

Abstract
CD8 T cells specific for self-antigens are present in the peripheral lymphoid system and can contribute to autoimmunity or transplant rejection. Whether recognition of Ag leads to full activation, or to induction of tolerance, depends upon availability of cytokine at critical stages of the response. Signals provided by IL-12 and/or IFN-α/β are required for activation of naïve CD8 T cells, and IL-2 is needed to sustain and further expand the effector cells if Ag persists. These critical signaling requirements provide new insights into the factors that regulate the CD8 T cell contributions to development of autoimmunity or rejection of transplants.
Keywords: CD8 T lymphocyte, activation, differentiation, anergy, peripheral tolerance
1. Introduction
Central tolerance resulting from negative selection in the thymus eliminates the majority of self-reactive CD8 T cells, but is incomplete. Despite the presence of the AIRE system that provides for presentation of many self-Ags in the thymus [1], some self-reactive T cells do escape negative selection and enter the periphery. Thus, mature naïve CD8 T cells that can respond to self-Ag are present in the lymphoid organs where they can potentially contribute to autoimmunity and transplant rejection. On the positive side, however, these cells also have the potential to respond to self Ags expressed on tumor cells, and are the target of vaccine approaches to cancer immunotherapy. Thus, understanding the mechanisms that can result in the induction of peripheral tolerance in CD8 T cells is important for developing strategies to avoid responses that contribute to autoimmunity or transplant rejection, as well as for devising ways of circumventing the induction of tolerance to Ags on tumor cells.
Mechanisms responsible for establishing peripheral tolerance can be broadly classified into three categories; ignorance, deletion and anergy. Clonal ignorance is the simplest form of peripheral tolerance; the naïve T cells circulating in the lymphoid system fail to gain access to the peripheral site where Ag is present, and sufficient Ag is not being carried to the lymph nodes draining the site to result in T cell activation in the nodes [2]. Tolerance due to ignorance is tenuous, however, because some peripheral self-Ags do reach the lymph nodes via migrating dendritic cells (DC), and pathological insults can lead to increased levels of these Ags becoming available for T cell recognition. Peripheral tolerance can still occur, however, due to clonal deletion or anergy that can result when the T cell recognizes Ag but the ensuing TCR-initiated cell division and differentiation are incomplete, and lead to either death of the Ag-activated cells (deletion), or to the induction of a long-lived non-responsive state (anergy) [3].
For CD8 T cells, there is now considerable evidence that the mechanisms responsible for tolerance induction by deletion or establishment of anergy involve the recognition of Ag in the absence of additional signals that are required to support development of full responsiveness, and the molecular basis for these additional signaling requirements is beginning to be clarified. It appears that these additional requirements represent ‘checkpoints’ in the CD8 T cell response that help to insure that a productive response occurs only under appropriate circumstances, e.g. upon infection with a pathogen or when help from CD4 T cells is available. When Ag is recognized and a checkpoint is reached, but the additional signal is not available, the result is establishment of peripheral tolerance in the cells specific for the Ag.
2. Activation and differentiation of naïve CD8 T cells
In most cases, naïve CD8 T cells first encounter Ag being cross-presented by DC in lymph nodes [4,5], and the outcome depends on the maturation state of the DC [6]. Presentation by immature, quiescent DC results in tolerance induction by a mechanism involving deletion, induction of non-responsiveness (anergy), or both. In contrast, the T cells make a productive response if the DC have been activated to mature as a result of either engagement of their Toll-like receptors (TLR) by pathogen-derived polymers (adjuvants), or by a CD40-dependent interaction with CD4 T helper cells [7-9]. Thus, the DC must be ‘licensed’ or ‘conditioned’ to present Ag in a manner that stimulates a productive response and avoids tolerance induction. This is often attributed to increased expression of Ag and/or costimulatory ligands B7-1 (CD80) and B7-2 (CD86) on the activated DC, but there is no direct evidence that these quantitative changes are responsible for the different outcomes. Furthermore, there are reports of DC failing to activate productive CD8 T cell response even when they are expressing high levels of costimulatory ligands [10-12]. Considerable evidence has accumulated to indicate that survival and differentiation of CD8 T cells responding to Ag and costimulation require activation of a signaling pathway(s) distinct from those activated by the TCR and CD28 receptors, and that cytokines produced by mature DC can provide the ‘third signal’ needed to activate this pathway.
2.1. A ‘three signal’ model for CD8 T cell activation
Activation of T cells requires both Ag engagement by the TCR and signals provided by a costimulatory receptor, usually CD28 binding to its B7 ligands [13-16]. These classical ‘two signals’ are necessary for activation of naïve CD8 T cells, but alone are not sufficient to support strong clonal expansion, development of effector functions, or establishment of a long-lived, responsive memory population. Instead, a third signal is needed, and can be provided by either IL-12 or Type I IFN (IFN-α/β). This was initially suggested by in vitro studies showing that highly purified naïve CD8 T cells could not expand and develop cytolytic activity in response to artificial APC presenting just Ag and B7, but responded strongly if either IL-12 [17] or IFN-α/β [18] was present. In contrast, CD44hi memory cells bearing the same TCR responded to the artificial APC presenting Ag and B7 by undergoing strong clonal expansion and developing effector functions [17]; they did not require a third signal. Earlier work had shown that IL-12 can augment generation of cytotoxic T lymphocyte (CTL) responses [19,20] and that IFN-α/β could promote survival [21] and IFN-γ [22] production by CD8 T cells, but the critical role of these cytokines in determining whether Ag recognition leads to tolerance versus full activation and development of a memory population has only recently been realized.
