

Abstract
Background
Several studies have shown an elevated prevalence of coeliac disease (CD) in sibs of coeliac patients (risk 8–12%).
Aim and method
We evaluated the risk that sibs of children with CD will also develop CD. This cohort of 188 Italian families was composed of probands with CD, at least one sib and both parents. CD status was determined and human leucocyte antigen (HLA)‐DQ genotyping performed in all family members. The study also used a dataset of Italian triads (127 probands and both their parents) also genotyped for HLA‐DQ.
Results
The overall risk that a sib of a CD patient will develop the disease was estimated at 10% in this sample. The risk estimate ranged from 0.1% to 29% when HLA‐DQ information of the proband, parents and sib was considered. We found a negligible risk (lower than 1%) for 40% of the sibs of probands, a risk greater than 1% but less than 10% for 30%, and finally a high or very high risk (above 25%) in one‐third of families.
Conclusion
These results make it possible to provide more accurate information to parents with a child with CD about the real risk for another child. An antenatal estimate of the order of risk of CD is now possible. Specific follow‐up can thus be offered for babies at high risk.
Coeliac disease (CD) is an immune mediated enteropathy caused by permanent sensitivity to gluten in genetically susceptible individuals.1 CD has a strong genetic association with human leucocyte antigen (HLA). Most CD patients (90–95%) express the heterodimer DQA1*05/DQB1*02.2,3 Among DQA1*05/DQB1*02 heterodimer carriers, the risk of disease is greater in individuals homozygous for DQB1*02.4,5
Using a large samples of patients from four European countries, Margaritte‐Jeannin et al6 recently showed that the genetic risk that individuals will develop CD can be stratified into five classes according to their HLA‐DQ genotype. DQ2 carriers can be divided into three groups, according to whether they have two copies of DQB1*02 (group G1), one copy of DQB1*02 acting in trans with DQA1*05 (group G2) or one copy of DQB1*02 acting in cis with DQA1*05 (group G3). DQ2 non‐carriers are divided into two groups: one with two copies of a DQB1*02, DQ8 or one copy of each (group G4), and one with other DQ genotypes (group G5). In all populations, the risk is highest for group G1, but the relative risks for the other genotypes vary from one population to another. In the Italian population, the relative risks for individuals belonging to groups G2, G3, G4 and G5 are 0.68, 0.23, 0.10 and 0.02, respectively.
Several studies have shown a higher prevalence of CD in sibs of CD patients compared with the general population, with risk estimates ranging from 8% to 12%.7,8,9,10,11,12,13 However, the meaning of these estimates are unclear as the CD phenotype is not often precisely defined and may or may not include different forms of CD. The ESPGHAN criteria14,15,16 distinguish three different forms: the latent (or potential) form, defined only by the presence of specific antibodies; the silent form, defined by the presence of specific antibodies and villous atrophy of the small intestine; and the symptomatic form, defined by the presence of specific antibodies, villous atrophy and clinical symptoms.
The aim of the present study was to evaluate the risk in the Italian population that a sib of a symptomatic patient will develop any of the three forms of CD, and to provide to the parents of a child with CD the most precise estimate of the risk for any future child. We estimate this risk according to familial and genetic information. In some situations we are able to predict the risk antenatally while in other situations it is necessary to specify the risk by genotyping after birth. Thus special attention to weaning and monitoring sibs with higher risks can begin as soon as possible.
Materials and methods
Family samples
A cohort of 188 Italian nuclear families included a symptomatic CD patient (the proband), at least one sib and both parents. Both parents were available for typing for nearly all probands (184/188). All probands were confirmed to have symptomatic CD, diagnosed according to the revised ESPGHAN protocol, with positive antiendomysial or antitransglutaminase antibodies, or both, and lesions more serious than Marsh 1 classification on biopsy of the small intestines.17 Samples were taken from 798 subjects: 184 CD probands and 614 of their first degree relatives—246 sibs and 368 parents (184 fathers, 184 mothers). They were enrolled in a 3 year follow‐up programme from January 2001 to December 2003 to check their CD status.
