

Abstract
Context
The P450 enzyme aromatase (CYP19) plays a crucial role in the endocrine and paracrine biosynthesis of estrogens from androgens in many diverse estrogen-responsive tissues. Complete aromatase deficiency has been reported in a small number of 46,XX girls with genital ambiguity and absent pubertal development, but it is unknown whether non-classic phenotypes exist.
Objective
The objective of the study was to determine whether variant forms of aromatase insufficiency can occur in humans.
Patients
Four patients (46,XX) from three kindred with variable degrees of androgenization and pubertal failure.
Methods
Mutational analysis of CYP19 and assay of enzyme activity.
Results
Aromatase insufficiency resulting in genital ambiguity at birth, but with variable breast development at puberty (B2-B4), occurred in 46,XX patients from two kindred who harbored point mutations or single codon deletions (R435C, F234del). Absent puberty with minimal androgenization at birth was found in one girl with a deletion involving exon5 of CYP19 (exon5del), which would be predicted to lead to an in-frame deletion of 59 amino acids from the enzyme. Functional studies revealed low residual aromatase activity in the cases where breast development occurred.
Conclusions
These studies demonstrate that aromatase mutations can produce variable or “non-classic” phenotypes in humans. Low residual aromatase activity may be sufficient for breast and uterine development to occur at puberty, despite significant androgenization in utero. Such phenotypic variability may be influenced further by modifying factors, such as non-classic pathways of estrogen synthesis, variability in co-regulators, or differences in androgen responsiveness.
Keywords: aromatase, CYP19, steroidogenesis, ambiguous genitalia, puberty, breast development, uterus, aromatase inhibitors
Introduction
Aromatase (CYP19A1, 15q21.2) is a cytochrome P450 enzyme (P450arom) that plays a crucial role in the biosynthesis of estrogens (C18 steroids) from androgens (C19 steroids) in all vertebrate species (1). In humans, aromatase (CYP19) expression is regulated by different tissue-specific promoters in the placenta, ovary, breast, bone, adipose tissue, vascular endothelium, and brain, resulting in systemic (gonadal/ovarian) and local (extragonadal) estrogen production in these tissues (1,2). Aromatase has important biological effects at different stages of development. For example, aromatase expression in the fetally-derived placenta protects the mother from the potentially androgenizing (virilizing) effects of fetal adrenal androgens during pregnancy whereas ovarian (endocrine) and breast (paracrine/intracrine) aromatase expression is necessary for estrogen-dependent breast development at puberty (1). In addition, aromatase mediates uterine growth and bone maturation during adolescence, and influences bone mineralization, lipid metabolism and cardiovascular risk through into adult life (1,3).
Although targeted deletion of the gene encoding aromatase (Cyp19) in mice is providing fascinating insight into the role of this enzyme in endocrine function, metabolism, cardiovascular function, and fertility (4-7), our understanding of the role of aromatase in human biology has been furthered by several individual case reports of patients with complete aromatase deficiency (OMIM: 107910). Karyotypic (46,XX) females with this condition (n=6) present with androgenization of the external genitalia at birth, elevated androgens and undetectable estrogens, and complete lack of breast development at puberty (8-14). Males (46,XY) with aromatase deficiency (n=7) usually present after puberty with prolonged linear growth and tall stature, reduced bone mineralization, impaired fertility, insulin insensitivity, and dyslipidemia (3, 15-20). Maternal virilization during pregnancy occurs with fetuses of both sexes, following the transplacental passage of fetal androgen precursors into the maternal circulation due to a lack of fetal placental aromatase action. Furthermore, polymorphic variability within the aromatase (CYP19) locus has been reported in association with variations in bone mineral density and fracture risk in both sexes (21-23), hyperandrogenism in younger females (24), and survival in patients with metastatic prostate cancer (25), suggesting an important modulatory role for this enzyme in endocrine and metabolic function within the wider population.
Despite the potential deleterious consequences of aromatase depletion, aromatase inhibitors are emerging as important pharmacological strategies for treating growth disorders (26), endometriosis (27), and breast cancer (28-31). Thus, the identification and characterization of patients with aromatase insufficiency provides useful structural and functional information about the role of this enzyme in humans.
Here we report the clinical, biochemical and genetic features of variable aromatase insufficiency in a series of four 46,XX patients from three families with point mutations or deletions within the CYP19 gene.
Methods
DNA sequence analysis
After obtaining Institutional Review Board approval and informed consent from the patients and parents, DNA was extracted from patients' blood leukocytes or saliva using standard methods. All nine coding exons and proximal splice sites of CYP19 were PCR amplified as described previously (12), and sequenced with a MegaBACE1000™ capillary DNA sequencer (Amersham Biosciences).
Microdeletion detection
Following an inability to PCR-amplify exon 5 of CYP19 in subject III:2 using different primers and conditions (12), a long-range PCR strategy was adopted to determine whether a small genomic deletion encompassing exon 5 had occurred. Primer pairs were designed at increasing intervals around exon 5. A 3.58 kb PCR product was eventually obtained from the patient's DNA with a forward primer in intron 4 (“Int4F.1” [c.452-2780_c.452-2761]: 5'-CCT CTA CCC TGA CAT GCA AG-3') and reverse primer in intron 5 [“Int5R.1” (c.628+2208_c.628+2227]: 5'-CAT TGT TCC TCC GCC TGG AG-3'). A corresponding 5.18 kb PCR product was obtained in wild-type DNA. The genomic deletion in the patient was confirmed by direct sequencing of the PCR product and by the generation of a predicted 352 base pair PCR product using primers located on either side of the deletion (“Int4F.2” [c.452-846_c.452-821]: 5'-GTGTAGGCCACCTACCATCAGGACCC-3' and “Int5R.2” [c.628+908_c.628+930]: 5'-CCAGGTTAGTGTGTGGATAAAGG-3'), but absence of a 279 base pair product using primers close to exon 5 (“Int4F.3” [c.452-63_c.452-43]: 5'-TGCATGATTGTGGTGTGTGCC-3' and “Int5R.3” [c.628+20_c.628+39]: 5'-GGACAGATGGTCAAGATGTG-3').
Endocrine assessment and bone densitometry
Follicle stimulating hormone (FSH) and luteinizing hormone (LH) concentrations were measured using standard radioimmunoassay kits. Testosterone and estradiol were measured by chemiluminescence (Bayer ACS 180, DPC Immulite 2000, Roche Modular). Androstenedione was measured by RIA (Coat-A-Count, DPC). Bone mineral density was assessed at the lumbar spine (L1–L4) using dual-electron x-ray absorptiometry (Hologic QDR-1000) and expressed as a z-score.
