantitative analysis of free thiols, and has been shown to exhibit a high degree of specificity to cysteine residues5; 14; 15; 16. In the current studies, disulfide labeling experiments were analyzed after running SDS gels on the treated cell lysates. As described below, an analysis of the resulting gels supported the veracity of the procedure employed.
The assay revealed an abundance of protein disulfide bonds in P. aerophilum cell lysates (Fig. 1). In order to quantify the abundance of disulfide bonding relative to the total cysteine content in the cellular proteins, the same assay was performed, but with the initial blocking step omitted; this leads to labeling of all cysteine residues. Fluorescence measurements of CPM-labelled proteins in the gels revealed that approximately 47% of the cysteine residues in the P. aerophilum lysate are involved in disulfide bonds, as opposed to approximately 8% in E. coli, which was used as a control. Estimates for the uncertainties in these measured values are 5% and 3%, respectively, based on variations between triplicate experiments; these values account for random but not systematic errors, and so represent lower bounds on the true errors. For E. coli, the measured value for disulfide abundance likely includes contributions from extra-cytosolic (e.g. periplasmic) proteins, which are known to contain disulfide bonds; P. aerophilum cells do not have a periplasmic space.
Figure 1.
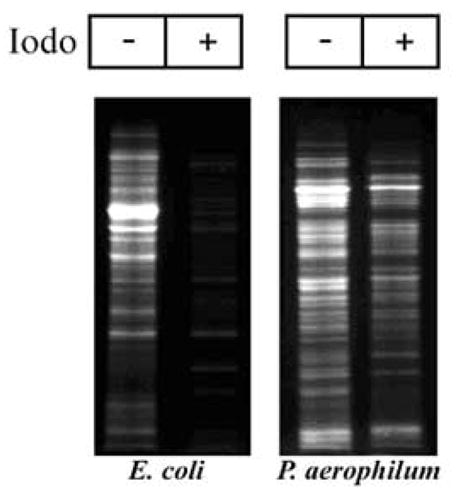
Abundance of disulfide-bonded proteins in P. aerophilum, as detected by fluorescent labeling. Whole cell lysate was reacted with iodoacetamide (+) to block free (thiol) cysteines. Following blocking, any disulfide bonds present were cleaved by reduction with TCEP and fluorescently labeled with CPM. When iodoacetamide is omitted (−) all cysteines are labeled. A comparison of corresponding lanes shows that a large fraction of P. aerophilum proteins contain disulfide bonds. E. coli cells serve as a control.
Two kinds of control experiments – the comparison of P. aerophilum to E. coli and the omission of the blocking reagent – were used to address concerns about the efficiency and selectivity of the disulfide labeling protocol. Some of the labeling observed in the gels could reflect non-specific binding of the fluorescent reagent, but the relatively low fluorescence measured for E. coli (8%) suggests that non-specific binding could account for only a few percent of the estimated (47%) disulfide abundance in P. aerophilum. Another concern is that some of the labeling could be to cysteine residues that were not involved in disulfide bonds, but that existed in the free thiol form and were simply not fully blocked in the initial step. It is difficult to eliminate this potential scenario entirely, but two points are noteworthy. First, the cysteine residues in the E. coli proteins were blocked almost completely, as judged by the low overall fluorescent labeling. Second, in the case of P. aerophilum the two lanes in the gel (with and without blocking) show very different patterns. In particular, in the lane in which no blocking was performed, numerous bands are visible that are effectively absent from the lane in which free cysteines were initially blocked by iodoacetamide. The effective disappearance of numerous labeled bands gives a measure of the effectiveness of the blocking step.
These new experimental results on whole cell lysates support earlier claims that disulfide bonding is widespread in P. aerophilum proteins5; 6; 17. The results also suggested the value of further experiments aimed at analyzing individual P. aerophilum proteins and the nature of their disulfide bonds.
Identification of disulfide-bonded protein complexes by 2D diagonal gel electrophoresis
An experimental method employing 2-dimensional diagonal gel electrophoresis (2D-DGE) was developed for identifying protein complexes in P. aerophilum that might be held together by disulfide bonding between protein chains (Fig. 2A). Similar methods have been described for identifying ribosomal proteins with non-native intermolecular disulfide bonds18, identifying protein targets of DsbA11, investigating oxidative folding in the endoplasmic reticulum19; 20, and most recently for identifying cytosolic proteins that form stable disulfide bonds under oxidative stress21; 22. In 2D-DGE, proteins are initially separated (in the horizontal dimension) by SDS-PAGE under non-reducing conditions that preserve any existing disulfide bonds. Prior to separation in the second (vertical) dimension, proteins within the gel are chemically reduced, cleaving any disulfide bonds that are present. Proteins that do not contain disulfide bonds migrate at the same rate in both dimensions, creating a prominent diagonal line in the 2D gel. Spots below the diagonal arise from proteins that were held together in disulfide-bonded complexes within the cell. Such proteins migrate more rapidly in the second dimension because the complexes in which they were held during the first electrophoresis have been separated into their smaller components. In addition, some individual proteins bearing intramolecular disulfide bonds appear above the diagonal. Such proteins can have mobilities that are measurably higher in the first dimension owing to a lower average extension of the protein chain when constrained by an intramolecular disulfide bond. As shown in Figure 2A, 2D-DGE analysis of a P. aerophilum lysate revealed numerous off-diagonal spots corresponding to disulfide bonded proteins. In particular, a large number of protein-protein complexes apparently held together by intermolecular disulfide bonds were observed below the diagonal. As expected, control experiments with E. coli revealed very few proteins off the diagonal in the 2D gel (Fig 2B).
Figure 2.
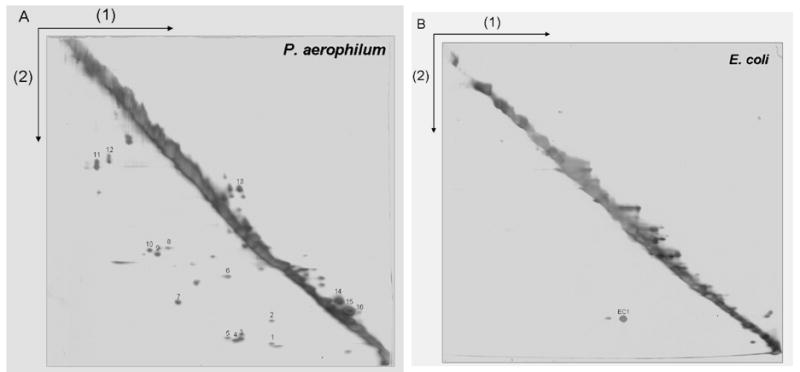
A 2-D diagonal gel electrophoresis method for identifying intermolecular disulfide bonded protein complexes. The first separation (1) is performed under non-reducing conditions so that disulfide bonds remain intact. Disulfide bonds are cleaved by reduction with DTT prior to the second electrophoretic separation (2). Proteins involved in intermolecular disulfide bonds appear as spots below the prominent diagonal, while spots above the diagonal mark certain intramolecularly disulfide-bonded proteins whose mobilities are retarded by reduction. Numerous disulfide bonded protein-protein complexes are visible in a cell lysate from (A) P. aerophilum, but not in (B) E. coli used as a control. P. aerophilum protein spots identified by mass spectrometry are numbered as in Table 2.