In vivo studies also provide strong support for a model in which naïve CD8 T cells require a third signal to develop effector functions and transition to a memory population. Immunization with a CD8 epitope peptide results in tolerance induction unless an adjuvant is given along with the peptide [23-25]. Co-administration of either IL-12 [26,27] or IFN-α [28-30] along with peptide replaces the requirement for adjuvant, and supports generation of strong primary clonal expansion, development of effector functions, and establishment of a long-lived, responsive memory population. It appears likely that the major role of adjuvants in supporting CD8 T cell responses to Ag, in addition to increasing expression levels of costimulatory ligands, is that of inducing IL-12 and/or Type I IFN production by DC and other cells through engagement of TLR.
Experiments examining the responses of CD8 T cells lacking the IL-12 receptor (IL-12R) or the Type I IFN receptor (IFN-IR) further support a three-signal model for CD8 T cell activation. Antigen-specific CD8 T cells that lack the IFN-IR exhibit a greater than 99% reduction in their ability to respond to lymphocytic choriomeningitis virus (LCMV) infection [28,29]. In contrast, responses of the IFN-IR-deficient cells to vaccinia virus (VV), vesicular stomatitis virus or Listeria monocytogenes (LM) are much less impaired [28,31]. We have found that Ag-specific CD8 T cells lacking the IL-12R exhibit a stronger reduction in response to VV and LM than do cells lacking the IFN-IR, and that generation of memory populations is only eliminated when the cells are deficient in both receptors (Xiao et.al., manuscript in preparation). Thus, it appears that either IL-12 or Type I IFN can support in vivo responses to pathogens, and that the levels of the cytokines produced in response to a particular pathogen will dictate which makes the greater contribution. In contrast, CD8 T cell recognition of a self-Ag, which may normally occur under conditions where these cytokines are not being produced, will result in an abortive proliferative response leading to tolerance. Alternatively, productive CD8 T cell responses to self-Ag that contribute to autoimmunity or transplant rejection may result in cases where IL-12, IFN-α/β, or potential alternate third signals, are present during the initial period of interaction with the Ag. One way that this may occur is by CD4 T helper cells stimulating production of these cytokines by DC.
2.2 CD4 T helper cells condition DC to provide signal 3
One of the ways in which CD4 T cells can provide help for a CD8 T cell response is by interacting with DC that are cross-presenting class I Ag to ‘condition’ these DC to effectively activate naïve CD8 T cells, and this requires binding of CD40 ligand (CD40L; CD154) expressed on the CD4 T cell to CD40 on the DC [7-9]. Engagement of CD40 on DC stimulates the cells to produce IL-12 [32], suggesting that ‘conditioning’ of the DC by CD4 T cell might involve stimulating the production of this cytokine so that the DC can provide the critical third signal for CD8 T cell activation. Hernandez et.al. [12] provided evidence consistent with this possibility by demonstrating that IL-12 could bypass a requirement for CD4 T cells in a CD8-dependent diabetes model. Generation of the CD8 response and development of disease depended on CD40L-dependent CD4 T cell help, but both occurred in the absence of CD4 help when IL-12 was administered to the mice.
The involvement of IL-12 production by DC in response to CD4 T cell help for a CD8 T cell response was directly demonstrated in a study by Filatenkov et.al. [33] examining an ectopic heart transplant model. In this model, donor mice expressed a membrane form of ovalbumin (mOVA) as a transgene under the control of an actin promoter, so that OVA was expressed in all tissues, including the heart. Recipient mice first received, by adoptive transfer, TCR transgenic OT-I CD8 and OT-II CD4 T cells, specific for H-2Kb/OVA257-264 and I-Ab/OVA323-339 respectively, and subsequently received heterotopic mOVA heart transplants. Thus, mOVA served as a model minor Ag, and the use of adoptively transferred CD4 and CD8 T cells allowed direct visualization of the responses of these cells to the graft. Rapid graft rejection required the presence of both OT-I and OT-II cells, and required that the OT-II cells express CD40L. When both OT-I and OT-II cells were present, OT-I cells clonally expanded, migrated to the graft, expressed high granzyme B and IFN-γ levels, and mediated rapid graft rejection.
In contrast, OT-I cells underwent some clonal expansion and migrated to the graft in the absence of OT-II cells, but expressed low levels of granzyme B and IFN-γ, and did not cause rapid rejection, suggesting that a signal 3 cytokine was not available to support their response. Administration of IL-12 replaced the requirement for CD4 help, causing the OT-I cells to express high granzyme B and IFN-γ and mediate rapid graft rejection. Furthermore, even when OT-II cells were present, OT-I cells that lacked the IL-12R failed to upregulate the expression of effector proteins or mediate graft rejection. It was also shown that DC from mice having OT-II cells produced IL-12, but only if the OT-II cells expressed CD40L. Thus, in this model of minor Ag-mediated transplant rejection, the evidence very strongly suggests that CD4 T cells provide help for CD8 T cells, at least in part, by upregulating IL-12 production by DC so that they can provide the third signal to the CD8 T cells to support a productive response. The CD4 T cells may also upregulate expression of costimulatory ligands on the DC, but this does not appear to be essential, since administration of IL-12 was sufficient to replace the need for help.
Under most circumstances, peripheral CD8 T cells with the potential to contribute to autoimmunity or graft rejection will undergo tolerance induction if they encounter the self-Ag they recognize, as a result of failing to receive a third signal needed to support their differentiation. However, if the encounter happens at a time when there is sufficient IL-12 and/or IFN-α/β present to provide the third signal, then strong clonal expansion and development of effector functions may result to contribute to disease. This can occur if CD4 helper cells are ‘conditioning’ DC to produce the necessary signal 3 cytokine, but may also occur if a concomitant infection induces production of these cytokines triggered by TLR ligands produced by the pathogen.