Structural modeling
The positions of the point mutations within the tertiary structure of aromatase were predicted using a three-dimensional homology model of human aromatase based on CYP2C9 (PDB code 1TQA) (32). Images were generated with MidasPlus software on a Silicon Graphics (Mountain View, CA) Octane workstation.
Generation of mutant cDNA constructs
Site-directed mutagenesis (Stratagene) was used to create the aromatase point mutants (R435C, F234del) using wild-type (WT) human aromatase cDNA in a pcDNA3.1(+) vector as a template. The exon5del mutant was constructed using an overlapping PCR strategy to create a novel AfeI restriction site in the cDNA template. This site was then used to ligate exon 4 of the cDNA to exon 6 of the cDNA within the pcDNA3.1(+) vector, allowing deletion of exon 5 (codons 151-209) whilst leaving the reading frame and amino-acid sequence unaltered. The entire cDNA sequences of all mutant constructs were verified prior to further studies.
RT-PCR of aromatase from transfected cells
RNA from transiently transfected COS7 cells was extracted using the Trizol methods and RTPCR was performed (Access Quick RT-PCR system, Promega) for aromatase using a forward primer located within exon 4 (“Ex4F”: 5'-GCATCGGTATGCATGAGAAAGG-3') and a reverse primer located within exon 7 (“Ex7R”: 5'-CAGGTCACCACGTTTCTCTGC-3'). Placental RNA (with high aromatase expression) and GAPDH were used as positive controls.
Functional studies of aromatase activity
COS7 cells were transiently transfected with wild-type or mutant cDNA (0.8 μg/well) using Lipofectamine (Invitrogen). A β-galactosidase expression plasmid (pSV-β-galactosidase control vector, Promega) was co-transfected in a molar ratio of 1:2. Aromatase activity was determined 24 hr later by the production of 3H2O from the substrate [1β-3H]androstenendione (Perkin Elmer), using methods described previously (33). In one set of studies, a saturated point assay was performed by incubating cells in 80 nM final concentration of [1β-3H]androstenendione in serum free medium for 6 hr. In another set of studies, a saturated curve assay was performed by incubating transfected cells with 0, 5, 10, 20, 50, 100, 200 nM final concentration of [1β-3H]androstenendione in serum free medium for 3 hr. Aromatase activity was calculated by measuring radioactivity with a scintillation counter and adjusting for transfection efficiency using β-galactosidase activity, and total protein expression as determined by standard Bradford assay (Bio-Rad). Aromatase expression and protein size was confirmed by immunoblot (Western) analyses with a mouse anti-human aromatase antibody (directed to codons 376-390) (Serotec). All experiments were performed in triplicate, on at least three separate occasions. Kinetic constants were calculated from plots of 1/v versus 1/[S].
Enzyme stability and localization
WT and mutant aromatase cDNAs were cloned into a pAcGFP-C1 vector (Clontech) to produce a protein with GFP fused in-frame to the aminoterminus of aromatase. Empty vector, WT and mutant aromatase constructs (0.8μg) were transfected in COS7 cells using Lipofectamine 2000 (Invitrogen). After 24 hours, cells were fixed and nuclear counterstaining performed with Vectashield containing DAPI (Vector Laboratories). Cells were visualized using a Zeiss Axioskop microscope and camera. Endoplasmic reticulum immunofluorescence was performed using a primary polyclonal anti-calnexin antibody (kindly provided by Dr Gao Bin, UCL Institute of Child Health) at 1:500 dilution, and a secondary rhodamine TRITC porcine anti-rabbit antibody (DakoCytomation) at a dilution of 1:100.
Results
Case histories
Kindred I, Subject 1 (I:1) (R435C)
Subject I:1 (Fig. 1A) is a Turkish girl who presented with ambiguous genitalia (Prader stage IV) at birth. A history of maternal voice changes was noted during pregnancy. At 13.5 yr of age she was referred for further evaluation and clitoroplasty, and was found to have androgenized external genitalia (Prader IV), Tanner stage 2 breast development and Tanner stage 3 pubic hair. Karyotype was 46,XX, basal gonadotropin (FSH, LH) and androgen (androstenedione, testosterone) concentrations were elevated, and estradiol was low but detectable (Table 1). Adrenal steroidogenic defects were excluded. Pelvic ultrasound revealed a 6.3cm uterus with thin endometrial stripe and bilateral cystic ovaries (mean uterine length for B2, 4.1±0.3 cm). Bone age was delayed by 2.5 yr. Her breast development did not progress further than Tanner stage 2 and she complained of facial hair at 14.8 yr of age. Thus, ethinylestradiol was used to fully induce breast development and cyproterone acetate was given to prevent further hair growth.
Figure 1.

Aromatase insufficiency due to a R435C mutation in CYP19. A) Kindred I. B) The proband is homozygous for a C to T transversion resulting in an arginine to cysteine mutation at codon 435. C) The affected amino acid is a highly conserved residue in the heme-binding region of aromatase from other species, as well as from other cytochrome P450 enzymes. h, human; r, rat; m, mouse; c, chicken.
Table 1.
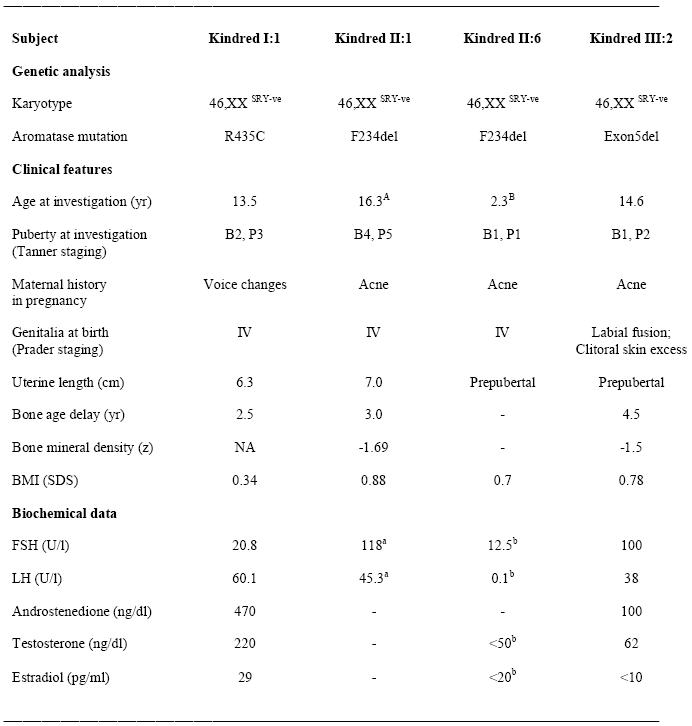
Genetic, clinical and biochemical features of patients with variable aromatase insufficiency.