Two fasting blood samples were collected in the morning from all 798 individuals, all of whom provided written informed consent. The research was approved by the Ethics Committee of the School of Medicine, University of Naples “Federico II”, Italy, and was in accordance with the principles of the Helsinki II declaration.
All were screened for antiendomysial and antitransglutaminase antibodies, and their class II HLA (DQ‐DR) genotypes determined.
To increase the size of the proband sample for the HLA‐DQ distribution after the initial stage of analysis and thus improve the robustness of the estimate, the final analysis also included a dataset of Italian triads (127 probands and both parents) studied by Margaritte‐Jeannin et al as part of the European genetics cluster on CD.6
Disease status
Serological tests
Serum samples were centrifuged before use for antibody evaluation. Antiendomysium IgA antibodies were detected by indirect immunofluorescence microscopy on rhesus monkey oesophagus substrate (Eurospital Trieste Italy).
Antitissue transglutaminase IgA antibodies were analysed by ELISA with human recombinant tissue transglutaminase as antigen (DIA Medix Corp.; Ivax Diagnostics, Florida, USA). Total serum IgA was evaluated by a nephelometric assay (BN ProSpec System; Behring, Marburg Germany).
Small intestinal biopsy
Participants positive for antiendomysium IgA antibodies and antitissue transglutaminase IgA antibodies underwent a small intestinal biopsy, 17 with a Watson capsule and 9 by endoscopy. Mucosal lesions were graded according to Marsh17: MI, more than 30% increased intraepithelial lymphocyte infiltration (lymphocytic enteritis); MII, MI with crypt hyperplasia; MIIIA, MII with partial villous atrophy; MIIIB, MII with subtotal villous atrophy; MIIIC, MII with total villous atrophy.
Genotyping
DNA extraction
Genomic DNA was extracted from the EDTA+ blood samples with a commercially available Kit (Nucleon BACC 2; Amersham Biosciences Europe, Milan, Italy).
HLA DQ typing
HLA‐DQ typing for coeliac susceptibility was performed in a three step procedure. The first step used the single sequence specific primer‐PCR home based method previously described to detect the presence of the HLA heterodimer DQA1*0501‐DQB1*0201.18 The second step used a sequence specific oligonucleotide‐PCR based method to type the HLA DQB1* locus (Dynal Biotech Ltd, Bromborough, UK) to confirm the presence/absence of the DQB1*02 allele or to verify the presence of the other DQB1*03 risk allele. This step was followed by the sequence specific primer‐PCR technique (Dynal Biotech Ltd UK) intended to resolve the DQB1*03 locus, showing the presence or absence of the DQB1*0302 allele.
HLA DR typing
HLA‐DRB1 typing was performed by a sequence specific oligonucleotide‐PCR based method (Dynal Biotech LTD UK) to determine the phased DR‐DQ genotypes of all individuals.
Statistical analysis
Notation
Following Margaritte‐Jeannin et al,6 we consider five DQA1‐DQB1 haplotypes and five DQ genotypic groups. The five haplotypes are:
H1: DQA1*05‐DQB1*02, denoted: α5β2
H2: None(DQA1*05)‐DQB1*02, denoted: α5β2
H3: DQA1*05‐none(DQB1*02), denoted: α5β2
H4: DQA1*(301‐DQB1*)302, denoted: α3β3
H5: other haplotypes, denoted: αβ
Three of the five genotypic groups cover the DQ2 heterodimer carriers:
Group 1 (G1): H1/H1 and H1/H2 (with a double dose of β2)
Group 2 (G2): H2/H3 (heterodimer encode in trans)
Group 3 (G3): H1/H3, H1/H4 and H1/H5 (heterodimer encode in cis)
The other two genotypic groups are:
Group 4 (G4): H2/H2, H2/H4 and H4/H4
Group 5 (G5): other genotypes
Statistical methods
Estimation of empirical risks for sibs of probands
In families with a child with CD (hereafter called the proband), the risk R that another child will have CD may be estimated by the ratio of the number of affected sibs over the total number of sibs.