To identify which proteins participate in disulfide bonding, off-diagonal protein spots were excised, subjected to in-gel trypsin digestion, and analyzed by liquid chromatography-tandem mass spectrometry. Identifications based on genomic sequence data were successful for 16 spots (Table 2), with the predicted molecular weights of identified proteins consistent with the observed molecular weight of the associated spot. All identified proteins contained at least one cysteine residue, while proteins predicted to form intramolecular disulfide bonds contained at least two cysteines. Ten of the proteins were annotated in the NCBI database as hypothetical or conserved hypothetical, making it difficult to assign cellular functions to them. However, an analysis of homologous sequences23 from other organisms showed that the majority of the cysteines in the identified proteins are not conserved. This suggests that the cysteines residues are not involved in enzymatic functions, but rather serve structural roles as anticipated. The major spot observed in the E. coli gel (EC1) was identified as an alkyl hydroperoxide reductase (AhpC), a homodimeric enzyme known to form an intermolecular disulfide bond during reduction of organic hydroperoxide24.
Table 2.
Disulfide bonded P. aerophilum proteins and complexes identified from 2D gels.
| Gene | Protein | Spot No. | Cysteines | Likely Disulfides# |
|---|---|---|---|---|
| PAE2842 | Hypothetical protein | 1 | 3 (57, 131, 144) | Potential heterocomplex |
| PAE3103 | NusG, transcription antitermination | 2 | 1 (6) | Probable heterocomplex |
| PAE3173 | Conserved hypothetical | 3 | 1 (130) | |
| PAE3144 | Conserved hypothetical | 4 | 1 (116) | |
| PAE2072 | Conserved protein | 5 | 1 (120) | Potential heterocomplex |
| PAE2576 | Hypothetical protein | 6 | 5 (29, 33, 67, 143, 177) | Potential heterocomplex |
| PAE2254 | Hypothetical protein | 7 | 2 (145, 154) | |
| PAE3406 | Conserved hypothetical | 8 | 3 (40, 133, 147) | |
| PAE0797 | Short chain dehydrogenase | 9 | 1 (259) | Homodimer |
| PAE2075 | Nitrilase, conjectural | 10 | 2 (141, 257) | Homodimer (257–257′) |
| PAE1689 | Citrate synthase | 11 | 2 (19, 394) | Homodimer (19, 394) |
| PAE2310 | Proline dehydrogenase | 12 | 1 (408) | |
| PAE0210 | Conserved hypothetical | 13 | 6 (75, 100, 107, 201, 265, 292) | Intramolecular |
| PAE2489 | Acetyl-CoA acyltransferase-associated | 14 | 6 (58, 61, 72, 75, 137, 173) | Intramolecular |
| PAE2701 | Conserved hypothetical | 15 | 2 (89, 114) | Intramolecular |
| PAE3161 | Hypothetical protein | 16 | 4 (23, 25, 106, 138) | Intramolecular |
See Figure 1A for corresponding spot numbers. Cysteine residue numbers are given in parenthesis.
Complexes were assigned as likely homomeric vs. heteromeric based on spot positions in the gel and the presence or absence of likely partners related vertically. Where possible, assignment of likely disulfide connectivity was based on analysis of conservation in multiple sequence alignments, with conserved cysteines presumed to be functionally active rather than disulfide bonded.
A number of the weaker spots in the P. aerophilum 2D gels could not be identified by mass spectrometry. Furthermore, there are likely additional disulfide bonded protein complexes in P. aerophilum that are present at concentrations too low to be visualized in the 2D gels. The proteins identified (Table 2) therefore represent only a subset of those that form disulfide bonded complexes in P. aerophilum. Conversely, based on the amino acid sequences and molecular mass data from the gels, none of the proteins identified as being in disulfide bonded complexes appear to be false positives. The method employed here may be useful in the future for identifying disulfide bonded complexes in other hyperthermophilic organisms. However, the feasibility of broad-based studies is limited at the present time by the general difficulty associated with culturing such organisms.
One of the protein spots observed in the P. aerophilum 2D-DGE analysis was identified as citrate synthase. Citrate synthase has been developed as a model protein for structural studies of thermophilic adaptation25, with crystal structures described for psychrophilic, mesophilic, thermophilic, and hyperthermophilic homologues26; 27; 28; 29; 30. Among the numerous homologues studied, none contain disulfide bonds, making the P. aerophilum citrate synthase (PaCS) the first shown to utilize this mechanism. In an effort to further characterize the enzyme and analyze the stabilizing effects of its disulfide bonds, PaCS was cloned, expressed, and purified for biochemical and structural studies. The recombinant protein was found to form a homodimeric complex, as anticipated from the 2D gel analysis and in agreement with the known structures of homologous enzymes from other organisms. In addition, SDS-PAGE analysis in the presence and absence of reducing reagent confirmed that the dimeric state was dependent on the presence of disulfide bonds (Fig. 5C), consistent with the 2D-DGE observations.
Figure 5.