2.3 Signaling pathways for IL-12 and IFN-α-dependent differentiation
Brief interaction of CD8 T cells with Ag and costimulation provides sufficient signals to program the cells to proliferate; allowing cells to interact in vitro with artificial APC for even a few hours, followed by removal of the stimulus, results in multiple rounds of division over the next three days [34]. Under these conditions, however, survival is limited so that clonal expansion is minimal by day 3 despite extensive cell division, the cells produce little IFN-γ upon re-stimulation, and they fail to develop cytolytic effector function [35]. When either IL-12 or IFN-α are present, the rate of cell division does not change significantly but survival, and thus clonal expansion, are increased, and effector functions develop by day 3 .
In contrast to the brief interaction required for Ag and costimulation to program the cells to divide, optimum survival and development of effector functions requires more prolonged signaling. In the case of IL-12, we found that optimal clonal expansion by 72 hr required that Ag and B7 were present for 24 hr, and that IL-12 was present from about 15 to 30 hr [35]. More prolonged exposure to IL-12, up to 60 hr, was required for optimum development of effector functions. The timing requirements for stimulation by IFN-α have not been examined, but are likely to mirror those seen for IL-12. These results suggest that varying levels of tolerance may result for CD8 T cells responding to Ag, depending not only on whether a signal 3 cytokine is available, but also on the duration of its availability. Thus, two signals are sufficient to rapidly program cell division, but the response is abortive in the absence of a third signal. Only when the signals provided by IL-12 or IFN-α/β are present for a prolonged period are the cells fully programmed to survive and become effectors.
Recognition of Ag by the TCR activates the Ras-MAPK pathway, and costimulation provided by CD28 activates the phosphatidyl inositol-3-kinase pathway and synergizes to augment TCR induced transcriptional regulation mediated by AP-1, NF-AT and NF-κB. In contrast to signaling by the TCR and CD28, the Jak/Stat (Janus kinase/signal transducer and activator of transcription) pathways play a major role in signaling by cytokine receptors. Upon cytokine binding, Jaks are activated and phosphorylate the receptor to form docking sites for STATs, latent transcription factors present in the cytosol. Upon docking to the phosphorylated receptor the Stats are phosphorylated by Jaks, resulting in dimerization and nuclear translocation, where the STATs then regulate gene transcription by binding to specific DNA motifs and recruiting other transcription factors and coactivators.
The IL-12 receptor, a hetrodimeric receptor composed of IL-12Rβ1 and β2 chains, recruits JAK2 and TYK2, leading to phosphorylation of STAT-4 to form homodimers, and this constitutes the major IL-12 signaling pathway. Mice lacking the IL-12R have defects in cell mediated immunity and Th1 CD4 helper cell development, and Stat-4-deficient mice have a very similar phenotype [36,37]. Thus, not surprisingly, provision of the ‘third signal’ to naïve CD8 T cells by IL-12 requires Stat-4. When wild type BalbC cells are stimulated in vitro with anti-TCR mAb and B7-1 on artificial APC, addition of IL-12 supports development of cytolyic function and the ability to produce IFN-γ. In contrast, these effector functions fail to develop when naïve CD8 T cells from Stat-4-deficient mice are stimulated in the same way [18].
IFN-α/β strongly activates Stat-1 and Stat-2 upon binding to the IFN-IR, but can variably activate other Stats, including Stat-3, 4 and 6, depending on the cell type being examined. Earlier studies suggested that IFN-α could activate Stat-4 in human T cells, but that Stat-4 activation was impaired in murine CD4 T cells due to a mini-satellite insertion in murine Stat-2 that resulted in impaired recruitment of Stat-4 to the receptor [38]. However, a subsequent study demonstrated that during an LCMV infection, IFN-α signaling led to Stat-4 being more strongly activated in CD8 T cells than in CD4 T cells, and promoted Stat-4-dependent IFN-γ production by the CD8 T cells [22]. Using naïve CD8 T cells from wild type and Stat-4 deficient mice, we found that IFN-α-dependent provision of signal 3 was largely dependent on Stat-4 [18], as assessed by strongly decreased development of cytolytic activity and ability to produce IFN-γ, although loss of function was not as complete as in the case of stimulation with IL-12.
While development of effector functions demonstrated similar Stat-4-dependence in response to IL-12 and IFN-α, more recent work suggests that the signaling pathways involved in promoting CD8 T cell differentiation may differ substantially for these two cytokines (Agarwal and Mescher, unpublished results). Artificial APC presenting anti-TCR mAb and B7-1 were used to stimulate wild type and Stat-4-deficient naïve CD8 T cells in the absence or presence of either IL-12 or IFN-α for 72 hr, and oligonucleotide microarray analysis was then done. In comparison to cells stimulated in the absence of a cytokine, IL-12 stimulated the up- or down-regulation of about 200 genes, and regulation of all but 2 was eliminated in the Stat-4-deficient cells. In contrast, IFN-α stimulated the up- or down-regulation of about 300 genes, and about a third of these showed similar regulation in the STAT-4-deficient cells. Genes regulated in common by both IL-12 and IFN-α are likely candidates for contributing importantly to differentiation, since functional outcomes are very similar with the two cytokines, and there were approximately 35 such genes in wild type cells. Of this commonly regulated subset, regulation of all of the genes was Stat-4-dependent in response to IL-12, but regulation of only about one-third of them was Stat-4-dependent in response to IFN-α. Thus, these preliminary results examining the ‘third signal’ pathways at the level of gene expression regulation suggest that while Stat-4 activation contributes in both cases, activation of additional Stats is likely to make an important contribution to the response to IFN-α. In addition, the results suggest that while either cytokine can support development of effector functions and a memory population, the phenotypes of the resulting cells may differ.