a data obtained 18 months after gonadectomy;
b data obtained at 2.3 years of age at a time of relative quiescence of the hypothalamic-pituitary-gonadal axis. B, breast stage (Tanner I-V); P, pubic hair development (Tanner I-V); Prader staging of genitalia, I (female) to V (male); FSH, follicle-stimulating hormone; LH, luteinizing hormone; NA, not available; BMI, body mass index; SDS, standard deviation score. Normal ranges: FSH 1.1-5.6 U/l (2 yrs old), 0.6-6.8 U/l (pubertal B2), 1.6-10.5 (adult women); LH <0.1 U/l (2 yrs old), <0.1-4.3 U/l (pubertal B2), 1.9-26 (ovulatory adult women); androstenedione 60-290 ng/dl (pubertal); testosterone 5-90 ng/dl (pubertal); estradiol, <5-80 pg/ml (pubertal B2), 20-290 pg/ml (adult women). Mean uterine length at B2, 4.1±0.3cm; mean uterine length at B4, 6.2±0.4cm. Conversion to SI units: androstenedione ng/dl × 0.0349 for nmol/l; testosterone ng/dl × 0.0347 for nmol/l; estradiol pg/ml × 3.67 for pmol/l.
Mutational analysis of CYP19 revealed a homozygous R435C (CGT to TGT) mutation in the patient (Fig. 1B). Her parents are heterozygous for this change. This arginine at position 435 forms part of the crucial heme-binding region motif and is absolutely conserved in P450 aromatase from different species as well as other cytochrome P450 enzymes (fig. 1C). Although the crystal structure of aromatase has not yet been solved, modeling of the heme-binding region based on the crystal structure of CYP2C9 (PDB code 1TQA) predicts that the arginine at position 435 plays an important role in domain stabilization as part of a network of arginine residues (R115, R145, R375 and R435), which form hydrogen bonds with the propionate groups of the heme moiety (32). This salt-bridge bonding would likely be lost following replacement of the arginine at 435 with a non-charged cysteine residue (Fig. 2). Of note, a homozygous mutation (R375G) involving an arginine residue in this network is one of the few other missense mutations in aromatase reported to date (11) and this R435C mutation has been reported previously in a compound heterozygous state with C437Y (Fig. 2) (9).
Figure 2.
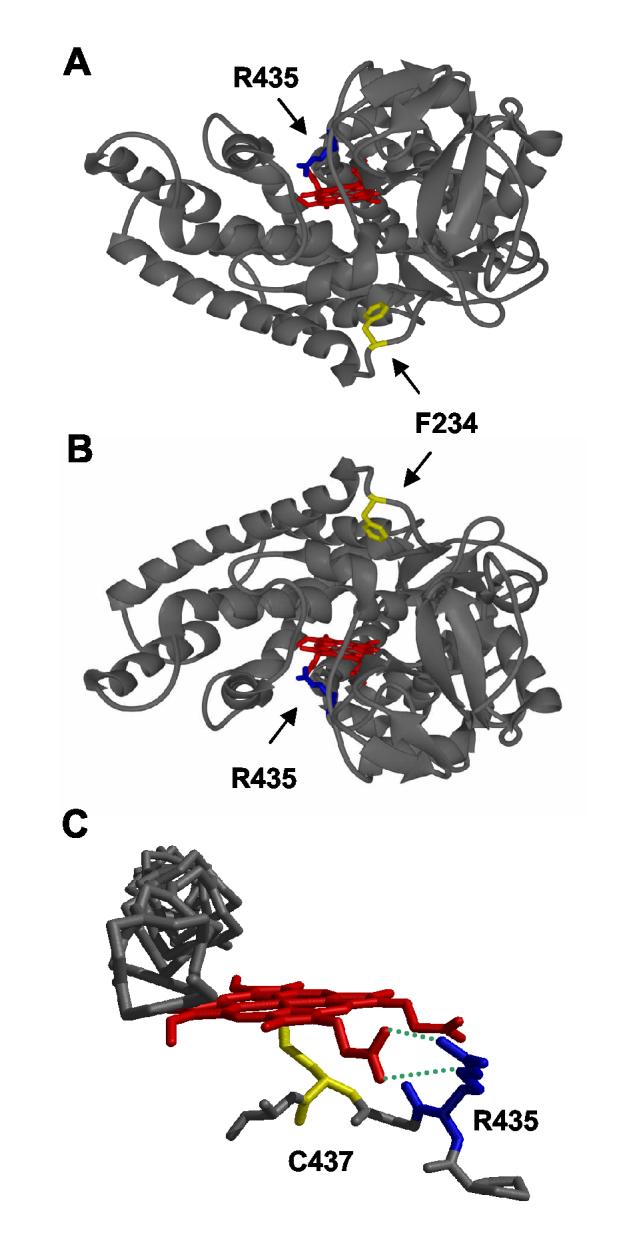
Position of the point mutations causing aromatase insufficiency with breast development (R435C, F234del) using a three-dimensional homology model of human aromatase based on the crystal structure of CYP2C9 (PDB code 1TQA) (32). A & B) The R435C mutation affects a residue (shown in blue) that interacts directly with the heme propionate (red). The phenylalanine deletion (shown in yellow) affects a residue that lies between the F- and G-helices, in a region that may be involved in membrane tethering of the enzyme. C) Localized view of the interaction between the charged arginine residue at codon 435 and the heme-moiety through hydrogen bonding to a carboxylate group. Substitution of this arginine for a non-polar cysteine is predicted to destabilize heme binding, resulting in reduced enzyme activity. A model of the exon 5 deletion was not generated as we feel that the large structural alterations caused by such a deletion are beyond the capabilities of available methods to generate a reasonable model.
Kindred II: Subjects 1 & 6 (II:1, II:6) (F234del)
Subjects II:1 and II:6 are from a consanguineous Pakistani family (Fig. 3A). The proband (II:1) was born with phallic enlargement and hypospadias (Prader IV), following a history of maternal acne during pregnancy. Karyotype was found to be 46,XX and a uterus and ovaries were identified on pelvic ultrasound (Table 1). However, he was raised male, reported male gender identity and exhibited male gender role behavior. Gynecomastia developed at the time of puberty, which progressed into Tanner stage 4 breast development by 14 yr of age (normal adult breast development, stage 4-5). This situation caused profound psychological distress; he wore tight clothing and ultimately attempted self-mutilation. Thus, salpingoophorectomy and hysterectomy was performed in an attempt to reduce secretion of sex hormones and arrest breast development. Histology revealed cystic ovarian tissue, a well-estrogenized uterus (7 cm length; mean uterine length for B4, 6.2±0.4 cm) and tortuous Fallopian tubes. He was referred for further assessment at 15 yr of age, prior to mastectomy. A 46,XX karyotype was confirmed, adrenal steroidogenic defects were excluded, and gonadotropin concentrations were elevated consistent with prior gonadectomy (Table 1). He had a 4cm phallus. Bone age was delayed by 3 yr, bone densitometry showed a z-score of −1.69 and a borderline low HDL-cholesterol was detected 18 months after oophorectomy (HDL-cholesterol 33 mg/dl [>35 mg/dl, males]). Breast reduction surgery was performed and he was commenced on testosterone supplementation, with no significant increase in phallic length.