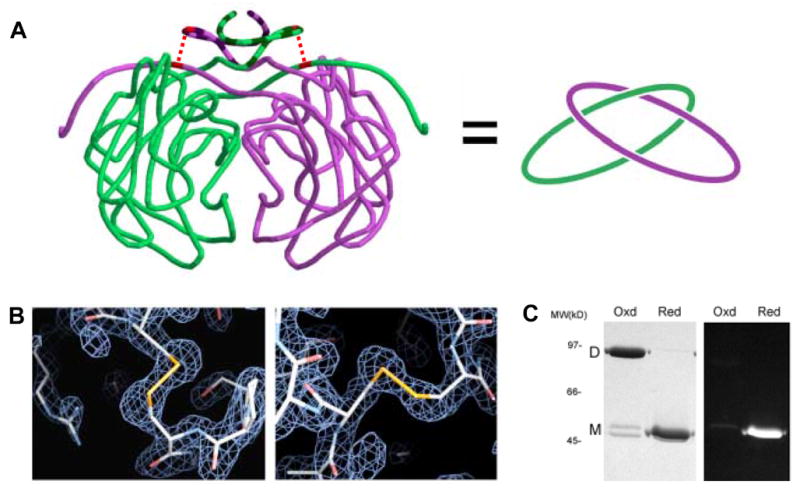
Intramolecular disulfide bonds leading to topological linkage or catenation of protein chains in P.aerophilum citrate synthase (PaCS). (A) Cartoon representation of interlinked PaCS chains. On the left, the protein backbone of the PaCS dimer is shown in a smoothed form to help clarify the chain topology. The region of the N-terminus (residues 1–24) that differs structurally in comparison to mesophilic homologs is indicated in darker striping. The topological connectivity is illustrated on the right. (B) Close up view of the disulfide bonds between Cys19 and Cys394 for both chains. Electron density maps calculated from diffraction data (based on phases from an omit-model) are shown in blue wireframe. (C) SDS-PAGE gel of purified recombinant PaCS. Under non-reducing (‘Oxd’) conditions, PaCS migrates at a molecular weight consistent with the dimeric form. Following treatment with the chemical reductant TCEP, the reduced (‘Red’) PaCS migrates as a monomer. A minor doublet in the oxidized lane likely corresponds to a mixture of linear and cyclized forms of the monomer. Fluorescent labelling with CPM indicates that no free thiols are present in the oxidized, dimeric form of PaCS.
Crystal structure of citrate synthase
The protein was crystallized and its structure was determined to a resolution of 1.6Å. The PaCS structure is dimeric, with each protein subunit consisting of 18 α-helices and 8 β-strands (Fig 3A). The structure overlaps well with previously determined citrate synthase structures, consistent with the strong conservation of the protein from bacteria to mammals. Each subunit is composed of one large domain (helices C-M and S), one small domain (helices N-R), and additional structural features at the termini (Fig. 3B–C). The active site of citrate synthase, identified through conserved active site residues, resides between the large and small domains of each subunit. The C-terminal region of each PaCS subunit wraps across the opposite subunit in a manner reminiscent of domain swapping31, with residues from the C-terminal arm contributing to the active site of the opposing subunit. Thus, stability of the dimer is necessary for enzyme activity.
Figure 3.
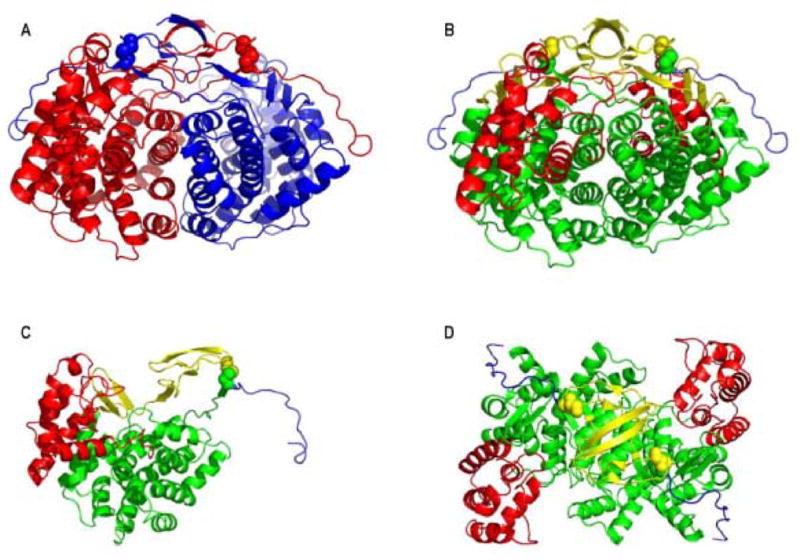
Crystal structure of P. aerophilum citrate synthase (PaCS). (A) The PaCS homodimer illustrated with the individual subunits colored red and blue. The arrangement of domains is illustrated for the (B) PaCS dimer, (C) individual subunit, and (D) PaCS dimer rotated 90° from B. The N-terminal β-sheet domain is colored yellow, the large domain (helices C-M and S) is colored green, the small domain (helices N-R) is colored red, and the C-terminal domain is colored blue. Disulfide-bonded cysteins are illustrated as spheres.
The N-terminal extension
Despite the general similarity of PaCS to homologues from other species, structural and sequence comparisons reveal unique features in PaCS (Fig. 4). In particular, PaCS contains an N-terminal extension not present in any of the archaeal or bacterial structures currently known. The pig and chicken citrate synthases also contain an N-terminal extension, but the sequence and structural features do not resemble those of PaCS. The PaCS N-terminal extension encodes three β-strands (strands 1–3). An intermolecular antiparallel β-sheet is formed between strand 1 and strand 1’ from the other subunit, while strands 2 and 3 interact intramolecularly to form another small antiparallel β-sheet within each subunit. These interactions suggest that the N-terminal extension serves to stabilize the dimer against thermal denaturation. In addition, the loop between strands 2 and 3 contains one of the cysteine residues (Cys19) involved in the key disulfide bond.
Figure 4.
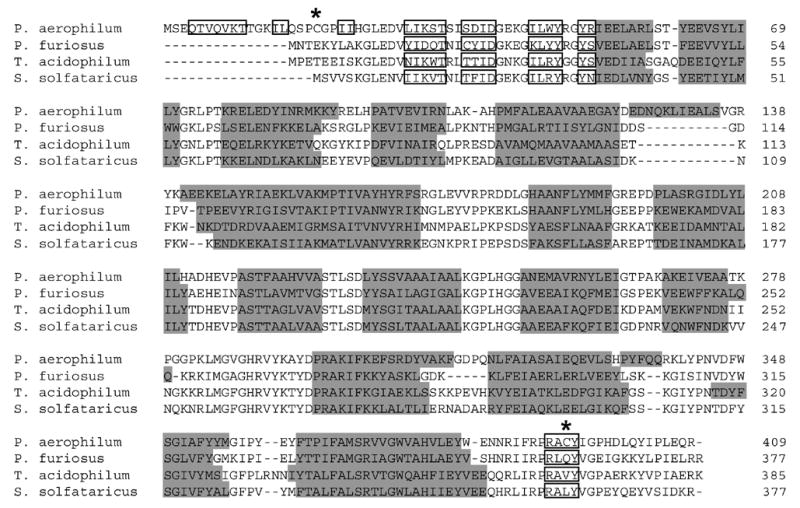
A multiple sequence alignment of citrate synthase homologues from four thermophilic organisms, illustrating the unusual N-terminus in P. aerophilum. Overall conservation in the remainder of the protein is highlighted with α-helices shaded in grey and β-strand segments boxed. Disulfide-bonded cysteines in P. aerophilum are indicated by asterisks (*).