Estimation of control DQA1‐DQB1 haplotype frequencies (Hi)
As families in both samples were identified through single probands, the parental haplotypes not transmitted to the proband are representative of the general population (affected family based controls) and provide unbiased estimates of Hi frequencies.19 These frequencies are estimated from 622 non‐transmitted parental haplotypes (311 families).
Estimation of the genotypic group risks
Given the Hi frequencies estimated in the population, we may calculate the risk of CD (Fij) for an individual with genotype HiHj.
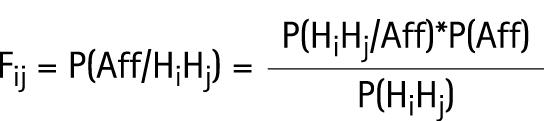 |
P(HiHj) is the probability that an individual in the population has the genotype HiHj. The computation of this probability is based on control haplotype frequencies that assume Hardy–Weinberg proportions. P(Aff) is estimated by the probability of CD in the population (ie, disease prevalence). P(HiHj/Aff) is estimated by the proportion of each genotype HiHj in the 311 affected probands.
Estimation of the familial correlation not due to HLA
Given R, Hi frequencies and Fij, it is possible to estimate λ, the familial correlation not due to HLA. We assume λ is a multiplicative factor independent of the HLA HiHj genotypes (see appendix).
Estimation of risks for sibs of probands
According to Mendelian segregation and assuming no recombination in the DR‐DQ haplotypes, we can compute the risk Rij that a sib will be affected according to the known HiHj genotype of the affected proband. If the parents' HLA is typed, we may have a more precise risk estimate (Rikjl) for the sib, depending on the parental genotypes. In some situations in which parental typing yields a wide range of risk for the fetus, postnatal typing may be proposed to refine the risk according to the infant's own genotype. Thus we further define the estimate in computing the risk Kij for sibs according to their own HiHj genotype (see appendix). Based on these risk estimates and on the probability that parents of a proband have a given genotype, it is possible to provide the expected distribution of the genetic groups in sibs.
Results
Estimation of risks for sibs of probands
Table 1 reports the distribution of the three forms of CD among the additional cases in this cohort. Fifteen first degree relatives (eight parents and seven sibs) were diagnosed with symptomatic CD before the study (based on the presence of antibodies, MII‐MIII lesions and clinical symptoms). Another 26 (9 parents and 17 sibs) of the 614 healthy relatives had elevated levels of antiendomysial and transglutaminase antibodies but were symptom‐free; 20/26 had previously tested negative. All underwent a biopsy: one parent had an MI lesion and eight showed greater degrees of damage (MIII). Of the 17 sibs, 12 had mucosal atrophy (grade 3, MIIIA; grade 4, MIIIB; grade 5, MIIIC) and 5 various intermediate grades MI/MII. Close follow‐up of the six relatives without frank mucosal damage, but with positive serology, began because they are considered to be latent (potential) CD patients.
Table 1 Distribution of coeliac disease forms in first degree relatives.
| Parents (n = 368) | Sibs (n = 246) | |
|---|---|---|
| Latent: no symptoms, MI–MII * | 1 | 5 |
| Silent: no symptoms, MIII* | 8 | 12 |
| Symptomatic: MII–MIII* | 8 | 7 |
| Total | 17 | 24 |
*Graded according to Marsh.17
The familial recurrence risk (R)—that is, the risk that a proband's sib will develop CD (latent + silent + symptomatic)—is estimated by:
R = 24/246 = 9.8% [6.1; 13.4].
This estimation is consistent with the value of ∼10% reported in the literature.7,8,9,10,11,12,13
Estimation of control DQA1‐DQB1 haplotype frequencies
Table 2 presents the frequency of each control DQA1‐DQB1 haplotype, with the corresponding DR for each.