Figure 3.
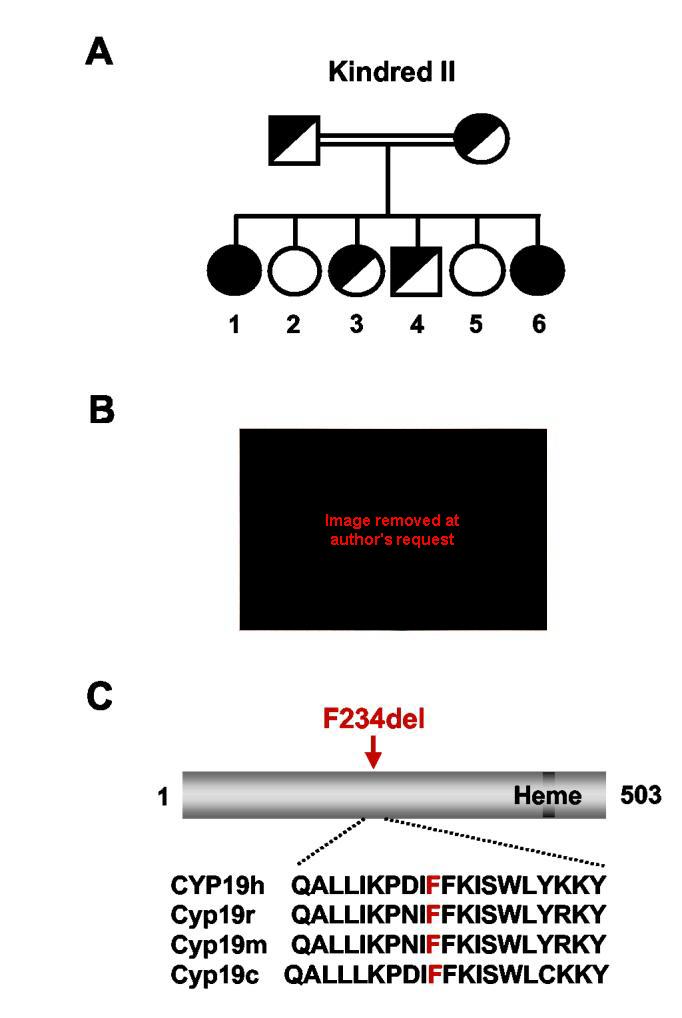
Aromatase insufficiency due to an F234del mutation in CYP19. A) Kindred II showing the affected individuals, II:1 and II:6. B) Both children with the homozygous F234del mutation presented with ambiguous genitalia at birth. The older child (II:1) was raised male but experienced significant breast development (Tanner stage IV) at puberty requiring oophorectomy and mastectomy (images not shown). The younger child presented with ambiguous genitalia (shown here) with a 2cm phallus and scrotalized labia, and was raised female. She remains prepubertal. C) The phenylalanines at position 234-235 is highly conserved in aromatase from other species, but not conserved in other P450 enzymes. The exact biological function of this region is not known, although it is likely involved in membrane tethering of the enzyme. h, human; r, rat; m, mouse; c, chicken.
A younger sibling (II:6, Figure 3A) was also found to have significant androgenization at birth (Prader IV). A 46,XX karyotype was found and an infantile uterus was identified on pelvic ultrasound. She was raised female. Maternal acne had occurred in this pregnancy, but not in intervening pregnancies (II:2 to II:5). She was noted to have a 2cm phallus and partial labial fusion on examination at this time (Fig. 3B).
Mutational analysis of CYP19 revealed a homozygous deletion of one of two phenylalanine residues at position 234-235 (TTCTTT to TTT; F234del) in both affected individuals (II:1, II:6). Parents were heterozygous for this change (Fig. 3A), and unaffected siblings were heterozygous or wild-type. These paired phenylalanine residues are highly conserved in P450 aromatase from other species but there is little homology with other P450 enzymes (Fig. 3C). Homology modeling of human aromatase suggests that these phenylalanine residues lie between the F- and G-helices, in a region that is likely to be involved in membrane tethering of the enzyme to the endoplasmic reticulum (Fig. 2) (32).
Kindred III: Subject 2 (III:2) (exon5del)
Subject III:2 is the second child of consanguineous Sri Lankan parents born following a history of maternal acne during the third trimester of pregnancy (Fig. 4A). She was noted to have labial fusion and an excess of clitoral skin at birth (Fig. 4B), and underwent surgery at 2 yr of age. She developed bilateral slipped femoral epiphyses in early adolescence and was referred for further evaluation as she failed to enter puberty by 14.6 yr of age. She had no breast development and a bone age delay of 4.5 yr. Her clitoral size was within normal limits and a urogenital sinus with a single opening was found on examination. Her gonadotropins were elevated, serum estradiol was undetectable and relatively low concentrations of circulating testosterone were measured (Table 1). She had mild dyslipidemia (fasting total cholesterol 211 mg/dl [88-192]; HDL-cholesterol 42 mg/dl [46-65, females]; LDL-cholesterol 150 mg/dl [<135]) and a lumbar spine bone mineral density z-score of −1.5. Karyotype was 46,XX, from blood, skin and genital skin. Pelvic ultrasound revealed a prepubertal uterus but could not easily visualize ovaries. An MRI scan was performed that showed the presence of small ovaries. Ethinylestradiol treatment was started, resulting in normalization of the lipid profile.
Figure 4.

