

Abstract
Little is known about how gene expression variation within a given species controls phenotypic variation under different treatments or environments. Here, we surveyed the transcriptome response of seven diverse Arabidopsis thaliana accessions in response to two treatments: the presence and absence of exogenously applied salicylic acid (SA), an important signaling molecule in plant defense. A factorial experiment was conducted with three biological replicates per accession with and without applications of SA and sampled at three time points posttreatment. Transcript level data from Affymetrix ATH1 microarrays were analyzed on both per-gene and gene-network levels to detect expression level polymorphisms associated with SA response. Significant variation in transcript levels for response to SA was detected among the accessions, with relatively few genes responding similarly across all accessions and time points. Twenty-five of 54 defined gene networks identified from other microarray studies (pathogen-challenged Columbia [Col-0]) showed a significant response to SA in one or more accessions. A comparison of gene-network relationships in our data to the pathogen-challenged Col-0 data demonstrated a higher-order conservation of linkages between defense response gene networks. Cvi-1 and Mt-0 appeared to have globally different SA responsiveness in comparison to the other five accessions. Expression level polymorphisms for SA response were abundant at both individual gene and gene-network levels in the seven accessions, suggesting that natural variation for SA response is prevalent in Arabidopsis.
INTRODUCTION
Variation in transcript abundance, termed expression level polymorphism (ELP) (Doerge, 2002), influences quantitative phenotypic variation in organisms. Investigation of ELPs using global gene expression methodologies in defined mapping populations has recently enabled progress toward understanding the potential role of gene expression variation in quantitatively inherited traits (Wang et al., 1999; Carrol, 2000; Brem et al., 2002; Kliebenstein et al., 2006a, 2006b; West et al., 2007). In plants, naturally occurring ELPs have been associated with phenotypic traits such as developmental changes during maize (Zea mays) domestication (Wang et al., 1999), flowering time control in Arabidopsis thaliana (Johanson et al., 2000; Caicedo et al., 2004; Werner et al., 2005), qualitative resistance to pathogens (Grant et al., 1995; Gassmann et al., 1999; Borevitz et al., 2003), and Arabidopsis insect resistance and secondary metabolism (Kliebenstein et al., 2001, 2002; Lambrix et al., 2001). Typically, these ELP-controlled quantitative trait loci (QTL) are associated with large phenotypic effects, and their potential interactions with environment or treatment have not been characterized. Most genes for which an ELP is associated with a phenotypic-based QTL contain cis-regulatory polymorphisms. It is possible that the genes controlling QTL that interact with environments and treatments are different genes from those controlling QTL that are stable across environments and treatments (Via et al., 1995; Sultan, 2000). Identifying the genes that regulate QTL × environment and QTL × treatment interactions is necessary to understand the molecular basis of phenotypic plasticity.
Differential regulation of gene expression is a well-characterized response to changes in environments and treatments; variation within this process may be the basis for ELPs exhibited only under specific environmental or treatment conditions. For example, if two individuals had a sequence polymorphism affecting a cis-DNA regulatory element required for a gene's response to a specific treatment, then these two individuals would display an ELP for this gene only in the presence of the treatment. Alternatively, if the sequence polymorphism occurred in a transcription factor required for response to the treatment, it could act in trans to generate numerous environment/treatment-dependent ELPs. Since environmentally stable ELPs are known to control some phenotypic-based QTLs (Wang et al., 1999; Kliebenstein et al., 2001; Lambrix et al., 2001), ELPs exhibiting interactions with environments or treatments may be a contributing factor toward genotype × environment interactions.
Salicylic acid (SA) is an endogenous plant compound that is an important signaling molecule and regulator of plant defense responses (Gaffney et al., 1993; Delaney et al., 1995). The genetic control of response to SA within the Columbia (Col-0) accession of Arabidopsis has been extensively studied and provides a baseline for investigating conditional ELPs in other Arabidopsis accessions. Our previous analysis of transcript level variation in a replicated factorial Affymetrix ATH1 microarray experiment with seven diverse Arabidopsis accessions focused on the analysis of stable ELPs (i.e., ELPs that did not exhibit a treatment interaction) (Kliebenstein et al., 2006a). The (factorial) design of the experiment included the presence and absence of exogenous SA: therefore, it is also possible to examine these data for treatment-dependent or conditional ELPs (i.e., ELPs that significantly differ among accessions for their response to a treatment). While the presence or absence of SA is not typically considered as an environmental fluctuation, it can serve as a model for the range of biological responses that may underlie complex environmental fluctuations and their effects on ELP × environment/treatment and genotype × environment/treatment interactions.
Here, we analyzed data from the seven-accession factorial Affymetrix microarray experiment described by Kliebenstein et al. (2006a) to investigate conditional ELPs. We first used analysis of variance to detect expression differences on a per-gene level to test the frequency of individual genes that exhibit treatment-dependent ELPs. We also used a priori–defined gene networks of coexpressed genes to test for the presence of treatment-dependent variation at the level of gene networks and were able to successfully separate ELP variation into individual gene and gene-network levels. We anticipate that this knowledge about ELP variation will contribute toward understanding how genotype × environment/treatment interactions affect and control quantitative trait phenotypes.
RESULTS
Pairwise Accession Comparisons for SA-Responsive Genes
We investigated transcript-level variation among all 21 pairs of seven accessions using split plot analyses of variance (ANOVA) and t tests to detect genes exhibiting either stable or conditional ELPs. This statistical analysis identified an average of 1267 genes per accession pair whose transcript accumulation was responsive to SA (range = 802 to 1744 genes; Table 1). On average, 689 of these genes showed differential SA response between the two accessions (range 411 to 1013 genes; Table 1). This is slightly more than half of all SA-responsive genes within an accession pair that showed differential SA responses; these are the genes exhibiting treatment-dependent or conditional ELPs. For the entire collection of pairwise comparisons, ∼95% (3620 out of a total of 3837 SA-responsive genes) of the transcripts with an SA response showed evidence of a genotype × treatment interaction. The number of conditional ELPs is approximately one-third of the number of stable ELPs (i.e., ELPs that do not exhibit a treatment interaction), which have been described previously (Kliebenstein et al., 2006a).
Table 1.