2.4. Alternate ‘third signals’
The phenotype and functions of T cells can be modified in response to a wide array of cytokines, but IL-12 and IFN-α are distinguished by the fact that they can be essential for productive Ag-dependent activation of naïve CD8 T cells, i.e. they can act as the ‘switch’ that determines whether a response to Ag will result in tolerance versus full activation leading to formation of a responsive memory population. This, together with the fact that their action is mediated through activation of a signaling pathway(s) distinct from those activated by the TCR and CD28, provide the rationale for viewing them as a ‘third signal’. A number of other cytokines have been found to lack signal 3 activity based on their inability to support development of effector functions in naïve CD8 T cells stimulated in vitro with artificial APCs presenting Ag and B7-1, including IL-1, -2, -4, -6, -7, -15, -18, -TNF-α and IFN-γ [18]. Some of these cytokines can modify responses of developing CD8 T cells. IL-4, for example, can skew effector cells to an IL-4-producing Tc2 phenotype [39], and IL-2 can promote proliferation and clonal expansion (see below). IFN-γ has been referred to as a signal 3 cytokine [40] based on its ability to promote optimal expansion of CD8 T cells responding to LCMV [41,42] or immunization with peptide and LPS [43], and on the fact that it does so by acting directly on the CD8 T cell, at least in the case of the LCMV response [42]. While IFN-γ can clearly modify the extent of proliferation/clonal expansion, it does not support development of effector functions by naïve CD8 T cells in response to signals 1 and 2 ([18] and unpublished results). We would suggest that the term ‘signal three’ be reserved for cytokines or ligands that act comparably to IL-12 and IFN-α, i.e. cytokines that act directly on naïve CD8 T cells responding to Ag and costimulation to support development of effector functions in vitro, and to support productive activation and development of a memory population in vivo. Thus, signal 3 cytokines would be defined as those that initiate the CD8 T cell differentiation program, rather than simply modifying the program once it is initiated.
While IL-12 and/or IFN-α appear to be the major contributors to several responses, as discussed above, it is possible that other cytokines or cell surface ligands may have a comparable function. We have found that IL-21 [44], a member of the common gamma chain family of cytokines that is produced by activated CD4 T cells, promotes clonal expansion and supports development of cytolytic effector function by naïve CD8 T cells stimulated in vitro with Ag and B7-1 (Casey and Mescher, manuscript submitted), suggesting that it may act as a signal 3 cytokine. Work is in progress to determine whether IL-21 also acts directly on CD8 T cells in vivo to support productive activation, avoidance of tolerance and development of memory. If so, it could provide a means by which CD4 T cells provide help to initiate a CD8 T cell response, in addition to the CD4-dependent conditioning of DC discussed in the following section.
2.5 Long-term fate of CD8 T cells activated in the absence of signal 3
Tolerance of CD8 T cells can involve deletion of the cells, induction of anergy, or both, depending on the response being examined [3]. The tolerance that results when naïve CD8 T cell respond to Ag in the absence of signal 3 also appears likely to involve both of these outcomes. IL-12 and IFN-α promote survival of the responding cells as they become effector cells, at least in part by upregulating expression of Bcl-3 [45]. Thus, the death of the majority of the responding cells that occurs following the peak of clonal expansion is more rapid and profound in the absence of a third signal ([26] and unpublished results). Nevertheless, some of the cells that respond in the absence of a third signal can survive long term, at least in the case of immunization with peptide, and these cells are anergic in that they do not respond to re-challenge with a potent stimulus of Ag and adjuvant [26,27]. What determines the balance between deletion and survival of anergic cells is not clear, but there is considerable evidence to suggest that the ‘strength’ of interaction with Ag (TCR affinity, Ag dose and duration) may be an important factor, as Redmond and Sherman [3] have recently discussed.
3. Activation-induced non-responsiveness (AINR)
When costimulation and a signal 3 cytokine are available, naïve CD8 T cells begin to divide about 24 hr after initially encountering Ag, and proceed through multiple rounds of division so that clonal expansion peaks at about 72 hr, and the cells have developed effector functions by this time. A second form of tolerance can occur at this stage of the response, however, if the resulting effector cell population is insufficient to clear the Ag. In the face of persistent Ag, the activated CD8 T cells undergo a gradual decline in number and functional capacity, as demonstrated in studies examining responses to HY Ag [46] and LCMV [23,47]. There is considerable evidence to suggest that this results from another checkpoint in the CD8 T cell response.
Effector CD8 T cells develop AINR
When CD8 T cells are stimulated in vitro they develop effector functions by day 3, but are anergic [48]. Signaling through the TCR still occurs, as evidenced by killing of Ag-bearing target cells and upregulation of IFN-γ production, but the cells can no longer produce IL-2 to support further proliferation in response to Ag and costimulation. This resembles the classical anergy induced in CD4 T cells when they are stimulated by TCR engagement in the absence of costimulation [16,49,50]. It clearly differs, however, in that the CD8 T cell anergy develops even when costimulation and a signal 3 cytokine are present along with the Ag stimulus. Thus, we have termed this ‘activation-induced non-responsiveness’ (AINR) to distinguish it from classical CD4 T cell anergy. Although CD8 T cells that have developed AINR cannot produce IL-2, they can still respond to the cytokine by proliferating. Furthermore, when exogenous IL-2 is provided in vitro to drive proliferation of effector CTL for one to two days, the AINR state is reversed and the cells regain the ability to produce IL-2 in response to Ag [51]. The AINR state that develops during a response by naïve CD8 T cells resembles the ‘split anergy’ described by Otten and Germain for cloned CTL lines [52].