Aromatase insufficiency associated with a homozygous 1600 bp deletion encompassing exon 5 of CYP19. A) Kindred III showing the affected individual, III.2. B) Labial fusion and excess clitoral skin was noted at birth. C). PCR-amplification of CYP19 failed to produce a product for exon 5 in the patient, III.2. D) Long-range PCR using primers in intron 4 (Int4F.1) and intron 5 (Int5R.1) produced a 5.18 kb product in control DNA, a 3.58 kb product in the patient, and both 5.18 kb and 3.58 kb products in the father. E) Cartoon of the genomic structure of CYP19 showing deletion of exon 5 in the patient. Direct sequencing of the PCR product showed a deletion between two ACT nucleotide sequences, designated c.452-621_c.628+803. F) A PCR-based strategy using primers located inside and outside the deleted region was used to confirm the deletion in the patient's DNA (lymphocytes, saliva) and in samples from her father and control (WT). Primer positions are indicated in the right hand panel. Amplification using primers around the deletion (Int4F.2 and Int5R.2) generated the predicted 352 base pair product in DNA samples from the proband and her father, but not from control WT DNA (lanes 1-5). Primer pairs in close proximity to exon 5 (Int4F.3 and Int5R.3) generated the predicted 279 base pair product in the father and control.
Aromatase (CYP19) was considered to be a candidate gene in this patient given the history of maternal acne in pregnancy, mild virilization at birth and evidence of estrogen deficiency (slipped femoral epiphyses, severely delayed bone age, absent breast development). No CYP19 mutations were detected, but exon 5 failed to amplify by PCR (Fig. 4C). Thus, a long-range PCR strategy with primer pairs located at increasingly greater intervals was used in an attempt to detect small deletions in this region of 15q21.2. Ultimately, it was possible to amplify a product and show deletion of a 1600 bp fragment encompassing exon 5 (Fig. 4D). This deletion was confirmed to be between c.452-621 and c.628+803 using direct sequencing and PCR-based strategies (Fig. 4E & 4F). The patient (III:2) appeared to be homozygous for this change and her father appeared to be heterozygous. Her mother could not be tested, but the parents were known to be consanguineous, and 11 polymorphic markers within the CYP19 locus are all homozygous (data not shown). This 1600 bp deletion is predicted to remove exon 5, resulting in the in-frame deletion of 59 amino acids from the P450 aromatase enzyme between codons 151-209 (Fig. 4E).
Aromatase expression and localization
In vitro studies of wild-type and mutant aromatase expression showed no differences in RNA stability or protein translation using RT-PCR (Fig. 5A) and immunoblot/Western analysis (data not shown). GFP-aromatase fusion proteins appeared to be expressed normally and localized to cytoplasm of the cell, largely within the endoplasmic reticulum (Fig. 5B).
Figure 5.

Aromatase activity assays. A) RT-PCR using RNA extracted from COS7 cells transfected with empty vector, wild-type or mutant aromatase cDNA showed stable RNA transcripts of the correct predicted size. Placental RNA was used as a positive control (lane 1). B) GFP-aromatase fusion proteins showed stable expression and appropriate cytoplasmic localization. DAPI staining was used to identify the nucleus and an anti-calnexin antibody was used to localize the endoplasmic reticulum. C) Velocity versus concentration curves of wild-type and mutant aromatase activity. COS7 cells were transiently transfected with wild-type ( ), F234del (■), R435C, (▲) or exon5del (data not shown) expression vectors. Twenty-four hours later these cells were incubated for 3 hours with increasing concentrations of the substrate, [1β-3H]androstenendione. Aromatase activity at each point was calculated by measuring 3H2O released from the substrate. Data represent the mean±SEM for triplicate experiments. The entire study was performed on three occasions and typical data are shown. At saturation, the F234del mutant showed 16% of wild-type activity, and the R435C mutant showed 0.7% activity. The exon5del mutant showed complete loss-of-function (data not shown). Lineweaver-Burke transformation generated kinetic parameters that showed that the F234del mutant protein had low-level residual function consistent with partial aromatase activity, but preserved substrate binding (see results). D) Wild-type and mutant aromatase activity in a saturated point tritiated androstenedione assay (80 nM [1β-3H]androstenedione, 6 hour incubation). Representative data are shown as mean± SEM for triplicate experiments. Percentage of wild-type activity is shown in parentheses.
), F234del (■), R435C, (▲) or exon5del (data not shown) expression vectors. Twenty-four hours later these cells were incubated for 3 hours with increasing concentrations of the substrate, [1β-3H]androstenendione. Aromatase activity at each point was calculated by measuring 3H2O released from the substrate. Data represent the mean±SEM for triplicate experiments. The entire study was performed on three occasions and typical data are shown. At saturation, the F234del mutant showed 16% of wild-type activity, and the R435C mutant showed 0.7% activity. The exon5del mutant showed complete loss-of-function (data not shown). Lineweaver-Burke transformation generated kinetic parameters that showed that the F234del mutant protein had low-level residual function consistent with partial aromatase activity, but preserved substrate binding (see results). D) Wild-type and mutant aromatase activity in a saturated point tritiated androstenedione assay (80 nM [1β-3H]androstenedione, 6 hour incubation). Representative data are shown as mean± SEM for triplicate experiments. Percentage of wild-type activity is shown in parentheses.
Aromatase activity assays
Tritiated androstenedione assays were used to assess mutant aromatase activity with both 3 hr saturated curve assays (0-200 nM [1β-3H]androstenedione) and 6 hr saturated point assays (80 nM [1β-3H]androstenedione) (Fig. 5C&D) (33). The R435C change was found to have 0.7-1.5% wild-type activity, whereas the F234del mutant was found to have 16-19% wild-type activity (Fig. 5A&B) (Vmax = 667 (F234del) vs. 3330 (wild-type) pmol/mg protein·3h; Km = 83 (F234del) vs. 170 (wild-type) nM). The exon5del mutant was found to be completely inactive.
Discussion
Aromatase is well-established as the critical regulator of the conversion of androgens (C19) to estrogens (C18), but relatively few cases of aromatase insufficiency have been reported in humans to date. The description here of four patients with variable forms of aromatase insufficiency highlights the range of phenotypes that can be associated with mutations in this cytochrome P450 enzyme in humans.
The first patient (I:1) described has aromatase insufficiency due to a homozygous R435C mutation. This arginine forms part of a network that contributes hydrogen bonds to stabilize the heme-group structure (32, 34) and an R435C mutation has been reported previously in a compound heterozygous state (with C437Y) in a patient with complete aromatase insufficiency and a lack of pubertal development (9,10). Previous studies showed limited residual function of the R435C mutant enzyme (1.1%), similar to our observations (0.7-1.5%), whereas the C437Y mutation was essentially totally inactive (9). These cases confirm the importance of the heme-binding region for substrate conversion by cytochrome P450 enzymes. Furthermore, although the degree of aromatase activity might have been expected to track with the less severely disrupted allele (R435C) in the compound heterozygous patient (9, 10), our patient demonstrates that low levels of residual aromatase activity may be sufficient for limited breast development (Tanner stage B2) and uterine growth, with detectable circulating estradiol in the presence of elevated androgens.