Pairwise ANOVAs on a per-Gene Basis for Detection of Genotype × Treatment Interactions
| Accession 1 | Accession 2 | Parental Difference | SA Response | Interaction |
|---|---|---|---|---|
| Col-0 | Cvi-1 | 3109 | 1733 | 1013 |
| Col-0 | Est-1 | 2359 | 1744 | 931 |
| Col-0 | Kin-0 | 2238 | 1451 | 730 |
| Col-0 | Mt-0 | 1690 | 1360 | 588 |
| Col-0 | Tsu-1 | 2567 | 1586 | 847 |
| Col-0 | Van-0 | 2481 | 1236 | 642 |
| Cvi-1 | Est-1 | 2869 | 1450 | 857 |
| Cvi-1 | Kin-0 | 3170 | 1274 | 772 |
| Cvi-1 | Mt-0 | 3369 | 1186 | 764 |
| Cvi-1 | Tsu-1 | 2695 | 1282 | 776 |
| Cvi-1 | Van-0 | 3307 | 1141 | 674 |
| Est-1 | Kin-0 | 1970 | 1178 | 632 |
| Est-1 | Mt-0 | 1910 | 1433 | 672 |
| Est-1 | Tsu-1 | 1987 | 1267 | 674 |
| Est-1 | Van-0 | 1846 | 1036 | 524 |
| Kin-0 | Mt-0 | 1440 | 1010 | 448 |
| Kin-0 | Tsu-1 | 1732 | 1046 | 549 |
| Kin-0 | Van-0 | 1640 | 802 | 411 |
| Mt-0 | Tsu-1 | 1934 | 1348 | 764 |
| Mt-0 | Van-0 | 1988 | 1221 | 776 |
| Tsu-1 | Van-0 | 1490 | 833 | 415 |
| Total | 9222 | 3837 | 3620 |
The parental difference column indicates the number of genes showing a difference in expression between the two accessions. The SA response column indicates the number of genes showing an SA response in the pairwise ANOVA comparing the two accessions, either up- or downregulated. The interaction column indicates the number of genes that show a differential SA response between the accessions. These genes are also counted in both the parental difference and SA response columns. Total indicates number of unique genes per column.
The majority of SA-responsive genes showed an SA response in only one or a few accessions (Figure 1). Only 38 genes were SA responsive in the majority of accessions (Figure 1, inset). If the transcriptional response regulated by SA is critical to a plant's defense ability, a higher level of conservation across accessions would be expected in terms of the genes responding to SA. However, a gene-by-gene comparison may not be the most informative analysis since the expression of individual genes is controlled both by their own promoters and the signal transduction networks upstream of the promoter. Natural variation within the SA signal transduction networks may alter transcript accumulation for groups of genes and thereby minimize our ability to infer patterns from variation in individual genes.
Figure 1.

Variation in Gene Response to SA Treatment in Seven Accessions.
All genes that responded significantly to SA treatment at any time point (4, 28, and 52 hpt) were classified with respect to the number of accessions for which they showed a statistically significant response to SA treatment. The inset shows detail for the genes that responded in three to seven accessions.
If there is variation among accessions in the signal transduction response, some accessions may be either hyper- or hyporesponsive to the SA treatment. In hyperresponsive accessions, more genes would be expected to change their expression levels in response to SA treatment compared with hyporesponsive accessions. To test for altered responsiveness, we determined the number of genes responding to SA, either up or down, within each accession for each pairwise comparison. At 4 h posttreatment (hpt), accessions Col-0 and Mt-0 showed significantly more genes showing an SA response compared with the other accessions at 4 hpt (Figure 2A). Mt-0 had on average twice as many genes showing an SA response than the other accessions, while Col-0 had 1.5 times as many genes. By contrast, Cvi-1 had half the number of SA-responsive genes as the other accessions (Figure 2A). At 28 hpt, Van-0 had a significant enhancement in the number of SA-responsive genes, while the other accessions had similar ratios (Figure 2B). At all time points, Cvi-1 had the lowest level of SA-responsive genes (Figures 2A to 2C). Collectively, these results are consistent with natural variation among the accessions at the gene-network level for SA responses.
Figure 2.

Hypo- and Hyperresponses to SA Treatment as Measured by Individual Genes.
The number of genes responding to SA in each of the accessions (as detected in the six pairwise analyses) was divided by the number of genes responding in the other accession in the pairwise analysis. Each analysis was conducted independently at each of three time points posttreatment. t tests were used to test for statistically significant differences in the ratio of SA-responsive genes between the seven accessions. Bars represent se. Different letters represent statistically different groupings at P = 0.05.
(A) 4 hpt.
(B) 28 hpt.
(C) 52 hpt.
Variation in SA-Responsive Gene Networks
To examine gene-network level variation, we used 54 defense-related, coregulated groups of genes (i.e., putative gene networks) that were defined from public microarray data sets on the reference accession, Col-0 (see Methods). These 54 ad hoc–defined, defense-related gene networks allowed us to directly test for higher-order variation in SA response among the seven accessions. Individual statistical analyses (ANOVA) on each of the seven accessions resulted in 25 of the 54 gene networks having a significant SA response in at least one of the seven accessions (Table 2), with most of the differential SA responses being detected at 4 hpt (Table 2). This latter observation may have been due to the treatment consisting of a single exogenous application of SA whose effects diminished over time. Of the 25 SA-responsive gene networks, 14 gene networks were upregulated by SA (1400, 9000, 9300, 9400, 9403, a403, b000, b003, b300, b303, b400, b403, c000, and c003), and 11 gene networks were downregulated (1000, 1004, 1100, 1104, 2000, 2100, 3000, 5003, 7400, 740x, and z100). Interestingly, gene network 1100 is downregulated with SA treatment at 4 hpt in all accessions but upregulated at 28 or 52 hpt. The majority of gene networks showed evidence for differential expression among accessions, suggesting variation in gene-network level SA response within these seven accessions (Table 2).
Table 2.
Differential Gene-Network Expression in Response to SA Treatment in Seven Arabidopsis Accessions
| Col-0
|
Cvi-1
|
Est
|
Kin-0
|
Mt-0
|
Tsu-1
|
Van-0
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Network | 4 | 28 | 52 | 4 | 28 | 52 | 4 | 28 | 52 | 4 | 28 | 52 | 4 | 28 | 52 | 4 | 28 | 52 | 4 | 28 | 52 |
| 1000 | Dn | – | – | Dn | – | – | Dn | – | – | Dn | – | – | Dn | – | – | Dn | – | Up | Dn | – | – |
| 1004 | Dn | – | – | Dn | – | – | Dn | – | – | Dn | – | – | Dn | – | – | Dn | – | – | Dn | – | – |
| 1100 | Dn | – | Up | Dn | Up | – | Dn | Up | Up | Dn | – | Up | Dn | – | Up | Dn | – | Up | Dn | – | – |
| 1104 | Dn | – | Up | Dn | – | – | Dn | – | Up | Dn | – | – | Dn | – | Up | Dn | – | – | Dn | – | – |
| 1400 | – | – | Dn | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – |
| 2000 | Dn | – | Up | – | – | – | – | – | – | Dn | – | – | Dn | – | – | – | – | – | Dn | – | – |
| 2100 | Dn | – | Up | – | – | – | Dn | – | Up | Dn | – | – | Dn | – | – | – | – | – | Dn | – | – |
| 3000 | Dn | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | |||
| 5003 | Up | Dn | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – |
| 7400 | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | Dn | – | – | – | – | – |
| 740x | Dn | – | – | – | – | – | Dn | – | – | Dn | – | – | – | – | – | Dn | – | – | – | – | – |
| 9000 | Up | – | – | – | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – | – | – | – |
| 9300 | Up | – | – | Up | – | – | Up | – | Dn | Up | – | – | Up | – | – | Up | Up | – | Up | – | – |
| 9400 | Up | – | – | Up | – | – | Up | – | – | Up | – | – | – | – | – | Up | – | – | Up | – | – |
| 9403 | Up | – | – | Up | – | – | Up | – | – | – | – | – | Up | – | – | Up | – | – | Up | – | – |
| a403 | – | – | – | Up | – | – | Up | – | – | – | – | – | Up | – | – | – | – | – | – | – | – |
| b000 | Up | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – |
| b003 | Up | – | – | Up | – | – | Up | – | – | – | – | – | Up | – | – | Up | – | – | Up | – | – |
| b300 | Up | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – |
| b303 | Up | – | – | Up | – | – | Up | – | – | – | – | – | Up | – | – | Up | – | – | Up | – | – |
| b400 | Up | – | – | – | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – | – | – | – |
| b403 | Up | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – | Up | – | – |
| c000 | Up | – | – | – | – | – | Up | – | – | Up | – | – | – | – | – | – | – | – | – | – | – |
| c003 | Up | – | – | – | – | – | Up | – | – | – | – | – | Up | – | – | Up | – | – | – | – | – |
| z100 | Dn | – | – | – | – | – | – | – | – | – | – | – | – | – | – | Dn | – | – | – | – | – |
Cells designated with Dn or Up indicate gene networks with significantly different downregulation and upregulation, respectively, in response to SA treatment as detected by ANOVA for three sample time points (4, 28, and 52 hpt) per accession. The dashes indicate the comparisons for which no statistically significant differences between control and SA treatment were detected.