Development of AINR appears to be a normal part of the CD8 T cell differentiation program. This non-responsive state develops in CD8 T cells responding to LCMV virus [53] or allogeneic tumor [48], where Ag is cleared, or to syngeneic tumor [54] or persistant virus [55], where Ag persists. For responses to both syngeneic tumor [56] and persistent virus [57], administration of IL-2 stimulates the AINR cells to proliferate, and AINR is reversed so that the cells regain the ability to produce IL-2. Furthermore, we showed that CD4 T cells could provide the IL-2 needed to reverse AINR in CD8 T cells responding to syngeneic tumor [58]. Thus, it appears that AINR constitutes a CD4 helper T cell-dependent checkpoint in the CD8 T cell response; the effector CD8 T cells cannot continue to expand in number without IL-2 that can be provided by helper T cells.
In cases where Ag is successfully cleared by the initial response, AINR is reversed over time as some of the effector cells survive to become responsive memory cells. Results from Kaech et.al. [53] indicate that this occurs one to two weeks after the peak of the effector response. At the peak of the response to LCMV, the effector CD8 T cells were unable to make IL-2 or proliferate upon re-exposure to Ag, but the cells remaining two weeks later had regained this capacity. IL-7 plays a role in the transition of effector CD8 T cells to memory cells [59,60], and may be involved in reversal of AINR. We have found that in vitro culture of AINR CD8 effector cells in IL-7 stimulates their proliferation, and the AINR state is reversed within one to two days (Hammerbeck and Mescher, unpublished results). Thus, following Ag clearance and re-expression of the IL-7Rα receptor on some of the effector CD8 T cells ([61,62] and Hammerbeck and Mescher, manuscript submitted), the low levels of IL-7 present in vivo may stimulate slow proliferation of the effectors and reversal of AINR over one to two weeks. In contrast to Ag clearance and memory formation, if the effector CTL resulting from the initial phase of clonal expansion fail to clear the Ag, tolerance to the persisting Ag results if CD4 T cell help in the form of IL-2 is not available to reverse AINR. The numbers and functional capacity of the AINR CD8 T cells then decline over time. CD8 T cell responses limited by AINR could potentially be a mechanism that contributes to chronic graft rejection, with new thymic emigrants having specificity for the graft making an initial response and causing some tissue damage and inflammation, but being limited by the development of AINR and lack of CD4 T cell help so that the graft is not acutely rejected. The same mechanism may also be involved in chronic autoimmune diseases where CD8 T cells can contribute to the disease.
The molecular basis for the AINR state
While classical CD4 T cell anergy differs from AINR in CD8 T cells with respect to the mode of induction, lack of costimulation versus full activation, the consequences are similar; the cells can signal through the TCR to upregulate effector functions, but fail to produce IL-2 in response to TCR and CD28 signaling. The first signaling defects characterized in anergic CD4 T cells were blocks in activation of the ras-MAP-kinase pathway. Anergic CD4 T cell clones failed to activate p21 ras in response to TCR and CD28 engagement [63] with a resulting block in ERK and JNK upregulation [64,65]. Activated ERK and JNK act synergistically to transactivate AP-1, a transcription factor required for IL-2 production [66]. A third member of the MAPK family, p38, has also been implicated as being necessary for IL-2 production [67]. Defects in the ras-MAP-kinase pathway have also been demonstrated in CD8 T cells in the AINR state, although this has been much less extensively studied than in CD4 T cells.
Signaling in naïve and AINR CD8 T cells was studied in vitro using artificial APC having just peptide-MHC Ag complex or anti-TCR mAb immobilized alone on microspheres to provide signal 1, or along with immobilized B7-1 to provide signals 1 and 2 [68]. ERK was activated in response to just TCR engagement, and activation was increased little, if at all, when B7-1 was present (Fig. 1). Signal 1 alone also significantly increased phosphorylation of p38, but there was a substantial further increase when B7-1 was also present, as Zhang et.al. observed for mouse splenic T cells [67]. In contrast, JNK was only activated in response to both TCR and CD28 signals, as is the case for CD4 T cells [65,69].
Figure 1.
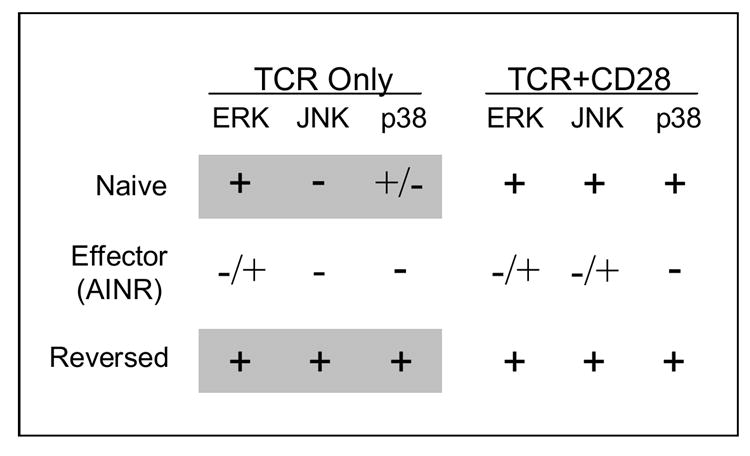
MAP kinase activation in naïve and effector (AINR) CD8 T cell, and cells that have been stimulated with IL-2 to reverse AINR. Cells were stimulated with either Ag (TCR) or Ag and B7-1 (TCR/CD28) and kinase activity measured for ERK and JNK, and phosphorylation measured for p38 [51]. Shaded regions highlight the ‘re-wiring’ that occurs upon reversal of AINR.