Table 2 Control haplotype frequencies.
| Notation | DQA1‐DQB1 haplotype | DR allele | Frequency |
|---|---|---|---|
| H1 | DQA1*05 ‐ DQB1*0201 | DR3 | 0.11 |
| H2 | DQA1*02 ‐ DQB1*0202 | DR7 | 0.12 |
| H3 | DQA1*05 ‐ DQB1*03 | DR5 | 0.30 |
| H4 | DQA1*0301 – DQB1*0302 | DR4 | 0.04 |
| H5 | Others | DRX | 0.43 |
HiHj distribution in probands
At this stage, to increase the accuracy of the estimate of the familial risk, we added the Italian families studied in our previous European study.6 They were not included in the preceding calculations. Table 3 gives the observed number and frequencies of the HiHj genotype for the 311 probands and expected frequencies in the general population.
Table 3 Number and frequencies of HiHj in 311 probands and expected frequencies in the general population.
| DQ genotypes | Corresponding DR genotypes | Genotypic group | Probands | Expected in control population |
|---|---|---|---|---|
| H1/H1 | DR3/DR3 | G1 | 74 (24%) | 4% |
| H1/H2 | DR3/DR7 | |||
| H2/H3 | DR5/DR7 | G2 | 117 (38%) | 7% |
| H1/H3 | DR3/DR5 | G3 | 79 (25%) | 17% |
| H1/H4 | DR3/DR4 | |||
| H1/H5 | DR3/DRX* | |||
| H2/H2 | DR7/DR7 | G4 | 13 (4%) | 3% |
| H2/H4 | DR7/DR4 | |||
| H4/H4 | DR4/DR4 | |||
| Others | Others | G5 | 28 (9%) | 69% |
*X, different from 3, 4, 5 and 7.
Risk of coeliac disease according to genotypic group
The risk of CD for individuals with genotype HiHj in the Italian population is given in table 4. The corresponding haplotype combinations and DR genotypes (based on typing) are also shown for each group.
Table 4 Population risk according to genotypic group.
| DQ genotypes | Corresponding DR genotypes | Genotypic group | Fij |
|---|---|---|---|
| H1/H1 | DR3/DR3 | G1 | 0.21 |
| H1/H2 | DR3/DR7 | ||
| H2/H3 | DR5/DR7 | G2 | 0.17 |
| H1/H3 | DR3/DR5 | G3 | 0.06 |
| H1/H4 | DR3/DR4 | ||
| H1/H5 | DR3/DRX* | ||
| H2/H2 | DR7/DR7 | G4 | 0.05 |
| H2/H4 | DR7/DR4 | ||
| H4/H4 | DR4/DR4 | ||
| Others | Others | G5 | 0.006 |
*X, different from 3, 4, 5 and 7.
Familial correlation λ not due to transmission of HLA Hi haplotypes
Given R (0.098), Hi frequencies and Fij, we can compute λ, the familial correlation not due to transmission of Hi haplotypes:
λ = 1.4.
Risk for sibs of probands according to the proband's DQ genotype
Table 5 reports the risk of CD for sibs of probands according to each possible proband DQ genotype. Results are classed by genotypic group.
Table 5 Risks for sibs of probands according to proband DQ genotype.
| Proand group | G1 | G1 | G2 | G3 | G3 | G3 | G4 | G4 | G4 | G5 | G5 | G5 | G5 | G5 | G5 |
| Proband genotype | H1H1 | H1H2 | H2H3 | H1H3 | H1H4 | H1H5 | H2H2 | H2H4 | H4H4 | H2H5 | H3H3 | H3H4 | H3H5 | H4H5 | H5H5 |
| Rij | 0.14* | 0.14* | 0.11* | 0.07† | 0.07† | 0.06† | 0.09† | 0.06† | 0.04‡ | 0.04‡ | 0.03‡ | 0.03‡ | 0.03‡ | 0.02‡ | 0.02‡ |
*Rij <0.05; †0.5 ⩽Rij ⩽0.1; ‡Rij >0.1.