Characterization of SA-Responsive Gene Networks
We investigated the potential biological function of the SA-responsive gene networks by conducting GOslim term functional categorization analysis. The biological process category “response to abiotic or biotic stimuli” was significantly overrepresented in both up- and downregulated gene networks (see Supplemental Figure 2 online). The category “electron transport or energy pathways” was overrepresented in the downregulated gene networks (see Supplemental Figure 2 online), which agrees with the analysis of cellular components showing an overrepresentation of “chloroplast” and “plastid” categories in these same gene networks (see Supplemental Figure 3 online). We performed a lower-level Gene Ontology (GO) term analysis and found that the upregulated gene networks were enriched in signaling and defense processes (see Supplemental Table 5 online), while the downregulated gene networks were enriched in photosynthetic processes (see Supplemental Table 6 online). We also investigated the potential bias in promoter elements within the identified SA-responsive gene networks. As expected from the GO analysis that identified photosynthetic genes as downregulated by SA, the significantly overrepresented promoter elements for the downregulated genes showed a prevalence of light-responsive promoter elements, while the upregulated genes showed a prevalence of stress-responsive promoter elements (see Supplemental Tables 7 and 8 online).
SA-Responsive Gene Networks: A Comparison of Data Sets
To determine if relationships among the 25 SA-responsive gene networks are similar between the 16ATH1 data set from Col-0 and our seven-accession natural variation data set, we subjected each of these two data sets to Local Context Finder (LCF) analysis to generate graphical relationships based on nonlinear dimension reduction (Katagiri and Glazebrook, 2003). Unlike hierarchical clustering, which is limited to single dimension relationships, LCF analysis provides information about the relationships among the gene networks in multiple dimensions. Similar gene-network relationships are evident in both data sets, with two larger groups of gene networks clustering together in both data sets (see Supplemental Figure 4 online, compare A and B to C and D). These two groups correspond to the SA upregulated and SA downregulated gene networks. For the control data sets (see Supplemental Figures 4A and 4C online), the 16ATH1 data set identified 87 connections, while the seven-accession data set identified 91 connections, with 52 connections overlapping. In the treatment data sets (see Supplemental Figures 4B and 4D online), 16ATH1 identified 90 connections, the seven-accession data set identified 89 connections, and there were 54 connections in the intersection of these analyses. Basically, we have illustrated that natural genetic variation among these seven accessions identifies similar, but not identical, gene-network relationships as within the Col-0 accession. Gene expression within the 25 SA-responsive gene networks had an average correlation of 0.176 within the seven-accession data, while a random sample of genes had an average correlation of only 0.018. The similarity in coregulation and network connections across data sets suggests that the gene networks defined within Col-0 are also behaving as networks within other accessions. Based on this information and these results, it appears that we can use the gene networks defined in Col-0 to analyze SA responses in the other accessions. However, this approach does not permit identification of gene networks that are specific to the other accessions.
Diversity of Gene-Network Expression among Accessions
Our gene-network analysis suggested that there are differential gene-network level responses to SA among the accessions (Table 2). There was also supporting evidence for differential gene-network responses from the individual gene responses within SA hyper- and hyporesponsive accessions (Figure 2). We used the 25 SA-responsive gene networks to determine whether there was evidence of hyper- and hyporesponsive accessions at the gene-network level. The average response of the 25 gene networks to SA treatment at 4 hpt was lowest in Cvi-1 and the greatest in Mt-0, while the other accessions had a similar average response (Figure 3). This result agreed with the per-gene analysis where Mt-0 had an elevated response, while Cvi-1 had a lower response in comparison to the other accessions (Figure 2).
Figure 3.

Hypo- and Hyperresponses to SA as Measured by Gene-Network Variation.
The 25 SA-responsive networks were classified as either up- or downregulated, and the two groups were analyzed separately. For each accession, the average network expression value was obtained for each network from both the control and SA treatments. Only the 4-hpt time point was used. For each accession, the average response of the SA-upregulated networks (A) or SA-downregulated networks (B), respectively, was determined by dividing the mean network expression value under SA by its mean expression value under control conditions. The average value across all networks is presented. t tests were used to detect significant differences between the means; different letters represent statistically different groupings at P = 0.05. Bars represent se.
To directly test for variation in gene-network level accession × treatment interactions, we used a mixed linear model (ANOVA) to test for pairwise differences between Col-0 and each of the other six accessions (see Supplemental Figure 5 online). This analysis identified instances in which a gene network showed differential transcript accumulation between Col-0 and the other six accessions in only the SA-treated samples, in only the control samples, or in both the SA-treated and control samples. Two examples of gene networks with treatment-dependent variation between the accessions are b300 and 1104 (Figures 4A and 4B). Most gene networks showed accession × treatment variation in only a few of the six pairwise comparisons, with the exception of one gene network (3000) that varied in all comparisons. Gene network 3000 had significantly reduced expression in Col-0 under both conditions in comparison to the other accessions (Figure 4C). These data and results indicate significant natural variation in gene-network level responses to SA within Arabidopsis.
Figure 4.

Gene-Network Expression Response to SA Treatment in Seven Accessions.
The average gene-network expression value under control and SA treatments at 4 hpt for the seven accessions is presented for three example gene networks. Bars represent se.
(A) Variable SA-mediated upregulation in network b300.
(B) Variable SA-mediated downregulation in network 1104.
(C) Gene network 3000 for which Col-0 is significantly different from the other accessions.