Defects in activation of all three of these MAPKs were seen in AINR CD8 T cells [68]. Initial activation of ERK (1 min) was comparable in normal and AINR cells, but activity declined much more rapidly in AINR cells. At 10 min, ERK activity remained high in normal cells, but had declined to almost basal level in AINR cells. JNK activity was reduced by greater than 50% in AINR cells at all time points, and p38 activation was strongly inhibited, with little or no phosphorylation detected at any time. Specific inhibitors of ERK and p38 (PD98059 and SB202190, respectively) caused a dosedependent decrease in IL-2 mRNA upregulation and proliferation of naïve CD8 T cells responding to TCR and CD28 signaling, suggesting that reduced activation of these kinases in AINR cells accounts, at least in part, for their inability to respond.
Activation of p21ras, which is upstream in the MAPK signaling pathway, is defective in anergic CD4 T cells [63], and this is likely to also be the case for CD8 T cells in the AINR state. Treatment of AINR cells with phorbol myristic acetate (PMA), an activator of ras and PKC, and ionomycin, a calcium ionophore, activated ERK, JNK and p38 and stimulated IL-2 production and proliferation by the cells, and the proliferation was inhibited by ERK and p38 inhibitors [68]. These results suggest that cells in the AINR state are unable to activate p21 ras via TCR and CD28 signaling, although this has not yet been directly demonstrated. Thus, it appears that the AINR state of CD8 T cells closely resembles that of anergic CD4 T cells with respect to defects in activation of the MAPK signaling pathway.
Uhlin et.al. [70] have recently shown that TCR engagement by anergic human CD8 T cells results in rapid degradation of lck, a src-family protein kinase involved in proximal TCR signaling. Inhibition of lck degradation, or transfection with an lck expression vector, restored responsiveness of the cells. Whether the cells examined in these experiments were in a state comparable to the AINR state seen in activated primary murine CD8 T cells is unclear, since development and maintenance of non-responsiveness in the human CD8 T cell lines was not affected by the exogenous addition of IL-2. A role for degradation of components of the T cell signaling pathway in CD4 T cell anergy has also emerged from results showing that a number of E3 ubiquitin ligases have important roles in the induction and maintenance of anergy [71], as discussed elsewhere in this volume. Whether degradation of signaling pathway components, by ubiquitination-dependent pathways or other pathways, has a role in the development or maintenance of the AINR state in primary CD8 T cells following an initial response to Ag and costimulatory signals remains to be determined.
As mentioned above, the AINR state can be reversed by culturing cells for one to two days in the presence of exogenous IL-2. Following reversal, the cells can again respond to Ag by upregulating mRNA expression and proliferating [51]. Furthermore, in contrast to naïve cells, the reversed cells do not require a costimulatory signal to respond; engagement of just the TCR is sufficient for IL-2 mRNA and protein upregulation, and IL-2-dependent proliferation to occur. This appears to result from some ‘re-wiring’ of the signaling pathways. While naïve cells require both Ag and costimulation to activate JNK, and to optimally activate p38, both are strongly upregulated in reversed cells in response to just TCR engagement, and B7-1 ligand makes no further contribution (Fig. 1). As is the case for naïve cells, the effects of pharmacological inhibitors indicate that both ERK and p38 contribute to IL-2 upregulation and proliferation in the reversed cells. The decreased dependence on costimulation following AINR reversal should allow the cells to more effectively continue to expand in response to Ag that is not expressed on professional APC. Memory cells also exhibit decreased dependence on costimulation, in comparison to naïve cells, raising the possibility that the ‘re-wiring’ that occurs upon reversal of AINR by IL-2 may also occur when AINR cells undergo transition to memory cells when Ag is no longer present.
Studies of signaling defects in AINR cells have primarily examined cells responding in vitro, but there is some evidence for defective activation of the MAPK signaling pathway in cells that have become AINR following an in vivo response to virus. As mentioned above, Kaech et.al. [53] showed that effector CD8 T cells present at the peak of the response to LCMV (day 8) are unable to make IL-2 or proliferate upon re-exposure to Ag, but regained responsiveness at later times. In this same study, it was shown that the effector cells at the peak of the response were defective in their ability to phosphorylate ERK in response to Ag, and thus activate the enzyme, and the ability to phosphorylate ERK increased in parallel with increased IL-2 production and proliferation as the cells transitioned to memory.
Whether development of AINR in effector CD8 T cells is intrinsic to the differentiation program initiated by TCR, CD28 and IL-12/IFN-α/β, or is, instead, the result of negative signaling through a surface receptor is unclear. CTLA-4 can deliver inhibitory signals upon binding to its B7 ligands, but blocking this interaction does not prevent or reverse development of the AINR state in vitro [48]. However, programmed death (PD)-1, another inhibitory member of the CD28 family, can negatively regulate T cell responses upon binding to its ligands PD-L1 and PD-L2. The PD-1 pathway mediates peripheral tolerance [72,73], and mice lacking PD-1 expression develop spontaneous autoimmune diseases [74,75]. The properties of PD-1-mediated inhibition of T cell responses resemble in many respects those of the AINR state. Barber et.al. [76] showed that blockade of the PD-1/PD-L1 pathway restored responsiveness of exhausted CD8 T cells present in mice chronically infected with LCMV. PD-1 mediated inhibition results in decreased IL-2 production and proliferation, but the cells remain responsive to IL-2 and exogenous IL-2 can overcome the inhibition and augment IL-2 production [77]. PD-1 expression is upregulated upon activation of CD8 T cells, and the similarities of PD-1-mediated inhibition and the AINR state suggest that PD-1 may mediate AINR. In the in vitro studies examining development of AINR in purified CD8 T cells using artificial APC, a PD-1 ligand was not present on the Ag-presenting surface [48,51,68]. However, in addition to expressing PD-1, activated T cells express the PD-1 ligand PD-L1 [78]. Thus, it is possible that PD-1 might mediate development of AINR under these conditions also, by acting in trans, i.e. by PD-1 on one T cell binding to PD-L1 on other cells in the culture. Whether this is the case remains to be determined.