Affected children in the second kindred showed significant pre-natal androgenization, yet the proband (II:1) reached Tanner stage 4 breast development at puberty. The homozygous deletion of a single phenylalanine residue at position 234-235 occurs between the predicted F and G helices and affects a region potentially involved in membrane tethering of the enzyme (32). Thus, substrate binding seems largely unaffected, but maximal enzymatic activity is reduced. Tritiated androstenedione assays show that this F234del mutant has between 16-19% wild-type activity, consistent with the significant uterine growth and breast development seen in the proband, but surprising given the degree of androgenization seen at birth and maternal history of acne during these two pregnancies. Of note, the oldest child (II:1) was raised male, reported a male gender identity and demonstrated male sex role behavior and gender dysphoria, which is unusual in severely androgenized 46,XX individuals with other steroidogenic defects (e.g., 21-hydroxylase deficiency) (35). Whether pre- and post-natal androgen exposure coupled with relative estrogen insufficiency – a unique feature of aromatase insufficiency – is important, or whether social and cultural influences are predominant, is unclear. It will be of interest to monitor gender identity and sex role behavior in the youngest child (II:6), who was raised female at birth, but with an apparently similar endocrine and genetic milieu.
Taken together, these cases of aromatase insufficiency show that low levels of residual aromatase activity can be associated with breast development and estrogen biosynthesis, especially when circulating androgenic substrate concentrations (androstenedione, testosterone) are elevated. Although it is possible that aromatase-independent pathways exist for the biosynthesis of alternative hormones with estrogenic activity from androgens (36), it is currently unknown whether these alternative pathways occur in humans. Alternatively, tissue specific (e.g., breast) upregulation of partially functional enzymes due to variability in tissue-specific co-regulators (e.g. P450 oxidoreductase) could have occurred in individual cases, the degradation of estrogenic or androgenic compounds might vary, and end-organ estrogen and/or androgen responsiveness may differ between cases. Clearly, these alternative mechanisms are not evident in the patients reported to date who have complete aromatase insufficiency, as they fail to show any evidence of estrogenization at puberty. Furthermore, the functional studies of aromatase activity shown here are generally consistent with the degree of estrogenization: 0.7-1.5% aromatase activity achieving Tanner breast stage 2 and moderate uterine development, and 16-19% aromatase activity allowing progression to breast stage 4 and full uterine growth. Nevertheless, individual differences in the synthesis and responsiveness to estrogens and androgens may have contributed to the limited breast development in Kindred I, and the unexpected degree of androgenization in both patients in Kindred II.
The fourth patient reported here (III:2) with severe aromatase insufficiency is the most challenging to our concepts of aromatase action, as her mother showed only mild virilization (acne) during the pregnancy, only limited androgenization of the child's genitalia was seen post-natally, and androgen concentrations were only moderately elevated at puberty. Nevertheless, there was a complete failure of pubertal development with small rather than enlarged ovaries at adolescence. The 1600 bp deletion encompassing exon 5 is predicted to result in an in-frame deletion of 59 amino acids from the enzyme. Functional studies showed complete lack of enzyme activity when the predicted mutant protein was expressed in an in vitro cell system suggesting that aromatase activity is severely disrupted in this individual. Unfortunately, it was not possible to confirm the presence of an altered transcript in the patient's RNA, although all our data supported the presence of this highly unusual homozygous genomic deletion in the proband and a heterozygous state in the father. Given this unusual combination of features, we cannot totally exclude an additional pathology, such as a form of partial ovarian dysgenesis, 46,XX gonadal dysgenesis with partial testicular differentiation, or a defect in androgen synthesis pathways. However, no unifying biochemical or genetic block can easily account for the range of pre- and post-natal features seen. Although aromatase may play a role in maintaining ovarian integrity in certain species (37-40), it is not appropriate to extrapolate these studies to humans at present. Thus, the typical ovarian phenotype of human aromatase insufficiency is an enlarged, cystic pattern, which has been seen in other 46,XX individuals reported to date including the other patients with variable aromatase insufficiency reported here.
Taken together, these cases show the phenotypic variability that can occur with aromatase insufficiency in humans. Low levels of aromatase activity seem to be sufficient for ongoing estrogenic stimulation of the breast and uterus, especially if androgen levels are elevated. Whilst these phenotypes may also reflect individual variability in alternative pathways of estrogen/androgen synthesis or end-organ responsiveness, it seems that in some individuals at least, a near complete aromatase blockade is necessary to prevent breast estrogenization. This finding could have important clinical implications, for example, in the treatment of perimenopausal breast cancer. Thus, these naturally-occurring models of partial aromatase insufficiency in patients who harbor CYP19 mutations have important translational messages for the pathogenesis and treatment of more common diseases within the population.
Acknowledgments
We are grateful to the patients and families, and to Peter Hindmarsh, Gerard Conway, Paul Rutland, Bryan Winchester, Caroline Brain, Terry Segal, Gudrun Moore and Maria Bitner-Glindzicz for useful discussions. A human aromatase cDNA construct was kindly provided by Evan Simpson and Colin Clyne, and anti-calnexin antibody by Gao Bin. Research at the Institute of Child Health and Great Ormond Street Hospital for Children NHS Trust benefits from R&D funding received from the NHS Executive. Funding for this project was provided by The Wellcome Trust and Child Health Research Appeal Trust. RJA is the recipient of a Clinical Scientist Award in Translational Research from the Burroughs Wellcome Fund (#1005954). JCA holds a Wellcome Trust Clinician Scientist Fellowship (068061) and a Wellcome Trust Senior Research Fellowship in Clinical Science (079666).
Footnotes
Disclosure statement: The authors have nothing to disclose. JCA holds a Wellcome Trust Clinician Scientist Fellowship (068061) and Senior Fellowship (079666) and RJA holds a Burroughs Wellcome Fund grant (1005954).
Publisher's Disclaimer: “This is an un-copyedited author manuscript copyrighted by The Endocrine Society. This may not be duplicated or reproduced, other than for personal use or within the rule of “Fair Use of Copyrighted Materials” (section 107, Title 17, U.S. Code) without permission of the copyright owner, The Endocrine Society. From the time of acceptance following peer review, the full text of this manuscript is made freely available by The Endocrine Society at http://www.endojournals.org/. The final copy edited article can be found at http://www.endojournals.org/. The Endocrine Society disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties. The citation of this article must include the following information: author(s), article title, journal title, year of publication and DOI.”