Interconnected Gene-Network Variation
The variation in gene networks did not show an obvious pattern across pairwise comparisons with Col-0 (see Supplemental Figure 5 online). To determine whether a higher-order pattern was present, we used the LCF relationships among the gene networks within Col-0 from the 16ATH data set (see Supplemental Figure 4D online) as a framework to indicate the node position for each network and its linkages to the other nodes. Subsequently, for each pairwise network ANOVA for our seven-accession data set, we colored in the network nodes that showed a statistically significant difference between Col-0 and the other accession; those network nodes with no significant difference were left blank (Figure 5). The majority of gene networks showing significant pairwise variation tended to be interconnected by one or more links (Figure 5). For example, in the Col-0 versus Mt-0 comparison, most of the variable gene networks are linked in the multidimensional space, suggesting that there may be higher-order signaling pathway polymorphisms controlling multiple gene networks (Figure 5). By contrast, polymorphisms that are relatively isolated in the multidimensional representation (i.e., b403 in Col-0 versus Cvi-1) are likely to be polymorphisms limited to a specific gene network (Figure 5).
Figure 5.

Variable Gene-Network Expression Responses in Pairwise Comparisons of Six Accessions with Col-0.
To investigate relationships between gene networks that showed variation in the pairwise ANOVAs, we used the LCF multidimensional gene-network relationship shown in Supplemental Figure 4D online as a framework for comparison of Col-0 to the other six accessions. We included only those networks that were induced by SA. For each pair of accessions, the gene networks with statistically significant variation are indicated as black, gray, or hatched nodes. Black and gray nodes indicate gene networks with differential expression between the two accessions after SA or control treatments, respectively. Hatched black/gray nodes indicate those networks with differential expression between the two accessions under both treatment conditions. Black lines connect nodes showing variable expression. Nodes for gene networks that were not significantly different are in white and connected by gray lines. The statistical identification of pairwise network differences is shown in Supplemental Figure 5 online.
DISCUSSION
A large factorial microarray analysis of transcript level variation in response to an SA treatment identified significant levels of natural variation among seven diverse Arabidopsis accessions. This variation included dramatic differences in SA treatment responses at both the individual-gene (transcript) level and the gene-network level (Table 2, Figures 1 to 5). Accession × treatment-dependent ELPs accounted for approximately one-third of all ELPs detected in this experiment, suggesting that Arabidopsis has a highly variable, largely plastic transcriptome. The SA response of a single accession's transcriptome is not highly predictive of other accessions nor is it predictive of the species.
Individual Gene Treatment-Dependent ELPs
More than 9000 genes displayed an ELP among these seven accessions (Table 1). Additionally, 3837 genes showed a significant response to SA treatment (Table 1), which is many more than previously identified in Arabidopsis studies employing mutants in primarily one genetic background, Col-0 (Glazebrook et al., 2003). Interestingly, the majority of SA-responsive genes did not respond similarly across accessions; ∼95% of SA-responsive genes exhibited genotype × treatment-dependent ELPs (Table 1, Figure 1). Individual genes showed various patterns of variation: SA responsive in one accession and constitutively high in another; SA responsive in one accession and constitutively low in another; and differentially SA responsive in a comparison of two accessions (data not shown).
There are two possible explanations for the observed genetic diversity in the ability of individual genes to respond to SA treatment. The first explanation is that there are few conserved SA-responsive genes, and the control of individual gene transcripts has evolved to respond to diverse signaling pathways. This model is supported by the idea that most signaling is buffered by the presence of multiple pathways, such as ethylene, jasmonic acid, reactive oxygen species, and SA for plant–pathogen interactions (Glazebrook et al., 2003). A gene that is regulated in response to pathogen attack may be controlled by multiple signaling pathways; if so, this gene would still be pathogen regulated even if its promoter lost the ability to respond to one specific signaling pathway. For instance, if a gene is regulated by SA and reactive oxygen species, it could vary for its SA response but not vary for its pathogen response as it would still be induced by reactive oxygen species. Testing this hypothesis would require a comparison of the level of genotype × treatment-dependent ELPs in this study to another study measuring natural variation in plant transcriptome responses to a pathogen.
The second potential explanation is that there are a large number of conserved SA-responsive genes but that their coregulation by signal transduction networks diminishes our ability to identify them. This would occur if genotype × treatment-dependent ELPs are predominantly caused by variation within the signaling response to SA. Given sufficient diversity in the signaling response, very few individual genes would respond similarly in all accessions. This idea is supported by our observations that there are hyper- and hypo-SA-responsive accessions (Figures 2 and 3) as well as genetic variation controlling gene networks (Figures 3 to 5). An expression QTL analysis examining the genetic position of loci controlling the genotype × treatment-dependent ELPs may have the potential to directly test this idea. If genotype × treatment-dependent ELPs are largely controlled by network level variation, then expression QTL hot spots will exist at the position of the genetic variation regulating the SA responsiveness of numerous genes.
Gene-Network Variation
In addition to identifying individual transcripts with treatment-dependent genetic variation, we detected significant variation for the genetic control of groups of genes (i.e., gene-network variation) (Figures 3 to 5). Twenty-four of 25 SA-responsive putative gene networks defined by coexpression in other studies showed evidence of significant variation within the seven accessions (Figure 5; see Supplemental Figure 5 online). This variation ranged from controlling large clusters of interconnected gene networks to altering individual, isolated networks (Figure 5). This result suggests that there can be genetic variation for signaling responses that lie within different levels of the signaling network controlling SA responses, ranging from a global response, as illustrated by Cvi-1 and Mt-0 (Figures 2 and 3), to individual gene responses, and potentially at all steps in between (Table 1). These results are expected for natural variation of signal transduction, given that many signal transduction pathways are typically branched; when a signal is initiated or perceived at a single point, the pathway branches out to control a number of genes. If the variation occurred in the beginning of the pathway, it would alter a large group of gene networks. However, if the variation occurred near the end of the pathway, it would affect a limited number of gene networks. Thus, genetic variation for SA response appears to be occurring at multiple levels of the treatment response pathway. Identifying which genes contain these polymorphisms will provide a first step in expanding our mechanistic understanding of responses to SA beyond the Col-0 reference accession. Associating the genetic variation for SA response within the SA signal transduction network will discern whether there is a bias as to where natural variation occurs within the network.
Hyper- and Hypo-SA Response Variation
A potential mechanism to control treatment-dependent gene-network variation is variability for global responses to a given level of the treatment. Altered global responses could be either an elevated (hyper) or subdued (hypo) response throughout the transcriptome and would alter the ability to identify a statistically significant response in individual genes within these accessions. Both the individual-gene and gene-network analyses identified Mt-0 as a hyper-SA-responsive accession. Mt-0 typically had greater upregulation of SA-inducible gene networks and more genes significantly responding to SA with a higher magnitude of change compared with the other accessions (Figures 2 and 3). Accession Cvi-1 was identified as hypo-SA-responsive at both levels. Cvi-1 was shown previously to be hyporesponsive to methyl jasmonate (MeJa) according to both transcript accumulation and ozone sensitivity (Rao and Davis, 1999; Rao et al., 2000). MeJa and SA signaling are generally assumed to be mutually antagonistic, so it is interesting to observe an accession that is hyporesponsive to both hormones (Kunkel and Brooks, 2002). The polymorphisms leading to the hyper- and hyporesponsiveness are likely close to the initial perception of SA given the global effects on individual gene and gene-network expression observed in Mt-0 and Cvi-1. Our data suggest that Cvi-1 may be more sensitive, and Mt-0 may be more resistant, to pathogens for which Arabidopsis uses an SA signal to initiate a resistance response. To test this hypothesis, these accessions would need to be challenged with pathogens to evaluate their actual responses.