Risk for sibs of probands according to parental DQ genotype
As shown in table 5, the risk for future sibs varies substantially according to the proband's HLA DQ. HLA typing may also be proposed for the parents for more detailed information. Figure 1 uses colour to illustrate the potential information available with parental genotyping: the risk of another baby may be negligible (<1% blue), moderate (1–10% green), intermediate (10–15% orange), high (15–20% yellow) or very high (>20% red). The orange and yellow boxes indicate that the risk estimate is associated with wide confidence intervals. In these families, an accurate estimate of the familial recurrence risk may require genotyping of the newborn.

Figure 1 Risk for a sib of a proband according to the DQ genotype of the parents.
For example, a H2H2 (DR7/DR7) mother who already has a child with CD has a 29% risk of another CD child if the father is H1H1 (DR3/DR3), but only a 7% risk if the father is H2H2 (DR7/DR7). Similarly, a H2H5 (DR7/DRX) father who already has a child with CD has a very small (2%) risk of another if the mother is H2H5 (DR7/DRX), but a higher (12%) risk if she is H3H3 (DR5/DR5).
Figure 1 also shows that the decision to genotype the newborn depends more on the range or variability of the risk for the child than on the mean expectation according to parental genotypes. For example, if parents are H1H1/H1H1, H2H4/H2H4 or H3H5/H4H5, there is no doubt about the child's risk and genotyping is unnecessary. If the parents are H2H4/H1H3 (in the orange area), any new child has a mean risk of 15%, but this risk ranges from 1 to 29%. In this situation, genotyping for the child is suggested to define the risk more precisely.
Risk for sibs of probands according to their own DQ genotype
Figure 2 gives the probability (on top of the box) that sibs of probands will be in G1 and their corresponding risk (inside the bar). It shows that approximately 40% of sibs are expected to belong to G5 and consequently will have a negligible risk (lower than 1%). Approximately 30%, however, will be in G1 or G2 and will have a predicted risk higher than 20%.

Figure 2 Probability for a sib of a proband to belong to G1 and the corresponding risk.
Discussion
Genetic counselling in families with a proband affected by a multifactorial disease is generally inappropriate, in view of the large uncertainty around the empirical risk of familial recurrence. Antenatal genetic counselling is neither required nor suggested for CD, but those who work with coeliac families are often asked about the recurrence risk for any subsequent children. We answer that the risk of recurrence is approximately 10%, but do not give any more detailed information.
In this study, we showed that sibs of CD probands have an average recurrence risk of 10%, but that this average can be broken down according to HLA DQ information from the proband. Depending on this information, the risk estimate for the sib ranges from 2% to 14%. However, better information is often available by also genotyping parental HLA. In other cases, for example, when parents are H2H4 and H1H3, postnatal genotyping may be advisable to clarify the infant's risk. Broadly, it is expected that approximately 40% of sibs of CD probands will have a negligible risk (lower than 1%) of developing any form of the disease. This procedure therefore provides substantial reassurance to 40% of families. Moreover, 30% of sibs are expected to have a risk lower than 10%, and greater than 1%.
There remain one‐third of families for which the risk of recurrence is high or very high (above 20%). We do share the information with the family and set up a plan to deal with this risk:
Breast feeding is strongly supported20: it does not prevent CD, but it does affect the phenotype, by delaying the onset of symptoms.21
Gluten containing foods should be introduced at weaning according to standard practices for infants from unaffected families as there is no evidence that time of gluten introduction affects disease incidence. It is also desirable to unmask the disease as early as possible and not delay it to an older age, when the risk of complications has increased.22 It has been suggested that there is a “window” for gluten introduction (4–6 months) during which the child is “protected” from developing the disease, but again this is likely to affect only the phenotype of this heavily genetic determined disease.23 A recent twin study suggests that unshared environment has little or no effect on the onset of CD24 while genetic factors play a major role. Gluten avoidance is environmental: it may completely stop disease onset, but it does not appear to be a realistic option. Who would agree to go on a gluten free diet for life because of the genetic risk—never higher than 30%—of developing a disease that can be brought into total remission by diet after onset?