Disulfide bond formation leads to catenation
Based on the biochemical results, it was anticipated that the disulfides present in PaCS should form intermolecular disulfide bonds, as the oxidized form of the protein ran as a dimer during eletrophoresis under denaturing conditions. An analysis of the structure confirmed the presence of disulfide bonds, but revealed that the disulfides are in fact intramolecular, between Cys19 and Cys394 from the same subunit (Fig. 5A and B). The formation of this internal bond within each subunit confers a topological connection between the subunits. Though not directly bonded, the two subunits are connected like links in a chain, making the dimer an example of what has been called a protein catenane. This interlinking of cyclized protein chains renders them inseparable even under denaturing conditions (Fig. 5C).
Interestingly, the N-terminal extension and disulfide-forming cysteines are conserved in the closely related Thermoproteus tenax and Pyrobaculum islandicum citrate synthases. These homologues have not been characterized biochemically or structurally, but the sequence conservation suggests that the topological linkage observed in P. aerophilum may be a feature common to this branch of archaeal homologues. Additional sequence and structure data should help clarify the evolutionary history of the unusual features of this enzyme.
Disulfide bonded catenation contributes to thermal stability
The stabilizing effect of the disulfide bonds was analyzed by comparing the native protein (PaCSnat) to a mutant version (PaCSmut) in which the two cysteines were replaced by serine residues. Due to the high stability of both proteins, addition of the denaturant guanidinium-HCl (GdnHCl) was required to partially destabilize the proteins in order to observe their unfolding transitions by circular dichroism. Although both constructs exhibited similar CD profiles at room temperature, PaCSnat exhibited a melting temperature (Tm) of 84°C, while PaCSmut exhibited a Tm of 73.5°C, a decrease of 10.5°C from the PaCSnat (Fig. 6). This shift in melting temperature suggests that the linkage created by the Cys19-Cys394 disulfide bond plays an important role in stabilizing the protein against thermal denaturation. Interestingly, enzymatic activity assays showed no difference between the reaction rates of PaCSnat and PaCSmut at temperatures up to 90°C (data not shown), indicating that other stabilizing factors, in addition to the disulfide bonds, contribute greatly to the high stability of the dimeric complex.
Figure 6.
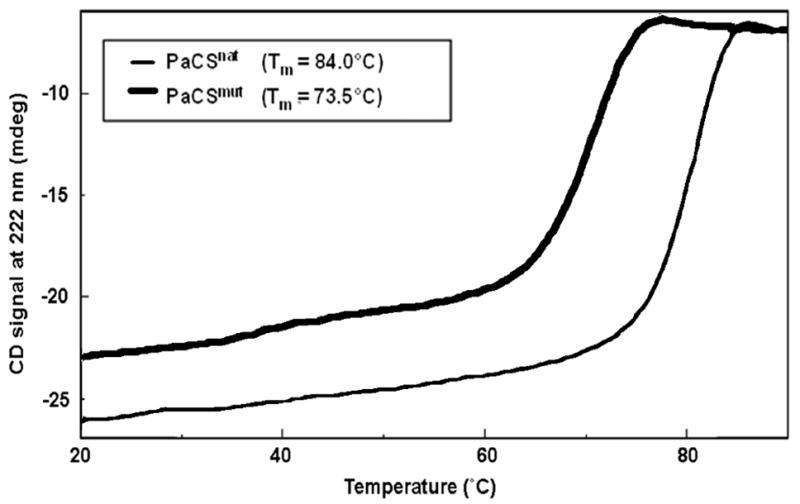
Thermal denaturation of native and mutant forms of P. aerophilum citrate synthase in 4.5M Gdn-HCl, monitored by circular dichroism. Experiments in the oxidized (thin curve) and reduced forms (thick curve) illustrate the stabilizing contribution from the linkage of the two protein chains.
Comparison of PaCS to homologous structures
Analysis of the native and mutant proteins revealed the stabilizing effect of the disulfide bond. However, the high stability of the disulfide-free mutant indicates that other factors are also important to the stability of the enzyme. Previous comparative studies of citrate synthase structures from distantly related organisms have identified several subtle trends correlated with thermophilicity, including decreased surface area and volume, shortening of loops, and increased ion pairing 25; 32. An analysis of these trends with respect to the PaCS structure revealed key differences in how certain stabilizing mechanisms are utilized between the various homologues. Table 3 summarizes several properties associated with thermal adaptation for a set of citrate synthase homologues. Previous studies highlighted an overall decrease in surface area and volume for thermophilic citrate synthases relative to the mesophilic pig citrate synthase (PigCS). Surprisingly, this trend does not hold for PaCS, as both the surface area and volume were found to be greater for PaCS than for all but the PigCS structure. The increase in size is partly due to the N-terminal extension and additional α-helices that are unique to the PaCS structure. The contributions of these elements to stability may override the potentially destabilizing effect of increased size. PaCS also exhibited a shortening of loops between α-helices. This decrease in disordered regions is likely to help counter the increase in size33. To take into account these differences in chain length, we analyzed the surface area and volume per residue. Interestingly, the PaCS still showed a greater surface area and volume per residue but, more significantly, the trend of decreased surface area and volume as a function of thermophilicity was no longer apparent, with the volume per residue actually greater in thermophilic homologues. These results show that decreased surface area and volume are not strongly correlated with increased thermal stability.
Table 3.
Comparison of structural features in homologues of citrate synthase.
| PaCS | PfCS | SsCSa | PigCS | ArCSb | |
|---|---|---|---|---|---|
| Optimal Temp (C°) | 100 | 100 | 85 | 37 | 31 |
| Sequence Identity (%)c | - | 46 | 44 | 27 | 35 |
| Sequence Length | 409 | 377 | 377 | 464 | 379 |
| ASA (×104 Å2) | 3.02 | 2.66 | 2.77 | 3.27 | 2.66 |
| ASA/residue (Å2) | 73.8 | 70.6 | 73.5 | 70.5 | 70.2 |
| Interface ASA (%) | 25.9 | 22.6 | 19.8 | 24.0 | 21.0 |
| Volume (×104 Å3) | 8.65 | 7.89 | 7.74 | 8.93 | 7.63 |
| Volume/residue (Å3) | 211.5 | 209.3 | 205.3 | 192.5 | 201.3 |
| Hydrophobic (%)d | 40.3 | 39.0 | 40.3 | 38.7 | 40.4 |
| Charged (%)e | 24.2 | 26.5 | 26 | 20.4 | 23.2 |
| Total Ion Pairs | 65 | 43 | 45 | 36 | 52 |
| Interface H-Bonds | 47 | 54 | 28 | 42 | 44 |
| Length of Loops | 101 | 106 | 109 | 116 | 114 |
From Sulfolobus solfataricus.