Biological Implications of Signaling Variation
A number of potentially interesting molecular observations concerning plant–pathogen interactions exist in this data set. At the individual gene level, the expression variation of R genes, both known and unknown, can potentially determine virulent versus avirulent host–pathogen interactions when a plant is attacked (Kliebenstein et al., 2006a). Some of the genes involved in regulating pathogen responses within accession Col-0, BAP1, NIMIN1, and ATR1, are among those with the most conserved SA responses across accessions (data not shown; http://elp.ucdavis.edu). However, the majority of the genes exhibiting SA responsiveness in the accessions did not have annotated functionalities.
In our view, the results of this study have important implications on the evolution of signal responses within a species. The plant's response to SA is an essential component of plant–pathogen interactions; a complete inability to respond is likely lethal to a plant in its natural environment. However, our study raises the possibility that the SA responses of Arabidopsis are more plastic than previously thought, given that genes known to be involved in SA/pathogen defense responses in the Col-0 accession (e.g., PR1, etc.) show dramatic variation at the transcript level in this collection of seven accessions (see Supplemental Table 1 online). This variation among accessions for gene expression suggests that this species as a whole contains more genes that respond to SA than does a single accession. Consequently, each individual accession is limited in its ability to mount a defense response against all pathogens, whereas the species has the capacity for a broader array of defense responses. This difference in SA responsiveness between individuals and the species suggests that there may be energetic costs of maintaining the ability for a full response within an individual (Cui et al., 2002, 2005; Raacke et al., 2006; Tang et al., 2006; Veronese et al., 2006; Zheng et al., 2006). Alternatively, a cost may arise when a defense response by the plant that defeats one pathogen enhances the ability of a different pathogen to attack the same plant. The difference between a species capacity and an individual's capacity to respond to pathogens is well understood at the qualitative level, where larger numbers of functional R genes exist within a species than within each individual genotype. Our results suggest that a similar genetic architecture of natural genetic variation may occur in a quantitative fashion at signaling events downstream of pathogen perception.
Implications for Studies Using Reference Accessions
The SA signaling pathway has been primarily studied in Col-0, a common reference accession, with mutants that cause major perturbations. This approach, while valuable, does not fully reveal the complete set of genes or gene networks that are SA responsive within the Arabidopsis species. For example, gene network 3000 was identified as significantly decreased in only Col-0 (Figure 4C). If this network is critical for plant–pathogen interactions, it will potentially be easier to study in the other accessions. There are likely to be signaling components and SA-responsive gene networks present in less-studied accessions that have been missed due to reliance on Col-0. Our data demonstrate the potential benefit of including a diverse genetic representation of a species in studies of key regulatory mechanisms and pathways.
Future Avenues
We identified a considerable level of genetic variation in transcript level responses to a key signaling molecule controlling plant–pathogen interactions. Does transcript variation lead to variation in plant defense responses at the protein and metabolite levels? Is this variation significant in determining the outcome of plant–pathogen interactions? Does this variation impact the fitness of the plant? To address these questions, it is essential to determine whether natural variation in SA responses of the transcriptome alters plant–pathogen interactions. Genetic approaches, such as analysis of defined genetic populations and association mapping within the species, can be used to identify genes controlling the gene-network variation. They also have potential to determine whether changes in the transcript levels of these genes alter the plants response to, and fitness under, pathogen attack. Well-established genetic approaches have the capability to expand our understanding of plant–pathogen interactions and how genetic variation and selection interact to modulate essential responses within a eukaryotic species.
METHODS
Plant Material and Experimental Conditions
Seeds of seven accessions of Arabidopsis thaliana (Col-0, Cvi-1, Est, Kin-0, Mt-0, Tsu-1, and Van-0) were kindly provided by J. Chory (Salk Institute, San Diego, CA). These accessions can be obtained from the European Arabidopsis Stock Center (http://arabidopsis.info) and from The Arabidopsis Information Resource (TAIR; www.arabidopsis.org).
We conducted a preliminary experiment to identify a level of exogenous SA application that caused transcript accumulation changes without visible phytotoxicity in any of the accessions tested (described in Kliebenstein et al., 2006a). The accessions varied in their sensitivity to exogenous SA application, with concentrations >0.40 mM SA causing symptoms of macroscopic phytotoxicity in some accessions (see Supplemental Figure 1 online). To identify physiologically relevant SA-mediated differences in transcript levels rather than differences in toxicity responses among the accessions, we used a 0.30 mM SA treatment. This treatment concentration was also found to induce pathogen-responsive genes (described in Kliebenstein et al., 2006a). Therefore, based on the results of the preliminary experiment, we chose to use two treatments, 0.30 mM SA plus a zero SA control. Using a factorial experimental design, we sampled three biological replicates each at three time points, 4, 28, and 52 hpt, to conduct global assays for ELPs among these seven accessions (126 samples). The sampling time points were spaced at 24-h intervals to avoid diurnal effects on measurement of transcript levels.
RNA Isolation and Microarray Analysis of the Transcriptome
RNA was purified from the 126 samples using the TRIzol extraction procedure followed by purification on RNeasy columns (Qiagen). Labeled cRNA was prepared and hybridized, according to the manufacturer's guidelines (Affymetrix), to whole genome Affymetrix ATH1 GeneChip microarrays containing 22,746 Arabidopsis transcripts. The GeneChips were scanned with an Affymetrix GeneArray 2500 scanner and data acquired via the Microarray Suite software MAS 5.0. The MAS 5.0 algorithm with default scaling was used to obtain gene expression levels for all data analyses. A quality control analysis was conducted to verify that each GeneChip was correctly matched to its corresponding biological sample, as described previously (Kliebenstein et al., 2006a). The data for the 126 GeneChips are publicly available from EBI ArrayExpress under accession number E-TABM-51 and from our project website (http://elp.ucdavis.edu/).
Statistical Analysis on a per-Gene Basis
A statistical analysis of transcript level abundance was conducted to test for differential gene expression, which represented potential gene ELPs among accessions, and to identify accessions with altered transcript levels resulting from SA treatment. A mixed linear model ANOVA was used to analyze the GeneChip data (Kliebenstein et al., 2006a). The mixed linear model partitions the transcript level variation (e.g., accession, treatment, time points, array, gene, and their respective interaction terms) for the purpose of testing differential transcript level accumulation (Craig et al., 2003). Our previous analysis of this data set showed that the presence of sequence polymorphisms that could possibly impact probe hybridizations was not a significant source of variation in gene expression estimates and that transcript accumulation differences among accessions were primarily due to actual ELPs (Kliebenstein et al., 2006a).