4. Conclusion
The requirement for a third signal to support differentiation and memory development, and the requirement for IL-2 to sustain effector CTL expansion if Ag persists, represent two distinct checkpoints in the CD8 T cell response (Fig. 4). CD4 T helper cells can provide the signals necessary for CD8 T cells to pass these checkpoints, by stimulating DC to produce signal 3 cytokines, IL-12 and/or IFN-α/β, and by producing IL-2 to reverse AINR and allow continued expansion. In the absence of CD4 T cell help, the first checkpoint can still be passed if DC become activated through engagement of TLR or other surface receptors to produce signal 3 cytokines. Whether alternatives pathways exist to pass the AINR checkpoint is not clear, although costimulatory receptors of the TNFR family (OX40, 4-1BB) may be able to stimulate the AINR cells that are unresponsive to CD28-mediated costimulation.
These checkpoints may be the major means by which self-reactive CD8 T cells are prevented from productively responding to cause autoimmune diseases or transplant rejection. Encounter with self-Ag in a non-inflammatory environment where DC are not activated will result in initial proliferation, but tolerance will ensue in the absence of a signal 3 cytokine. However, the self-reactive cells may successfully pass this initial checkpoint if they encounter Ag in an inflammatory environment, such as could occur during an infection. In this regard, the emerging roles of IFN-α in directly signaling to T cells to skew CD4 cells to the Th1 pathway and drive differentiation of Ag-activated CD8 T cells are particularly interesting. The importance of Type I IFN in the pathogenesis of autoimmunity has been appreciated for some time, and recent evidence is implicating genetic alterations that lead to Type I IFN overproduction as contributing to susceptibility to systemic lupus erythematosus and possibly other autoimmune diseases (reviewed in [79,80]). Type I IFNs have a broad and diverse impact on both innate and adaptive immune responses, and affect many cell types, but direct signaling to skew and activate CD4 and CD8 T cells is likely to be among their important roles in contributing to autoimmune diseases.
Even when CD8 T cells receive a full set of signals for initial activation and differentiation, AINR limits the extent to which they can expand in the face of persisting Ag. A CD8 T cell response to self-Ag or transplanted tissue under conditions where a full set of initial signals is available, but help in the form of IL-2 produced by CD4 T cells is not available to reverse AINR, may contribute to chronic autoimmune disease states or transplant rejection. As the effector CTL in the AINR state become exhausted, disease may wane, but may recur if newly emerged self-reactive CD8 T cells again encounter Ag under stimulatory conditions.
Signaling by cytokines clearly plays a critical role in initiating and maintaining CD8 T cell responses, and the Jaks and Stats involved in the signaling pathways employed by these cytokines are potential targets for immunosuppressive agents to treat autoimmunity or prevent transplant rejection [81]. Stat4 would appear to provide a particularly good target, given its important role in both skewing CD4 T cells to the Th1 pathway and signaling for differentiation of CD8 T cells.
Figure 2.
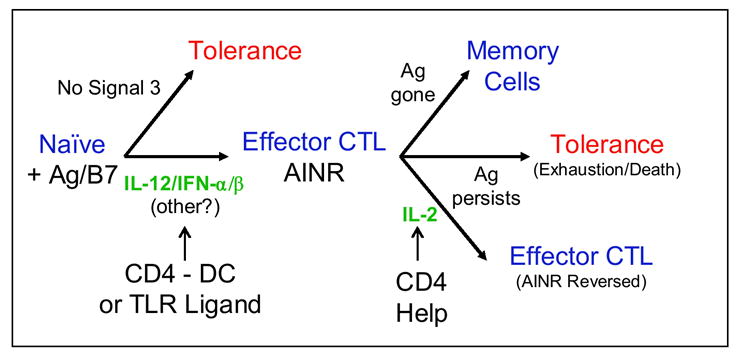
CD8 T cells require cytokine signaling to traverse checkpoints in the response and avoid tolerance.