From Antarctic bacterium DS2-3R.
Calculated as identity to PaCS.
Hydrophobic residues Ala, Ile, Leu, Met, Phe, Trp, Val
Charged residues Arg, Lys, Asp, Glu
Intermolecular ion-pairing and hydrophobicity at the dimeric interface have previously been implicated as stabilizing elements in the thermophilic citrate synthase homologues. An analysis of overall hydrophobic and charged amino acid content showed no correlation with thermophilicity, emphasizing the importance of three-dimensional spatial considerations in understanding thermal stability. Interestingly, a structural comparison of PaCS and PfCS revealed significant differences in how various stabilizing elements are utilized. Although ion-pairing was previously identified as a major stabilizing mechanism at the PfCS dimeric interface 25, it does not appear to play as great a role in the stabilization of the PaCS dimer; only 8% of the residues in the P. aerophilum interface are charged, while the corresponding value is 22% for P. furiosis. Ion pairs in PaCS seem to be primarily intramolecular, stabilizing each subunit. Instead, PaCS utilizes hydrophobic interactions as a primary mechanism for stabilizing the dimeric interface. In P. aerophilum, 62% of the residues in the dimeric interface are hydrophobic (following the definition in Table 3), while the corresponding value is only 32% for P. furiosis.
CONCLUSIONS
Topologically interlinked protein chains are extremely rare among natural proteins. The situation observed in PaCS – two cyclized chains linked together – has been observed only once before in the crystal structure of Pichia pastoris lysyl oxidase34. A similar case has been created synthetically by designing specific cysteine mutations into the tetramerization domain of the protein p5335. Stability studies of that construct revealed an increase in melting temperature of 59°C over the linear wild-type peptide. A different type of topological linkage was discovered in the bacteriophage HK97 capsid36. In that case, an isopeptide bond (between a lysine side chain in one polypeptide chain and an asparagine side chain in another) creates covalently connected rings of six subunits, with the rings topologically linked to each other. This “chain mail” organization presumably contributes to the stability of the unusually thin HK97 viral capsid. Another type of catenation has been noted in the protein 2-Cys peroxiredoxin. The crystal structure of that enzyme revealed two interconnected dodecameric rings of protein subunits37. However, the protein subunits are held together by non-covalent forces rather than covalent bonding, so the role of the interlocking rings in that case is less clear. Although the PaCS structure is just the second observed case of a natural protein forming a catenane through disulfide bonding, it seems unlikely that these are the only cases present in nature. As more structures of thermophilic proteins are determined, it is likely that other instances will be uncovered, or that other similar mechanisms of topological entanglement will be discovered.
A number of parallels can be drawn between linking and knotting in protein chains. Like linked proteins, knotted proteins are extremely rare; only five distinct protein folds have been identified in which the chain is judged to be deeply knotted38; 39; 40; 41. In some cases, the knot has been implicated in stabilizing or rigidifying the enzyme active site38; 41. As described here for P. aerophilum citrate synthase, linking appears to confer similar advantages. In addition, knotting and linking both lead to important questions about folding pathways or landscapes. Both kinds of structural features introduce potentially significant restrictions on the routes by which the folded state can be reached. In the case of the PaCS dimer, the correct native structure can only be realized if the N-termini of the two chains wind around each other in the proper configuration before the disulfide bonds cyclize the two chains. An examination of the structure suggests that the disulfide bonded cysteines from a single chain are brought into proximity by the native interactions between the two chains, thereby promoting the proper sequence of folding events. To the extent that disulfide bonds in P. aerophilum are thermodynamically stable, their presence in the PaCS dimer must stabilize the native structure by restricting the freedom of the chains to fully dissociate from each other; the interlocking of the chains effectively reduces the entropy of the unfolded state. Experimental studies on the thermodynamic and kinetic stabilities of knotted proteins have been initiated only recently42; 43. Further studies on knotted and linked proteins should provide insight into unusual mechanisms of protein folding and stabilization. In addition, understanding how nature has exploited topological linking could prove beneficial in engineering stable proteins and enzymes for therapeutic and industrial applications.
MATERIALS AND METHODS
Preparation of cell lysate
For both E. coli and P. aerophilum samples, the cell pellet was resuspended in lysate buffer (20 mM Tris pH 7.2, 10 mM NaCl, 1 mM EDTA, 20 mM iodoacetamide) to wash, and repelleted by centrifugation for 5 min at 20,000 x g. Washed cells were again resuspended in lysate buffer and lysed on ice by sonication for 3 × 1 min. Samples were centrifuged for 10 min at 20,000 x g to remove cell debris, with lysate quickly utilized according to one of the experimental protocols described below, in order to limit undesired modifications and proteolysis. In positive control samples where specified, a similar procedure is followed, with the exception of iodoacetamide in the lysate buffer.
Fluorescent-labeling of cysteines involved in native disulfide bonds
Lysate samples were denatured by heating to 95°C (2 min. for E. coli, 4 min. for P. aerophilum to ensure denaturation) in the presence of 1% SDS. Samples were treated with 20 mM iodoacetamide for 30 min. in the dark to block free cysteine thiols, and then dialyzed for 2 hours (3,000 M WCO) to remove excess iodoacetamide. Samples were reduced with 10 mM TCEP for 30 min. Following disulfide cleavage by reduction, samples were reacted with 25 μM CPM (7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin, Molecular Probes) for 30 min. for fluorescent labelling of free thiols. Preparation of fully labelled sample followed the same protocol, with the exception of iodoacetamide treatment. Proteins were separated by SDS-PAGE on a 12% acrylamide gel. Gels were imaged on AlphaImager 2200 (Alpha Innotech Corp.), and the levels of fluorescence were quantitated using AlphaEase v. 5.5 (Alpha Innotech Corp.).