The easy availability of point‐of‐care antitransglutaminase antibody tests makes it possible to suspect the onset of the disease long before the appearance of clinical symptoms.25 Secondary prevention may be put in place for subjects with a significant risk estimate: the vast majority of infants who will eventually develop the disease may be diagnosed before the disease becomes clinically manifest.
Accordingly, for these infants in CD families at high risk of recurrence, doctors should encourage breast feeding, ordinary weaning with gluten and careful monitoring after gluten introduction. A great amount of suffering, anxiety and healthcare resource use could thus be prevented.
Despite our intentions, ethical issues will inevitably arise from the interpretation of genetic risk tables. To reduce this risk, we did not assign exact point estimates to each cross of fig 1, but families in the red area may feel the burden of the increased risk estimate.
We do not use new markers here: we are simply improving the rough risk estimate that is already in daily use. Most families may feel reassured by this information. A few in the red area may feel depressed or discouraged. We do not yet have an exact estimate of the effects of this information on families: we have begun a 4 year European multicentre prospective study to gain this knowledge. A large cohort of approximately 1000 DQ2+ or DQ8+ newborns from at risk families (with one coeliac proband) will be followed‐up to estimate the incidence of new cases according to the familial as well as the individual genetic risk factors. The parental impact of this genetic information will be evaluated in real life.
Our professional experience suggests that families do prefer to have the best estimate of their recurrence risks. We do hope that not one child birth is prevented but an enhancement of childbirths is promoted by a refined information of the recurrence risk.
Other genetic information can then build on this solid risk estimation from HLA genotyping. Currently, other susceptibility genes for CD have been mapped either by linkage or by association studies,26,27,28,29 but this information cannot immediately be used in the risk estimation until susceptibility variants are clearly identified. Predisposing variants might be identified soon by currently running extensive association and prospective studies: a multivariable combination of these expected findings is likely to provide a powerful tool to really improve the risk estimate.
The combination of readily available serological tests with a robust estimate of the genetic risk is likely to significantly reduce the burden of clinical disease in at risk families.
Acknowledgements
We would like to thank the patients and their families.
This work was supported by: FRM (Fondation de la Recherche Médicale) for the doctoral funding of M Bourgey; ELFID (European Laboratory for the Investigation of Food‐Induced Diseases); CEINGE, Regione Campania Convenzione‐ Del G. R. 27/12/2002 N. 6276; Regione Campania (progetto finalizzato Ricerca Sanitaria, D.G 10 del 21/01/05, sistema B linea di ricerca 6); and MIUR (Italian Ministero dell'Istruzione, dell'Università e della Ricerca).
Abbreviations
CD - coeliac disease
HLA - human leucocyte antigen
Appendix
In this section, we detail the formulae used in the methods section.
We use the following notations:
Saff, affected sib
Paff, affected proband
Pij, proband with HiHj genotype
Cikjl, parents with HiHk and HjHl genotypes
Aff, affected individual
λ, familial correlation not due to HLA, on assumption that CD is a multiplicative factor independent of the HLA DQ genotypes.