For each pair of accessions (21 in total), we performed an ANOVA using a split-plot mixed linear model with a random array effect. In this model, yijkgr denotes the transcript level accumulation of gene g, measured from the accession i under SA treatment j at the time point k for the chip replication r. The ANOVA model for the log-transformed expression levels is as follows:
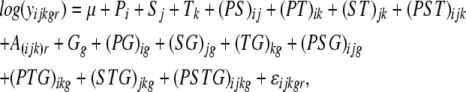 |
where i = 1, 2; j = 1, 2; k = 1, 2, 3; g = 1,…., 22,746; and r = 1, 2, 3. The main effects are denoted as P, S, T, and G and represent accession, SA treatment, time point, and gene, respectively. The array effect (A) is assumed to be distributed as a normal random variable with mean 0, and variance σA2; ɛijkgr represents the subplot error and is assumed to be normally distributed with mean 0 and variance σɛ2. All genes (22,746) were tested simultaneously in each pairwise ANOVA.
Two null hypotheses were employed to test for transcript level (gene expression) differences both within and between accessions. The first hypothesis (H01) tests if a gene was differentially expressed between treatments within an accession. Specifically, for each gene of each accession at each time point, H01: Sj + (SG)jg = Sj′ + (SG)j′g was tested. The second hypothesis (H02) was tested to identify potential ELPs. For each gene under each treatment condition at each time point, H02 tests differential expression between any two accessions: H02: Pi + (PG)ig = Pi′ + (PG)i′g. Comparisons of the t test results between accessions indicated if the gene significantly responded to the SA treatment in one accession but not in the other. For each of 21 accession pairs, t tests were used with the type I error adjusted for multiple comparisons using the false discovery rate (Holm, 1979; Benjamini and Hochberg, 1995). The accession analysis results were summarized based on the criterion of the total number of statistically significant differentially expressed genes as identified by one or both of the two hypotheses H01 and H02. We compared the number of statistically significant differentially expressed genes that satisfy this criterion for all 21 parental pairs to determine the maximum and minimum number of differences.
Analysis of Previously Reported SA-Responsive Genes
We used a collection of 37 genes shown previously to be responsive to SA treatment and/or pathogen/pest attack (see Supplemental Table 1 online) to test the SA response within our experiment. At a false discovery rate of 0.05 (Benjamini and Hochberg, 1995), nine of the 37 genes showed an SA response (i.e., transcript level differences). At a nominal 0.05 level, the number of genes identified as significantly SA responsive doubled, and the number of time points for which a gene was identified as SA responsive increased (see Supplemental Table 1 online). The results suggest that we are measuring SA responses of transcript levels within the seven accessions. However, not all of the 37 pathogen/SA-responsive genes identified in other experiments were detected as significantly SA responsive in our experiment. The difference may be due to the moderate level of exogenous SA used, the absence of pathogens, and/or the shorter time course employed in our experiment.
Defining Gene Networks by Responses of Col-0 to Different Treatments
We defined ad hoc defense-related gene networks a priori using publicly available Affymetrix ATH1 GeneChip Col-0 data generated from pathogen challenge experiments. The use of public data sets permitted the defining of defense-related groups of genes that were coregulated as putative gene networks independently from our own data set. GeneChip data from four experiments were considered: interactions with Pseudomonas syringae strains (Tao et al., 2003), interactions with Botrytis cinerea (data from the Integrated Microarray Database System; http://ausubellab.mgh.harvard.edu/imds/experiment_display.jsp?experiment_id=7), interactions with viruses (Whitham et al., 2003), and a treatment with oligogalacturonide (plant cell wall–derived molecule that stimulates defense gene expression during pathogenesis) (data from the Integrated Microarray Database System; http://ausubellab.mgh.harvard.edu/imds/experiment_display.jsp?experiment_id=1).
Gene classification was conducted in the following manner. In each experiment, characteristic expression profile patterns were identified using agglomerative hierarchical clustering (Eisen et al., 1998). Clusters were arbitrarily named, and genes were classified according to these expression patterns. When data for a particular gene were not available in a given experiment, the gene was classified into a pattern for genes with missing data. The expression pattern classes of gene groups were represented as a string of pattern names from different experiments (see Supplemental Table 2 online). For example, the class for a gene with expression pattern 2 in Experiment 1, pattern b in Experiment 2, and pattern 3 in Experiment 3 is designated as class 2b3. Note that the same pattern name from different experiments does not imply any biological relatedness. In total, 134 putative gene networks were identified as responsive to challenge by plant pathogens or selected elicitors. Given this definition of the gene networks, the average number of genes per network was 14.6, ranging from 2 to 385. We selected a subset of gene networks that contained a minimum of six genes (arbitrarily chosen) to minimize the influence of individual gene ELPs of large effect on a gene network (Kliebenstein et al., 2006b). Fifty-four defense-related putative gene networks (see Supplemental Table 3 online) resulted from this classification, with an average of 32 genes per gene network and a maximum of 385 genes.
Statistical Analyses of Gene Networks
We used a mixed linear model ANOVA in SAS to analyze the log2 transcript level (gene expression) data from our factorial experiment to determine which of the 54 defined gene networks showed a response to SA in at least one of the seven accessions. In this analysis, we used the expression values for the genes belonging to each of the gene networks. The transcript level of gene g from gene network n under SA treatment j at the time point k for the chip replication r is denoted as ygnjkr. The ANOVA model for the log2-transformed expression levels is as follows:
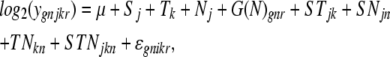 |
where g = 6, …,385; n = 1, …,54; j = 1, 2; k = 1, 2, 3; and r = 1, 2, 3. The main effects are denoted as G, N, S, and T and represent gene, gene network, treatment, and time point, respectively. The error, ɛgnikr, is assumed to be normally distributed with mean 0 and variance σɛ2.
For each of the three time points following treatment, the 95% confidence levels for each gene network were used to determine which gene networks were significantly different in expression between the control and SA treatments. The least-square means were used to determine whether the gene networks were up- or downregulated in response to SA treatment. This analysis indicated that the vast majority of the 25 SA-responsive gene networks were only responsive at the 4-hpt time point; therefore, only the data for 4 hpt was used for further analysis of the gene networks.
Subsequently, we used the 25 gene networks found to be SA responsive in at least one accession in our experiment to test for variation in gene-network responses among the accessions. A pairwise comparison was conducted between Col-0 and each of the other six accessions using the mixed linear model as described previously (i.e., genes nested within gene network), except that accession replaced time point as a main effect. Two different analyses using this same linear model were performed in SAS. The first analysis included the least-square means using 95% confidence intervals. Using an F-test, each gene-network × treatment effect was tested for a significant difference between pairs of accessions. We used the least-square means and 95% confidence intervals per gene network within each accession for each treatment condition to determine the accession for which the gene network was expressed at a higher level. As a second analysis, the least-square means and standard errors values were used to estimate the average response for the 25 SA-responsive gene networks.