2-D diagonal gel electrophoresis
The method used was adapted from Sommer, et al.44 Samples of whole-cell lysate were obtained as described above from either P. aerophilum or E. coli cells. Samples were treated with non-reducing SDS-PAGE sample loading buffer (12 mM Tris pH 6.8, 0.4% SDS, 0.02% bromophenol blue, and 5% glycerol) and run on a 10% acrylamide gel under denaturing conditions in the absence of reducing agents. Upon completion of the first-dimension run, the lane containing the separated sample was excised from the gel, and proteins within the gel were reduced by incubating the gel slice in loading buffer containing 10 mM DTT at 80°C for 10 min, followed by 37°C for 20 min. The reduced gel slice was placed horizontally across the top of a 10% acrylamide gel and sealed with 0.5% agarose. The gel was then run to completion. Protein spots were visualized by silver staining as described by Shevchenko, et al.45, or Sypro Ruby stain according to the manufacturer.
Identification of unknown proteins by LC-MS/MS
Off-diagonal spots of interest were excised from the 2D diagonal gels and subjected to in-gel digestion by trypsin45. Briefly, spots were destained, reduced with DTT and derivatized by treatment with iodoacetamide. Following alkylation, spots were subject to digestion with modified trypsin (Promega) for 18 hr. at 37°C. Peptides were recovered by sequential extraction with formic acid and acetonitirile, and pooled extracts were lyophilized.
Extracted peptides were subjected to tandem electrospray-ionization mass spectrometry for sequence identification. Lyophilized samples were reconstituted in 0.1 % formic acid, 5 % acetonitrile. Peptides were separated by reverse-phase liquid chromatography (Ultimate, LC Packings) and analyzed on a QSTAR XL hybrid quadrupole time-of-flight mass spectrometer (Applied Biosystems) using automated data-dependent tandem mass spectrometry with collision-activated dissociation. Proteins were identified by correlation of experimental tandem mass spectra to predicted mass values calculated from the P. aerophilum sequence database using the program Mascot (Matrix Science).
The molecular weights of the protein identifications were analyzed against the molecular weights observed by 2D-DGE. Where possible, likely disulfide connectivity was predicted based on a combination of 2D-DGE position (i.e. intramolecular when above diagonal, intermolecular when below), molecular weight, sequence analysis, and functional and structural comparison to homologues identified by BLAST23. The vertical alignment of spots 1 with 2 and 5 with 6 led to their interpretation as potential heterocomplex subunits. The identification of spot 2 as NusG, a transcriptional antitermination factor known to form hetero- but not homocomplexes in a number of genetically diverse organisms, led to its classification as a probable heterocomplex subunit. Spots 9 and 10 were classified as homodimers based upon analysis of homologous structures. Cys257 of spot 10 was implicated as the disulfide-forming residue based on conservation of cysteines.
Protein expression and purification
The Pyrobaculum aerophilum citrate synthase gene (PaCS) was cloned from P. aerophilum str. IM2 genomic DNA into a pETM11 vector containing an N-terminal His-tag followed by a TEV (tobacco etch virus) protease cleaveage site. PaCS was overexpressed in E. coli BL21 (DE3) Codon Plus RIL cells (Stratagene) by IPTG induction. Cells were resusupended in Tris pH 8.0 buffer and lysed by sonication for 3 × 1 min. Due to its thermophilic nature, the recombinant PaCS was heat purified by bringing the lysate to 75°C for 15 min., followed by centrifugation to pellet the precipitate. The soluble fraction was loaded onto a nickel Hitrap column (Amersham Pharmacia) for affinity purification of PaCS. Following elution of purified PaCS, the His-tag was cleaved by incubation with TEV protease for 3 hr at 37°C as described46. Cleaved PaCS was separated from uncleaved PaCS and cleaved His-tag by reloading the sample on a nickel Hitrap column and collecting the flow-through.
The PaCS-pETM11 plasmid was purified by QIAprep Miniprep Kit (Qiagen). A Cys19Ser/Cys394Ser double mutant (PaCS-mut) was cloned using the Quickchange Multi-Site Directed Mutagenesis Kit (Stratagene) with the PaCS-pETM11 plasmid as template. PaCS-mut was overexpressed and purified in a manner similar to that described for PaCS.
Thermal Denaturation Studies
Circular dichroism (CD) measurements were recorded using a Jasco J-715 spectrometer with a Peltier temperature-controlled cell holder (Jasco). The CD signal was recorded at 222 nm as the temperature was increased linearly at a rate of 1°C min−1 over a temperature range of 20–95°C. Samples contained 200 μg ml−1 protein in 30 mM sodium phosphate (pH 8.0), 10 mM sodium chloride and 4.5 M guanidinium hydrochloride.
Crystallization and structure determination of PaCS
The purified PaCS protein was concentrated to 18 mg ml−1 in a buffer containing 30 mM Tris pH 8.0 and 30 mM NaCl, and crystallized by hanging drop vapor diffusion at room temperature against a well solution of 100 mM sodium cacodylate pH 6.5, 20% (v/v) PEG 8000, and 200 mM magnesium acetate. Crystals grew within hours, but exhibited poor diffraction. A second phase of crystal growth routinely occurred within 1–2 weeks with crystals diffracting to high resolution. No additional cryoprotectant was necessary.
For structure determination, a 1.60Å native data set was collected on a single crystal at beamline 8.2.2. at the Advanced Light Source (ALS) in Berkeley, California. All data were processed using DENZO and SCALEPACK (HKL Research, Charlottesville, Virginia, United States). Phases were determined by molecular replacement using the program PHASER47 and the PDB coordinates 1VGP, corresponding to citrate synthase from Sulfolobus tokodaii. Electron density maps were generated and the model built using the program Coot48. Refinement of the model was carried out using the REFMAC5 in the CCP4 suite49. The quality of the structural model was evaluated using the programs ERRAT50 and PROCHECK51. Data collection and refinement statistics are summarized in Table 4.
Table 4.
Data collection and refinement statistics for the crystal structure of P. aerophilum citrate synthase.
| Data collection | |
|---|---|
| Space group | P212121 |
| Cell dimensions a, b, c (Å) | 64.42, 89.80, 146.33 |
| Resolution (Å) | 1.60 |
| Rsym (%) | 6.2 (64.9) |
| I / σI | 15.1 |
| Completeness (%) | 94.3 (87.3) |
| Redundancy | 6.9 (5.8) |
| Wavelength (Å) | 1.00 |
| Refinement | |
|
| |
| Resolution (Å) | 76.47–1.60 (1.66–1.60) |
| Total no. working reflections | 100,651 (6,756) |
| Total no. test reflections | 5286 (363) |
| Rwork/Rfree (%) | 16.1/19.1 |
| No. atoms | |
| Protein | 6,648 |
| Ion | 11 |
| Water | 815 |
| B-factors | |
| Protein | 15.1 |
| Ligand/ion | 27.8 |
| Water | 26.5 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.013 |
| Bond angels (°) | 1.322 |
Values in parentheses refer to the highest resolution shell
Rsym = Σ |I − <I>|2 / Σ I2.