References
- 1.Simpson ER, Clyne C, Rubin G, Boon WC, Robertson K, Britt K, Speed C, Jones M. Aromatase - a brief overview. Annu Rev Physiol. 2002;64:93–127. doi: 10.1146/annurev.physiol.64.081601.142703. [DOI] [PubMed] [Google Scholar]
- 2.Bulun SE, Rosenthal IM, Brodie AM, Inkster SE, Zeller WP, DiGeorge AM, Frasier SD, Kilgore MW, Simpson ER. Use of tissue-specific promoters in the regulation of aromatase cytochrome P450 gene expression in human testicular and ovarian sex cord tumors, as well as in normal fetal and adult gonads. J Clin Endocrinol Metab. 1994;78:1616–1621. doi: 10.1210/jcem.78.2.8106605. [DOI] [PubMed] [Google Scholar]
- 3.Bilezikian JP, Morishima A, Bell J, Grumbach MM. Increased bone mass as a result of estrogen therapy in a man with aromatase deficiency. N Engl J Med. 1998;339:599–603. doi: 10.1056/NEJM199808273390905. [DOI] [PubMed] [Google Scholar]
- 4.Fisher CR, Graves KH, Parlow AF, Simpson ER. Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc Natl Acad Sci U S A. 1998;95:6965–6970. doi: 10.1073/pnas.95.12.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robertson KM, O'Donnell L, Jones ME, Meachem SJ, Boon WC, Fisher CR, Graves KH, McLachlan RI, Simpson ER. Impairment of spermatogenesis in mice lacking a functional aromatase (cyp 19) gene. Proc Natl Acad Sci U S A. 1999;96:7986–7991. doi: 10.1073/pnas.96.14.7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci U S A. 2000;97:12735–12740. doi: 10.1073/pnas.97.23.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shim GJ, Warner M, Kim HJ, Andersson S, Liu L, Ekman J, Imamov O, Jones ME, Simpson ER, Gustafsson JA. Aromatase-deficient mice spontaneously develop a lymphoproliferative autoimmune disease resembling Sjogren's syndrome. Proc Natl Acad Sci U S A. 2004;101:12628–12633. doi: 10.1073/pnas.0405099101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shozu M, Akasofu K, Harada T, Kubota Y. A new cause of female pseudohermaphroditism: placental aromatase deficiency. J Clin Endocrinol Metab. 1991;72:560–566. doi: 10.1210/jcem-72-3-560. [DOI] [PubMed] [Google Scholar]
- 9.Ito Y, Fisher CR, Conte FA, Grumbach MM, Simpson ER. Molecular basis of aromatase deficiency in an adult female with sexual infantilism and polycystic ovaries. Proc Natl Acad Sci U S A. 1993;90:11673–11677. doi: 10.1073/pnas.90.24.11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conte FA, Grumbach MM, Ito Y, Fisher CR, Simpson ER. A syndrome of female pseudohermaphrodism, hypergonadotropic hypogonadism, and multicystic ovaries associated with missense mutations in the gene encoding aromatase (P450arom) J Clin Endocrinol Metab. 1994;78:1287–1292. doi: 10.1210/jcem.78.6.8200927. [DOI] [PubMed] [Google Scholar]
- 11.Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. 1995;80:3689–3698. doi: 10.1210/jcem.80.12.8530621. [DOI] [PubMed] [Google Scholar]
- 12.Mullis PE, Yoshimura N, Kuhlmann B, Lippuner K, Jaeger P, Harada H. Aromatase deficiency in a female who is compound heterozygote for two new point mutations in the P450arom gene: impact of estrogens on hypergonadotropic hypogonadism, multicystic ovaries, and bone densitometry in childhood. J Clin Endocrinol Metab. 1997;82:1739–1745. doi: 10.1210/jcem.82.6.3994. [DOI] [PubMed] [Google Scholar]
- 13.Ludwig M, Beck A, Wickert L, Bolkenius U, Tittel B, Hinkel K, Bidlingmaier F. Female pseudohermaphroditism associated with a novel homozygous G-to-A (V370-to-M) substitution in the P-450 aromatase gene. J Pediatr Endocrinol Metab. 1998;11:657–664. doi: 10.1515/jpem.1998.11.5.657. [DOI] [PubMed] [Google Scholar]
- 14.Belgorosky A, Pepe C, Marino R, Guercio G, Saraco N, Vaiani E, Rivarola MA. Hypothalamic-pituitary-ovarian axis during infancy, early and late prepuberty in an aromatase-deficient girl who is a compound heterozygote for two new point mutations of the CYP19 gene. J Clin Endocrinol Metab. 2003;88:5127–5131. doi: 10.1210/jc.2003-030433. [DOI] [PubMed] [Google Scholar]
- 15.Carani C, Qin K, Simoni M, Faustini-Fustini M, Serpente S, Boyd J, Korach KS, Simpson ER. Effect of testosterone and estradiol in a man with aromatase deficiency. N Engl J Med. 1997;337:91–95. doi: 10.1056/NEJM199707103370204. [DOI] [PubMed] [Google Scholar]
- 16.Deladoey J, Fluck C, Bex M, Yoshimura N, Harada N, Mullis PE. Aromatase deficiency caused by a novel P450arom gene mutation: impact of absent estrogen production on serum gonadotropin concentration in a boy. J Clin Endocrinol Metab. 1999;84:4050–4054. doi: 10.1210/jcem.84.11.6135. [DOI] [PubMed] [Google Scholar]
- 17.Herrmann BL, Saller B, Janssen OE, Gocke P, Bockisch A, Sperling H, Mann K, Broecker M. Impact of estrogen replacement therapy in a male with congenital aromatase deficiency caused by a novel mutation in the CYP19 gene. J Clin Endocrinol Metab. 2002;87:5476–5484. doi: 10.1210/jc.2002-020498. [DOI] [PubMed] [Google Scholar]
- 18.Maffei L, Murata Y, Rochira V, Tubert G, Aranda C, Vazquez M, Clyne CD, Davis S, Simpson ER, Carani C. Dysmetabolic syndrome in a man with a novel mutation of the aromatase gene: effects of testosterone, alendronate, and estradiol treatment. J Clin Endocrinol Metab. 2004;89:61–70. doi: 10.1210/jc.2003-030313. [DOI] [PubMed] [Google Scholar]
- 19.Mittre Herve MH, Kottler ML, Pura M. Human gene mutations. Gene symbol: CYP19. Disease: Aromatase deficiency. Hum Genet. 2004;114:224. [PubMed] [Google Scholar]