Network Clustering Analysis
The 25 gene networks that responded to SA treatment in at least one of the seven accessions in our study were used to conduct network analysis on our data using LCF (Katagiri and Glazebrook, 2003). LCF uses nonlinear dimensionality reduction for pattern recognition and translates the results into a graphical representation. To conduct LCF, the data were split into two groups: control and treatment. Per group, we obtained the log2-transformed expression level values of each gene at 4, 28, and 52 hpt across accessions and replicates and then we normalized them using a z-score transformation as described by Kliebenstein et al. (2006b). The z-transformation has been shown to minimize the impact of cis-controlled transcript polymorphisms on estimates of gene-network expression (Kliebenstein et al., 2006b). All time points were used to maximize the similarity of our experimental data to the Col-0 data that included longer time courses. We then averaged our normalized (z-score) data for all the genes in a given gene network, after which we subjected the averaged values for the 25 gene networks to LCF. We visualized and analyzed the network output of LCF using PAJEK (http://vlado.fmf.uni-lj.si/pub/networks/pajek; Batagelj and Mrvar, 2002). For visualization, the Fruchterman-Reingold three-dimensional option was used.
Using the 25 gene networks, an independent LCF analysis was conducted for the 16 publicly available Arabidopsis ATH1 data sets (http://affymetrix.arabidopsis.info) (see Supplemental Table 4 online). These data sets include biotic stress, elicitor, and stress hormone treatments of Arabidopsis accession Col-0. For brevity, the collective data from these 16 data sets are hereafter referred to as 16ATH1 data. All CEL files were quantile normalized using RMAExpress version 0.4 (http://rmaexpress.bmbolstad.com/), split into two groups, control (112 arrays) and treatment (195 arrays), and subsequently analyzed as described above for comparison to the results of LCF analysis on our data set.
GO Analysis
The GO analysis was performed at two levels: GOslim terms (a high-level GO term for functional categorization) and GO terms. We performed the analysis using the GO annotation (download 20051119) from the TAIR website (Berardini et al., 2004; http://www.arabidopsis.org/). The frequency of each classification was obtained for the completely sequenced genome and for the genes present in the 25 defined gene networks that were upregulated and downregulated with SA treatment, respectively (see Results). We tested each gene list for significant deviation (P ≤ 0.001) from the expected frequencies for the complete genome using χ2 analysis (Chen et al., 2005).
Promoter Analysis
We used the cis-regulatory element annotation of promoter sequences of annotated Arabidopsis genes from the AGRIS AtcisDB database (Davuluri et al., 2003; http://arabidopsis.med.ohio-state.edu/), downloaded on November 29, 2005. The frequency of each cis-regulatory element was obtained for the complete genome and for the lists of genes from the upregulated and downregulated gene networks, respectively. We tested each gene list for significant deviation (P ≤ 0.001) from the expected frequencies for the complete genome using χ2 analysis (Chen et al., 2005).
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Variable Morphological/Phytotoxic Response to Exogenously Applied SA in Arabidopsis Accessions.
Supplemental Figure 2. GO Biological Process Annotation Analysis of SA-Regulated Gene Networks.
Supplemental Figure 3. GO Cellular Component Annotation Analysis of SA-Regulated Gene Networks.
Supplemental Figure 4. LCF Comparisons of Connections among SA-Responsive Gene Networks.
Supplemental Figure 5. Naturally Variable Gene-Network Responses to SA Detected with Pairwise ANOVA.
Supplemental Table 1. Analysis of Known Pathogen-, SA-, JA-, Wounding-, and Ethylene-Responsive Genes and Literature References.
Supplemental Table 2. Description of Gene-Network Numbering System.
Supplemental Table 3. Gene Lists for Each of the 54 Gene Networks.
Supplemental Table 4. Microarray Data Sets Used for LCF Analyses.
Supplemental Table 5. GO Analysis of Upregulated Networks.
Supplemental Table 6. GO Analysis of Downregulated Networks.
Supplemental Table 7. Promoter Analysis of SA Upregulated Networks.
Supplemental Table 8. Promoter Analysis of SA Downregulated Networks.
Supplementary Material
Acknowledgments
This research was supported by the National Science Foundation 2010 Project, Grant MCB-0115109 to D.A.S., R.W.M., and R.W.D., National Science Foundation Grant MCB-0323759 to D.J.K., and USDA–National Research Initiative Grant 2004-35301-14525 to F.K. Technical assistance was provided by Carol Lam, Rebecca Walker, and Tanya Tang.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantcell.org) is: Dina A. St.Clair (dastclair@ucdavis.edu).
Online version contains Web-only data.
Open Access articles can be viewed online without a subscription.
References
- Batagelj, V., and Mrvar, A. (2002). Pajek - Analysis and visualization of large networks. Graph Drawing Notes in Computer Science 2265 477–478. [Google Scholar]
- Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate - A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B 57 289–300. [Google Scholar]
- Berardini, T., et al. (2004). Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiol. 135 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borevitz, J.O., Liang, D., Plouffe, D., Chang, H.S., Zhu, T., Weigel, D., Berry, C.C., Winzeler, E., and Chory, J. (2003). Large-scale identification of single-feature polymorphisms in complex genomes. Genome Res. 13 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem, R.B., Yvert, G., Clinton, R., and Kruglyak, L. (2002). Genetic dissection of transcriptional regulation in budding yeast. Science 296 752–755. [DOI] [PubMed] [Google Scholar]
- Caicedo, A.L., Stinchcombe, J.R., Olsen, K.M., Schmitt, J., and Purugganan, M.D. (2004). Epistatic interaction between Arabidopsis FRI and FLC flowering time genes generates a latitudinal cline in a life history trait. Proc. Natl. Acad. Sci. USA 101 15670–15675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrol, S.B. (2000). Endless forms: The evolution of gene regulation and morphological diversity. Cell 101 577–580. [DOI] [PubMed] [Google Scholar]
- Chen, W.J., Chang, S.H., Hudson, M.E., Kwan, W.-K., Li, J., Estes, B., Knoll, D., Shi, L., and Zhu, T. (2005). Contribution of transcriptional regulation to natural variations in Arabidopsis. Genome Biol. 6 R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig, B.A., Black, M.A., and Doerge, R.W. (2003). Gene expression data: The technology and statistical analysis. J. Agric. Biol. Environ. Stat. 8 1–28. [Google Scholar]