R is the probability that the sib of an affected individual P (proband) will be affected:
 |
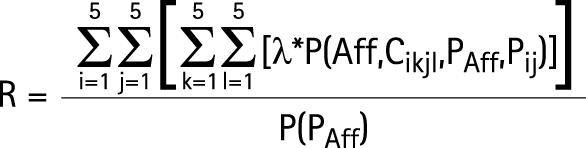 |
 |
As we can estimate R, Fij and the allelic frequencies of Hi haplotypes in our sample, we can also estimate λ, the familial correlation not due to HLA haplotypes:
 |
With:
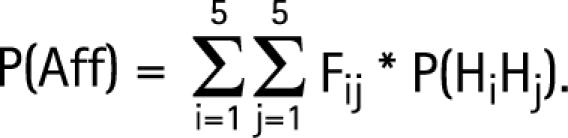 |
Given λ, Fij and the allelic frequencies of Hi haplotypes, it is possible to calculated Rij, the probability that a sib of the proband will be affected on condition that the proband has a genotype HiHj:
 |
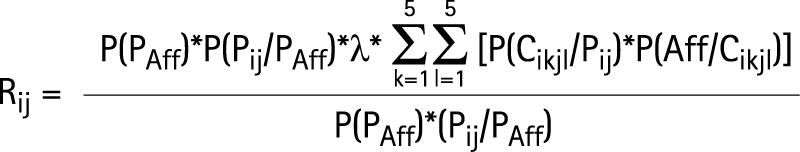 |
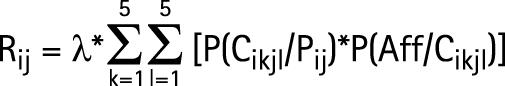 |
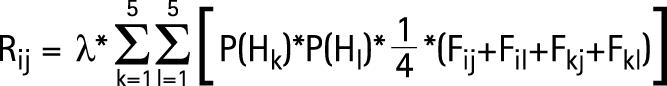 |
When the genotypes of parents are given, it is possible to define this risk in more detail by calculating the risk Rikjl, which is the probability that a sib of a proband will be affected on condition that the parents have genotypes HiHk and HjHl:
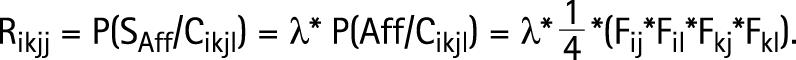 |
Footnotes
Competing interests: None.
References
- 1.Green P H, Jabri B. Coeliac disease. Lancet 2003362383–391. [DOI] [PubMed] [Google Scholar]
- 2.Sollid L M, Markussen G, EK J et al Evidence for a primary association of coeliac disease to a particular HLA‐DQ α/β heterodimer. J Exp Med 1989169345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sollid L M, Thorsby E. The primary association of coeliac disease to a given HLA‐DQ α/β heterodimer explains the divergent HLA‐DR association observed in various Caucasian populations. Tissue Antigens 199036136–137. [DOI] [PubMed] [Google Scholar]
- 4.Plosky R, Ek J, Thorsby E.et al On the HLA‐DQ associated susceptibility in coeliac disease: a possible gene dosage effect of DQB1*0201. Tissue Antigens 199341173–177. [DOI] [PubMed] [Google Scholar]
- 5.Clerget‐Darpoux F, Bouguerra F, Kastally R.et al High risk genotypes for coeliac disease. C R Acad Sci Paris III 1994317931–936. [PubMed] [Google Scholar]
- 6.Margaritte‐Jeannin P, Babron M C, Bourgey M.et al HLA‐DQ relatives risk for coeliac disease in European population: a study of the European Genetic cluster on coeliac disease. Tissue Antigens 200463562–567. [DOI] [PubMed] [Google Scholar]
- 7.Houlston R S, Ford D. Genetics of coeliac disease. Q J Med 199689737–743. [DOI] [PubMed] [Google Scholar]
- 8.Bonamico M, Mariani P, Mazzilli M C.et al Frequency and clinical pattern of coeliac disease among siblings of coeliac children. J Pediatr Gastroenterol Nutr 199623159–163. [DOI] [PubMed] [Google Scholar]
- 9.Farrè C, Humbert P, Vilar P.et al Serological markers and HLA‐DQ2 haplotype among first‐degree relatives of coeliac patient. Dig Dis Sci 1999442344–2349. [DOI] [PubMed] [Google Scholar]
- 10.Korponay‐Szabo I, Kovacs J, Lorincz M.et al Families with multiple cases of gluten‐sensitive enteropathy. Z Gastroenterol 199836553–558. [PubMed] [Google Scholar]
- 11.Trier J S. Coeliac Sprue. N Engl J Med 19913251709–1719. [DOI] [PubMed] [Google Scholar]
- 12.Hogberg L, Falth‐Magnusson K, Grodzinsky E.et al Familial prevalence of coeliac disease: a twenty year follow‐up study. Scand J Gastroenterol 20033861–65. [DOI] [PubMed] [Google Scholar]
- 13.Pittshieler K, Gentili L, Niederhofer H. Onset of coeliac disease: a prospective longitudinal study. Acta Paediatr 2003921149–1152. [DOI] [PubMed] [Google Scholar]