Rwork = Σ ||F| − |Fc|| / Σ|Fo|.
Rfree is as Rwork but calculated for a test set comprising reflections not used in the refinement.
The final model includes two chains with residues 3-409 of each chain, two acetate ions, two magnesium ions, and one chloride ion. The model was refined to 1.60Å to an R-factor of 16.0% and Rfree of 19.1%. The crystal structure contains two disulfide bonds, one per monomer, between cysteines 19 and 394.
Sequence and structure analysis
Citrate synthase sequences were aligned using the MUSCLE multiple alignment software52. Surface area was calculated using AREAIMOL in the CCP4 suite49. Volume was determined by VOIDOO53. Interface hydrogen bonds were determined using ACT in the CCP4 suite49. Interface surface area was calculated as the difference between the surface area of the dimeric and monomeric forms of the protein.
Acknowledgments
We thank Micahel Sawaya for technical assistance, beamline staff at ALS, Martin Phillips, Inna Pashkov, Imke Schröder and Cari del Casal for P. aerophilum cells, Julie Elbogen, Gunter Stier (EMBL, Heidelberg) for the pETM11 vector, Jason Forse, Sara Bassilian, Joseph Loo, Edward Marcotte, Jonathan Katz, and Parag Mallick. This work was supported by grants from the BER program of the Department of Energy Office of Science, and the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kumar S, Nussinov R. How do thermophilic proteins deal with heat? Cellular and molecular life sciences. 2001;58:1216–33. doi: 10.1007/PL00000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li WF, Zhou XX, Lu P. Structural features of thermozymes. Biotechnology advances. 2005;23:271–81. doi: 10.1016/j.biotechadv.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Petsko GA. Structural basis of thermostability in hyperthermophilic proteins, or “there’s more than one way to skin a cat”. Methods in enzymology. 2001;334:469–78. doi: 10.1016/s0076-6879(01)34486-5. [DOI] [PubMed] [Google Scholar]
- 4.Mallick P, Boutz DR, Eisenberg D, Yeates TO. Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds. Proc Natl Acad Sci U S A. 2002;99:9679–84. doi: 10.1073/pnas.142310499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beeby M, O’Connor BD, Ryttersgaard C, Boutz DR, Perry LJ, Yeates TO. The genomics of disulfide bonding and protein stabilization in thermophiles. PLoS biology. 2005;3:e309. doi: 10.1371/journal.pbio.0030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toth EA, Worby C, Dixon JE, Goedken ER, Marqusee S, Yeates TO. The crystal structure of adenylosuccinate lyase from Pyrobaculum aerophilum reveals an intracellular protein with three disulfide bonds. Journal of molecular biology. 2000;301:433–50. doi: 10.1006/jmbi.2000.3970. [DOI] [PubMed] [Google Scholar]
- 7.Fahey RC, Hunt JS, Windham GC. On the cysteine and cystine content of proteins. Differences between intracellular and extracellular proteins. J Mol Evol. 1977;10:155–60. doi: 10.1007/BF01751808. [DOI] [PubMed] [Google Scholar]
- 8.Branden C, Tooze J. Introduction to Protein Structure. Garland Publishing; New York: 1991. [Google Scholar]
- 9.Jakob U, Muse W, Eser M, Bardwell JC. Chaperone activity with a redox switch. Cell. 1999;96:341–52. doi: 10.1016/s0092-8674(00)80547-4. [DOI] [PubMed] [Google Scholar]
- 10.Kishigami S, Akiyama Y, Ito K. Redox states of DsbA in the periplasm of Escherichia coli. FEBS letters. 1995;364:55–8. doi: 10.1016/0014-5793(95)00354-c. [DOI] [PubMed] [Google Scholar]
- 11.Kadokura H, Tian H, Zander T, Bardwell JC, Beckwith J. Snapshots of DsbA in action: detection of proteins in the process of oxidative folding. Science. 2004;303:534–7. doi: 10.1126/science.1091724. [DOI] [PubMed] [Google Scholar]
- 12.Herbert B, Galvani M, Hamdan M, Olivieri E, MacCarthy J, Pedersen S, Righetti PG. Reduction and alkylation of proteins in preparation of two-dimensional map analysis: why, when, and how? Electrophoresis. 2001;22:2046–57. doi: 10.1002/1522-2683(200106)22:10<2046::AID-ELPS2046>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 13.Virden R, Watts DC. The role of thiol groups in the structure and mechanism of action of arginine kinase. The Biochemical journal. 1966;99:162–72. doi: 10.1042/bj0990162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Essex DW, Miller A, Swiatkowska M, Feinman RD. Protein disulfide isomerase catalyzes the formation of disulfide-linked complexes of vitronectin with thrombin-antithrombin. Biochemistry. 1999;38:10398–405. doi: 10.1021/bi990694s. [DOI] [PubMed] [Google Scholar]
- 15.Sippel TO. New fluorochromes for thiols: maleimide and iodoacetamide derivatives of a 3-phenylcoumarin fluorophore. J Histochem Cytochem. 1981;29:314–6. doi: 10.1177/29.2.7019305. [DOI] [PubMed] [Google Scholar]
- 16.Sippel TO. Microfluorometric analysis of protein thiol groups with a coumarinylphenylmaleimide. J Histochem Cytochem. 1981;29:1377–81. doi: 10.1177/29.12.7320496. [DOI] [PubMed] [Google Scholar]
- 17.Mallick P, Boutz DR, Eisenberg D, Yeates TO. Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds; Proceedings of the National Academy of Sciences of the United States of America; 2002. pp. 9679–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sommer A, Traut RR. Diagonal polyacrylamide-dodecyl sulfate gel electrophoresis for the identification of ribosomal proteins crosslinked with methyl-4-mercaptobutyrimidate. Proc Natl Acad Sci U S A. 1974;71:3946–50. doi: 10.1073/pnas.71.10.3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molinari M, Helenius A. Glycoproteins form mixed disulphides with oxidoreductases during folding in living cells. Nature. 1999;402:90–3. doi: 10.1038/47062. [DOI] [PubMed] [Google Scholar]
- 20.Molinari M, Helenius A. Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science. 2000;288:331–3. doi: 10.1126/science.288.5464.331. [DOI] [PubMed] [Google Scholar]
- 21.Cumming RC, Andon NL, Haynes PA, Park M, Fischer WH, Schubert D. Protein disulfide bond formation in the cytoplasm during oxidative stress. J Biol Chem. 2004;279:21749–58. doi: 10.1074/jbc.M312267200. [DOI] [PubMed] [Google Scholar]