- Cui, J., Bahrami, A.K., Pringle, E.G., Hernandez-Guzman, G., Bender, C.L., Pierce, N.E., and Ausubel, F.M. (2005). Pseudomonas syringae manipulates systemic plant defenses against pathogens and herbivores. Proc. Natl. Acad. Sci. USA 102 1791–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, J.P., Jander, G., Racki, L.R., Kim, P.D., Pierce, N.E., and Ausubel, F.M. (2002). Signals involved in Arabidopsis resistance to Trichoplusia ni caterpillars induced by virulent and avirulent strains of the phytopathogen Pseudomonas syringae. Plant Physiol. 129 551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davuluri, R.V., Sun, H., Palaniswamy, S.K., Matthews, N., Molina, C., Kurtz, M., and Grotewold, E. (2003). AGRIS: Arabidopsis Gene Regulatory Information Server, an information resource of Arabidopsis cis-regulatory elements and transcription factors. BMC Bioinformatics 4 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney, T.P., Friedrich, L., and Ryals, J.A. (1995). Arabidopsis signal transductionmutant defective in chemically and biologically induced disease resistance. Proc. Natl. Acad. Sci. USA 92 6602–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerge, R.W. (2002). Mapping and analysis of quantitative trait loci in experimental populations. Nat. Rev. Genet. 3 43–52. [DOI] [PubMed] [Google Scholar]
- Eisen, M.B., Spellman, P.T., Brown, P.O., and Botstein, D. (1998). Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney, T., Friedrich, L., Vernooij, B., Negrotto, D., Nye, G., Uknes, S., Ward, E., Kessmann, H., and Ryals, J. (1993). Requirement of salicylic acid for the induction of systemic acquired-resistance. Science 261 754–756. [DOI] [PubMed] [Google Scholar]
- Gassmann, W., Hinsch, M.E., and Staskawicz, B.J. (1999). The Arabidopsis RPS4 bacterial-resistance gene is a member of the TIR-NBS-LRR family of disease-resistance genes. Plant J. 20 265–277. [DOI] [PubMed] [Google Scholar]
- Glazebrook, J., Chen, W.J., Estes, B., Chang, H.S., Nawrath, C., Metraux, J.P., Zhu, T., and Katagiri, F. (2003). Topology of the network integrating salicylate and jasmonate signal transduction derived from global expression phenotyping. Plant J. 34 217–228. [DOI] [PubMed] [Google Scholar]
- Grant, M.R., Godiard, L., Straube, E., Ashfield, T., Lewald, J., Sattler, A., Innes, R.W., and Dangl, J.L. (1995). Structure of the Arabidopsis Rpm1 gene enabling dual-specificity disease resistance. Science 269 843–846. [DOI] [PubMed] [Google Scholar]
- Holm, S. (1979). A simple sequentially rejective multiple test procedure. Scand. J. Stat. 6 65–70. [Google Scholar]
- Johanson, U., West, J., Lister, C., Michaels, S., Amasino, R., and Dean, C. (2000). Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science 290 344–347. [DOI] [PubMed] [Google Scholar]
- Katagiri, F., and Glazebrook, J. (2003). Local Context Finder (LCF) reveals multidimensional relationships among mRNA expression profiles of Arabidopsis responding to pathogen infection. Proc. Natl. Acad. Sci. USA 100 10842–10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein, D., Lambrix, V., Reichelt, M., Gershenzon, J., and Mitchell-Olds, T. (2001). Gene duplication and the diversification of secondary metabolism: Side chain modification of glucosinolates in Arabidopsis thaliana. Plant Cell 13 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein, D., West, M., van Leeuwen, H., Loudet, O., Doerge, R., and St.Clair, D. (2006. b). Identification of QTLs controlling gene expression networks defined a priori. BMC Bioinformatics 7 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein, D.J., Pedersen, D., and Mitchell-Olds, T. (2002). Comparative analysis of insect resistance QTL and QTL controlling the myrosinase/glucosinolate system in Arabidopsis thaliana. Genetics 161 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein, D.J., West, M.A.L., van Leeuwen, H., Kyunga, K., Doerge, R.W., Michelmore, R.W., and St.Clair, D.A. (2006. a). Genomic survey of gene expression diversity in Arabidopsis thaliana. Genetics 172 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel, B.N., and Brooks, D.M. (2002). Cross talk between signaling pathways in pathogen defense. Curr. Opin. Plant Biol. 5 325–331. [DOI] [PubMed] [Google Scholar]
- Lambrix, V., Reichelt, M., Mitchell-Olds, T., Kliebenstein, D., and Gershenzon, J. (2001). The Arabidopsis epithiospecifier protein promotes the hydrolysis of glucosinolates to nitriles and influences Trichoplusia ni herbivory. Plant Cell 13 2793–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raacke, I.C., Mueller, M.J., and Berger, S. (2006). Defects in allene oxide synthase and 12-oxa-phytodienoic acid reductase alter the resistance to Pseudomonas syringae and Botrytis cinerea. J. Phytopathol. 154 740–744. [Google Scholar]
- Rao, M.V., and Davis, K.R. (1999). Ozone-induced cell death occurs via two distinct mechanisms in Arabidopsis: The role of salicylic acid. Plant J. 17 603–614. [DOI] [PubMed] [Google Scholar]
- Rao, M.V., Lee, H., Creelman, R.A., Mullet, J.E., and Davis, K.R. (2000). Jasmonic acid signaling modulates ozone-induced hypersensitive cell death. Plant Cell 12 1633–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan, S.E. (2000). Phenotypic plasticity for plant development, function and life history. Trends Plant Sci. 5 537–542. [DOI] [PubMed] [Google Scholar]
- Tang, D.Z., Ade, J., Frye, C.A., and Innes, R.W. (2006). A mutation in the GTP hydrolysis site of Arabidopsis dynamin-related protein 1E confers enhanced cell death in response to powdery mildew infection. Plant J. 47 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, Y., Xie, Z., Chen, W., Glazebrook, J., Chang, H.S., Han, B., Zhu, T., Zou, G., and Katagiri, F. (2003). Quantitative nature of Arabidopsis responses during compatible and incompatible interactions with the bacterial pathogen Pseudomonas syringae. Plant Cell 15 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veronese, P., Nakagami, H., Bluhm, B., AbuQamar, S., Chen, X., Salmeron, J., Dietrich, R.A., Hirt, H., and Mengiste, T. (2006). The membrane-anchored BOTRYTIS-INDUCED KINASE1 plays distinct roles in Arabidopsis resistance to necrotrophic and biotrophic pathogens. Plant Cell 18 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via, S., Gomulkiewwicz, R., De Jong, G., Scheiner, S.M., Schlichting, C.D., and van Tienderen, P.H. (1995). Adaptive phenotypic plasticity: Consensus and controversy. Trends Ecol. Evol. 10 179–220. [DOI] [PubMed] [Google Scholar]
- Wang, R.L., Stec, A., Hey, J., Lukens, L., and Doebley, J. (1999). The limits of selection during maize domestication. Nature 398 236–239. [DOI] [PubMed] [Google Scholar]
- Werner, J.D., Borevitz, J.O., Warthmann, N., Trainer, G.T., Ecker, J.R., Chory, J., and Weigel, D. (2005). Quantitative trait locus mapping and DNA array hybridization identify an FLM deletion as a cause for natural flowering-time variation. Proc. Natl. Acad. Sci. USA 102 2460–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, M.A.L., Kim, K., Kliebenstein, D.J., van Leeuwen, H., Michelmore, R.W., Doerge, R.W., and St.Clair, D.A. (2007). Global eQTL mapping reveals the complex genetic architecture of transcript-level variation in Arabidopsis. Genetics 175 1441–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitham, S.A., Quan, S., Chang, H.S., Cooper, B., Estes, B., Zhu, T., Wang, X., and Hou, Y.M. (2003). Diverse RNA viruses elicit the expression of common sets of genes in susceptible Arabidopsis thaliana plants. Plant J. 33 271–283. [DOI] [PubMed] [Google Scholar]
- Zheng, Z.Y., Abu Qamar, S., Chen, Z.X., and Mengiste, T. (2006). Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 48 592–605. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.