- 20.Jones ME, Boon WC, Proietto J, Simpson ER. Of mice and men: the evolving phenotype of aromatase deficiency. Trends Endocrinol Metab. 2006;17:55–64. doi: 10.1016/j.tem.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Masi L, Becherini L, Gennari L, Amedei A, Colli E, Falchetti A, Farci M, Silvestri S, Gonnelli S, Brandi ML. Polymorphism of the aromatase gene in postmenopausal Italian women: distribution and correlation with bone mass and fracture risk. J Clin Endocrinol Metab. 2001;86:2263–2269. doi: 10.1210/jcem.86.5.7450. [DOI] [PubMed] [Google Scholar]
- 22.Van Pottelbergh I, Goemaere S, Kaufman JM. Bioavailable estradiol and an aromatase gene polymorphism are determinants of bone mineral density changes in men over 70 years of age. J Clin Endocrinol Metab. 2003;88:3075–3081. doi: 10.1210/jc.2002-021691. [DOI] [PubMed] [Google Scholar]
- 23.Riancho JA, Zarrabeitia MT, Valero C, Sanudo C, Hernandez JL, Amado JA, Zarrabeitia A, Gonzalez-Macias J. Aromatase gene and osteoporosis: relationship of ten polymorphic loci with bone mineral density. Bone. 2005;36:917–925. doi: 10.1016/j.bone.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 24.Petry CJ, Ong KK, Michelmore KF, Artigas S, Wingate DL, Balen AH, de Zegher F, Ibanez L, Dunger DB. Association of aromatase (CYP 19) gene variation with features of hyperandrogenism in two populations of young women. Hum Reprod. 2005;20:1837–1843. doi: 10.1093/humrep/deh900. [DOI] [PubMed] [Google Scholar]
- 25.Tsuchiya N, Wang L, Suzuki H, Segawa T, Fukuda H, Narita S, Shimbo M, Kamoto T, Mitsumori K, Ichikawa T, Ogawa O, Nakamura A, Habuchi T. Impact of IGF-1 and CYP19 gene polymorphisms on the survival of patients with metastatic prostate cancer. J Clin Oncol. 2006;24:1982–1989. doi: 10.1200/JCO.2005.02.9439. [DOI] [PubMed] [Google Scholar]
- 26.Wickman S, Sipila I, Ankarberg-Lindgren C, Norjavaara E, Dunkel L. A specific aromatase inhibitor and potential increase in adult height in boys with delayed puberty: a randomised controlled trial. Lancet. 2001;357:1743–1748. doi: 10.1016/S0140-6736(00)04895-9. [DOI] [PubMed] [Google Scholar]
- 27.Attar E, Bulun SE. Aromatase and other steroidogenic genes in endometriosis: translational aspects. Hum Reprod Update. 2006;12:49–56. doi: 10.1093/humupd/dmi034. [DOI] [PubMed] [Google Scholar]
- 28.Swain SM. Aromatase inhibitors - a triumph of translational oncology. N Engl J Med. 2005;353:2807–2809. doi: 10.1056/NEJMe058273. [DOI] [PubMed] [Google Scholar]
- 29.Brueggemeier RW, Hackett JC, Diaz-Cruz ES. Aromatase inhibitors in the treatment of breast cancer. Endocr Rev. 2005;26:331–345. doi: 10.1210/er.2004-0015. [DOI] [PubMed] [Google Scholar]
- 30.Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, Castiglione M, Tu D, Shepherd LE, Pritchard KI, Livingston RB, Davidson NE, Norton L, Perez EA, Abrams JS, Therasse P, Palmer MJ, Pater JL. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med. 2003;349:1793–1802. doi: 10.1056/NEJMoa032312. [DOI] [PubMed] [Google Scholar]
- 31.Thurlimann B, Keshaviah A, Coates AS, Mouridsen H, Mauriac L, Forbes JF, Paridaens R, Castiglione-Gertsch M, Gelber RD, Rabaglio M, Smith I, Wardly A, Price KN, Goldhirsch A, Breast International Group (BIG) 1-98 Collaborative Group A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N Engl J Med. 2005;353:2747–2757. doi: 10.1056/NEJMoa052258. [DOI] [PubMed] [Google Scholar]
- 32.Favia AD, Cavalli A, Masetti M, Carotti A, Recanatini M. Three-dimensional model of the human aromatase enzyme and density functional parameterization of the iron-containing protoporphyrin IX for a molecular dynamics study of heme-cysteinato cytochromes. Proteins. 2006;62:1074–1087. doi: 10.1002/prot.20829. [DOI] [PubMed] [Google Scholar]
- 33.Ackerman GE, Smith ME, Mendelson CR, MacDonald PC, Simpson ER. Aromatization of androstenedione by human adipose tissue stromal cells in monolayer culture. J Clin Endocrinol Metab. 1981;53:412–417. doi: 10.1210/jcem-53-2-412. [DOI] [PubMed] [Google Scholar]
- 34.Grumbach MM, Auchus RJ. Estrogen: consequences and implications of human mutations in synthesis and action. J Clin Endocrinol Metab. 1999;84:4677–4694. doi: 10.1210/jcem.84.12.6290. [DOI] [PubMed] [Google Scholar]
- 35.Dessens AB, Slijper FM, Drop SL. Gender dysphoria and gender change in chromosomal females with congenital adrenal hyperplasia. Arch Sex Behav. 2005;34:389–397. doi: 10.1007/s10508-005-4338-5. [DOI] [PubMed] [Google Scholar]
- 36.Ishikawa T, Glidewell-Kenney C, Jameson JL. Aromatase-independent testosterone conversion into estrogenic steroids is inhibited by a 5 alpha-reductase inhibitor. J Steroid Biochem Mol Biol. 2006;98:133–138. doi: 10.1016/j.jsbmb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 37.Fenske M, Segner H. Aromatase modulation alters gonadal differentiation in developing zebrafish (Danio rerio) Aquat Toxicol. 2004;67:105–126. doi: 10.1016/j.aquatox.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 38.Vaillant S, Magre S, Dorizzi M, Pieau C, Richard-Mercier N. Expression of AMH, SF1, and SOX9 in gonads of genetic female chickens during sex reversal induced by an aromatase inhibitor. Dev Dyn. 2001;222:228–237. doi: 10.1002/dvdy.1190. [DOI] [PubMed] [Google Scholar]
- 39.Britt KL, Kerr J, O'Donnell L, Jones ME, Drummond AE, Davis SR, Simpson ER, Findlay JK. Estrogen regulates development of the somatic cell phenotype in the eutherian ovary. Faseb J. 2002;16:1389–1397. doi: 10.1096/fj.01-0992com. [DOI] [PubMed] [Google Scholar]
- 40.Couse JF, Hewitt SC, Bunch DO, Sar M, Walker VR, Davis BJ, Korach KS. Postnatal sex reversal of the ovaries in mice lacking estrogen receptors alpha and beta. Science. 1999;286:2328–2331. doi: 10.1126/science.286.5448.2328. [DOI] [PubMed] [Google Scholar]