- 22.Brennan JP, Wait R, Begum S, Bell JR, Dunn MJ, Eaton P. Detection and Mapping of Widespread Intermolecular Protein Disulfide Formation during Cardiac Oxidative Stress Using Proteomics with Diagonal Electrophoresis. J Biol Chem. 2004;279:41352–60. doi: 10.1074/jbc.M403827200. [DOI] [PubMed] [Google Scholar]
- 23.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 24.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends in biochemical sciences. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 25.Bell GS, Russell RJ, Connaris H, Hough DW, Danson MJ, Taylor GL. Stepwise adaptations of citrate synthase to survival at life’s extremes. From psychrophile to hyperthermophile. European journal of biochemistry/FEBS. 2002;269:6250–60. doi: 10.1046/j.1432-1033.2002.03344.x. [DOI] [PubMed] [Google Scholar]
- 26.Russell RJ, Gerike U, Danson MJ, Hough DW, Taylor GL. Structure. Vol. 6. London, England: 1998. Structural adaptations of the cold-active citrate synthase from an Antarctic bacterium; pp. 351–61. [DOI] [PubMed] [Google Scholar]
- 27.Remington S, Wiegand G, Huber R. Crystallographic refinement and atomic models of two different forms of citrate synthase at 2.7 and 1.7 A resolution. Journal of molecular biology. 1982;158:111–52. doi: 10.1016/0022-2836(82)90452-1. [DOI] [PubMed] [Google Scholar]
- 28.Karpusas M, Holland D, Remington SJ. 1.9-A structures of ternary complexes of citrate synthase with D- and L-malate: mechanistic implications. Biochemistry. 1991;30:6024–31. doi: 10.1021/bi00238a028. [DOI] [PubMed] [Google Scholar]
- 29.Russell RJ, Hough DW, Danson MJ, Taylor GL. Structure. Vol. 2. London, England: 1994. The crystal structure of citrate synthase from the thermophilic archaeon, Thermoplasma acidophilum; pp. 1157–67. [DOI] [PubMed] [Google Scholar]
- 30.Russell RJ, Ferguson JM, Hough DW, Danson MJ, Taylor GL. The crystal structure of citrate synthase from the hyperthermophilic archaeon pyrococcus furiosus at 1.9 A resolution. Biochemistry. 1997;36:9983–94. doi: 10.1021/bi9705321. [DOI] [PubMed] [Google Scholar]
- 31.Bennett MJ, Choe S, Eisenberg D. Domain swapping: entangling alliances between proteins; Proceedings of the National Academy of Sciences of the United States of America; 1994. pp. 3127–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar S, Nussinov R. Different roles of electrostatics in heat and in cold: adaptation by citrate synthase. Chembiochem. 2004;5:280–90. doi: 10.1002/cbic.200300627. [DOI] [PubMed] [Google Scholar]
- 33.Thompson MJ, Eisenberg D. Transproteomic evidence of a loop-deletion mechanism for enhancing protein thermostability. J Mol Biol. 1999;290:595–604. doi: 10.1006/jmbi.1999.2889. [DOI] [PubMed] [Google Scholar]
- 34.Duff AP, Cohen AE, Ellis PJ, Kuchar JA, Langley DB, Shepard EM, Dooley DM, Freeman HC, Guss JM. The Crystal Structure of Pichia pastoris Lysyl Oxidase. Biochemistry. 2003;42:15148–15157. doi: 10.1021/bi035338v. [DOI] [PubMed] [Google Scholar]
- 35.Blankenship JW, Dawson PE. Thermodynamics of a designed protein catenane. Journal of molecular biology. 2003;327:537–48. doi: 10.1016/s0022-2836(03)00115-3. [DOI] [PubMed] [Google Scholar]
- 36.Wikoff WR, Liljas L, Duda RL, Tsuruta H, Hendrix RW, Johnson JE. Topologically linked protein rings in the bacteriophage HK97 capsid. Science. 2000;289:2129–33. doi: 10.1126/science.289.5487.2129. [DOI] [PubMed] [Google Scholar]
- 37.Cao Z, Roszak AW, Gourlay LJ, Lindsay JG, Isaacs NW. Structure. Vol. 13. Cambridge, Mass: 2005. Bovine mitochondrial peroxiredoxin III forms a two-ring catenane; pp. 1661–4. [DOI] [PubMed] [Google Scholar]
- 38.Nureki O, Shirouzu M, Hashimoto K, Ishitani R, Terada T, Tamakoshi M, Oshima T, Chijimatsu M, Takio K, Vassylyev DG, Shibata T, Inoue Y, Kuramitsu S, Yokoyama S. An enzyme with a deep trefoil knot for the active-site architecture. Acta Crystallogr D Biol Crystallogr. 2002;58:1129–37. doi: 10.1107/s0907444902006601. [DOI] [PubMed] [Google Scholar]
- 39.Taylor WR. A deeply knotted protein structure and how it might fold. Nature. 2000;406:916–9. doi: 10.1038/35022623. [DOI] [PubMed] [Google Scholar]
- 40.Virnau P, L AM, Kardar M. Intricate Knots in Proteins: Function and Evolution. PLoS Comput Biol. 2006;2 doi: 10.1371/journal.pcbi.0020122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wagner JR, Brunzelle JS, Forest KT, Vierstra RD. A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature. 2005;438:325–31. doi: 10.1038/nature04118. [DOI] [PubMed] [Google Scholar]
- 42.Mallam AL, Jackson SE. Folding studies on a knotted protein. J Mol Biol. 2005;346:1409–21. doi: 10.1016/j.jmb.2004.12.055. [DOI] [PubMed] [Google Scholar]
- 43.Mallam AL, Jackson SE. Probing nature’s knots: the folding pathway of a knotted homodimeric protein. J Mol Biol. 2006;359:1420–36. doi: 10.1016/j.jmb.2006.04.032. [DOI] [PubMed] [Google Scholar]
- 44.Sommer A, Traut RR. Diagonal polyacrylamide-dodecyl sulfate gel electrophoresis for the identification of ribosomal proteins crosslinked with methyl-4-mercaptobutyrimidate; Proceedings of the National Academy of Sciences of the United States of America; 1974. pp. 3946–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Analytical chemistry. 1996;68:850–8. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 46.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein engineering. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 47.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta crystallographica. 2005;61:458–64. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 48.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 49.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta crystallographica. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 50.Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein science. 1993;2:1511–9. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 52.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr D Biol Crystallogr. 1994;50:178–85. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]