- 14.Walker‐Smith J A, Guandalini S, Schmitz J.et al Revised criteria for diagnosis of coeliac disease. Report of the Working Group of the European Society of Paediatric Gastroenterology and Nutricion. Arch Dis Child 199065909–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paparo F, Petrone E, Tosco A.et al Clinical, HLA, and small bowel immunohistochemical features of children with positive serum antiendomysium antibodies and architecturally normal small intestinal mucosa. Am J Gastroenterol 20051002294–2298. [DOI] [PubMed] [Google Scholar]
- 16.Fasano A. Clinical presentation of coeliac disease in the pediatric population. Gastroenterology 2005128S68–S73. [DOI] [PubMed] [Google Scholar]
- 17. When is a coeliac a coeliac? Report of a working group of the United European Gastroenterology Week in Amsterdam, 2001. Eur J Gastroenterol Hepatol 2001131123–1128. [DOI] [PubMed] [Google Scholar]
- 18.Sacchetti L, Tinto N. Calcagno G, et al. Multiplex PCR typing of the three most frequent HLA alleles in coeliac disease. Clin Chim Acta 2001310205–207. [DOI] [PubMed] [Google Scholar]
- 19.Thomson G. Mapping disease genes: family‐based association studies. Am J Hum Genet 199557487–498. [PMC free article] [PubMed] [Google Scholar]
- 20.Ivarsson A, Hernell O, Stenlund H.et al Breast‐feeding protects against coeliac disease. Am J Clin Nutr 200275914–921. [DOI] [PubMed] [Google Scholar]
- 21.Greco L, Auricchio S, Mayer M.et al Case control study on nutritional risk factors in coeliac disease. J Pediat Gastroenterol Nutr 19887395–399. [DOI] [PubMed] [Google Scholar]
- 22.Ventura A, Magazzu G, Greco L. Duration of exposure to gluten and risk for autoimmune disorders in patients with coeliac disease. SIGEP study group for autoimmune disorders in coeliac disease. Gastroenterology 1999117297–303. [DOI] [PubMed] [Google Scholar]
- 23.Norris J M, Barriga K, Hoffenberg E J.et al Risk of coeliac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA 20052932310–2312. [DOI] [PubMed] [Google Scholar]
- 24.Nistico L, Fagnani C, Coto I.et al Concordance, disease progression, and heritability of coeliac disease in Italian twins. Gut 200655803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korponay‐Szabo I R, Raivio T, Laurila K.et al Coeliac disease case finding and diet monitoring by point‐of‐care testing. Aliment Pharmacol Ther 200522729–737. [DOI] [PubMed] [Google Scholar]
- 26.Greco L, Babron M C, Corazza G R.et al Existence of a genetic risk factor on chromosome 5q in Italian coeliac disease families. Ann Hum Genet 20016535–41. [DOI] [PubMed] [Google Scholar]
- 27.Amundsen S S, Monsuur A J, Wapenaar M C.et al Association analysis of MYO9B gene polymorphisms with coeliac disease in a Swedish/Norwegian cohort. Hum Immunol 200667341–345. [DOI] [PubMed] [Google Scholar]
- 28.Naluai A T, Nilsson S, Samuelsson L.et al The CTLA‐4/CD28 gene region on chromosome 2q/33 confers susceptibility to coeliac disease in a way possibly distinct from that of type 1 diabetes and other chronic inflammatory disorders. Tissue Antigens 200056350–355. [DOI] [PubMed] [Google Scholar]
- 29.King A L, Moodie S J, Fraser J S.et al CTLA‐4/CD28 gene region is associated with genetic susceptibility to coeliac disease in UK families. J Med Genet 20023951–54. [DOI] [PMC free article] [PubMed] [Google Scholar]