Acknowledgments
This work was supported by NIH grants RO1 AI34824 and PO1 AI35296.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The Cellular Mechanism of Aire Control of T Cell Tolerance. Immunity. 2005;23(2):227–239. doi: 10.1016/j.immuni.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Ohashi P, Oehen S, Buerki K, Pircher H, Ohashi C, Odermatt B, Malissen B, Zinkernagel R, Hengartner H. Ablation of tolerance and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–17. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 3.Redmond WL, Sherman LA. Peripheral tolerance in CD8 T lymphocytes. Immunity. 2005;22:275–284. doi: 10.1016/j.immuni.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 4.Bevan MJ. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J Exp Med. 1976;143:1283–1288. doi: 10.1084/jem.143.5.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol. 2001;19:47–64. doi: 10.1146/annurev.immunol.19.1.47. [DOI] [PubMed] [Google Scholar]
- 6.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Ann Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 7.Bennett SRM, Carbone FR, Karamalis F, Flavell RA, Miller JFAP, Heath WR. Help for cytotoxic T-cell responses is mediated by CD40 signaling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 8.Ridge JP, DiRosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 9.Schoenberger SP, Toes REM, van der Voort EIH, Offringa R, Melief CJM. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 10.Albert ML, Jegathesan M, Darnell RB. Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nature Immunol. 2001;2:1010–1017. doi: 10.1038/ni722. [DOI] [PubMed] [Google Scholar]
- 11.Fujii S-i, Liu K, Smith C, Bonito AJ, Steinman RM. The Linkage of Innate to Adaptive Immunity via Maturing Dendritic Cells In Vivo Requires CD40 Ligation in Addition to Antigen Presentation and CD80/86 Costimulation. J Exp Med. 2004;199(12):1607–1618. doi: 10.1084/jem.20040317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernandez J, Aung S, Marquardt K, Sherman LA. Uncoupling of proliferative potential and gain of effector function by CD8(+) T cells responding to self-antigens. J Exp Med. 2002;196(3):323–33. doi: 10.1084/jem.20011612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allison JP. CD28-B7 interactions in T cell activation. Curr Opin Immunol. 1994;6:414–419. doi: 10.1016/0952-7915(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 14.Janeway CA, Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76:275–285. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 15.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Ann Rev Immunol. 1996;14:233. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 16.Mueller D, Jenkins M, Schwartz R. Clonal expansion vs functional clonal inactivation. Ann Rev Immunol. 1989;7:445–480. doi: 10.1146/annurev.iy.07.040189.002305. [DOI] [PubMed] [Google Scholar]
- 17.Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, Mescher MF. Inflammatory cytokines provide third signals for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162:3256–3262. [PubMed] [Google Scholar]
- 18.Curtsinger JM, Valenzuela JO, Agarwal P, Lins DC, Mescher MF. Cutting edge: Type I interferons provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol CE. 2005 doi: 10.4049/jimmunol.174.8.4465. in press. [DOI] [PubMed] [Google Scholar]
- 19.Gately MK, Wolitzky AG, Quinn PM, Chizzonite R. Regulation of human cytolytic lymphocyte responses by IL-12. Cell Immunol. 1992;143:127–142. doi: 10.1016/0008-8749(92)90011-d. [DOI] [PubMed] [Google Scholar]
- 20.Trinchieri G. Interleukin-12: a cytokine at the interface of inflammation and immunity. Adv Immunol. 1998;70:83. doi: 10.1016/s0065-2776(08)60387-9. [DOI] [PubMed] [Google Scholar]
- 21.Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med. 1999;189(3):521–30. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A, Gadina M, O’Shea JJ, Biron CA. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science. 2002;297(5589):2063–6. doi: 10.1126/science.1074900. [DOI] [PubMed] [Google Scholar]
- 23.Aichele P, Brduscha-Reim K, Zinkernagel R, Hengartner H, Pircher H. T cell priming versus T cell tolerance induced by synthetic peptides. J Exp Med. 1995;182:261–66. doi: 10.1084/jem.182.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aichele P, Kyburz D, Ohashi PS, Odermatt B, Zinkernagel RM, Hengartner H, Pircher H. Peptide-induced T-cell tolerance to prevent autoimmune diabetes in a transgenic mouse model. Proc Natl Acad Sci USA. 1994;91:444. doi: 10.1073/pnas.91.2.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kyburz D, Aichele P, Speiser D, Hengartner H, Zinkernagel R, Pircher H. T cell immunity after a viral infection versus T cell tolerance induced by soluble viral peptides. Eur J Immunol. 1993;23:1956–62. doi: 10.1002/eji.1830230834. [DOI] [PubMed] [Google Scholar]
- 26.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–1151. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt CS, Mescher MF. Adjuvant effect of IL-12: conversion of peptide antigen administration from tolerizing to immunizing for CD8+ T cells in vivo. J Immunol. 1999;163:2561–2567. [PubMed] [Google Scholar]
- 28.Aichele P, Unsoeld H, Koshella M, Schweier O, Kalinke U, Vucikuja S. Cutting Edge: CD8 T cells specific for lymphocytic choriomeningitis virus require Type I IFN receptor for clonal expansion. J Immunol. 2006;176:4525–4529. doi: 10.4049/jimmunol.176.8.4525. [DOI] [PubMed] [Google Scholar]
- 29.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C, Kalinke U, Tough DF. Direct stimulation of T cells by Type I IFN enhances the CD8+ T cell response during cross-priming. J Immunol. 2006;176:4682–4689. doi: 10.4049/jimmunol.176.8.4682. [DOI] [PubMed] [Google Scholar]
- 31.Thompson LJ, Kolumam GA, Thomas S, Murali-Krishna K. Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J Immunol. 2006;177:1746–1754. doi: 10.4049/jimmunol.177.3.1746. [DOI] [PubMed] [Google Scholar]
- 32.Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184(2):747–52. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Filatenkov AA, Jacovetty EL, Fischer UB, Curtsinger JM, Mescher MF, Ingulli E. CD4 T cell-dependent conditioning of dendritic cells to produce IL-12 results in CD8-mediated graft rejection. J Immunol. 2005 doi: 10.4049/jimmunol.174.11.6909. in press. [DOI] [PubMed] [Google Scholar]
- 34.van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. 2001;2(5):423–9. doi: 10.1038/87730. [DOI] [PubMed] [Google Scholar]
- 35.Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol. 2003;171:5165–5171. doi: 10.4049/jimmunol.171.10.5165. [DOI] [PubMed] [Google Scholar]
- 36.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 37.Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DA, Doherty PC, Grosveld GC, et al. Requirement for Stat4 in interleukin-12-mediated resposes of natural killer cells and T cells. Nature. 1996;382:171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- 38.Murphy TL, Geissal ED, Farrar JD, Murphy KM. Role of the Stat4 N domain in recptor proximal tyrosine phosphorylation. Mol Cell Biol. 2000;20:7121–7131. doi: 10.1128/mcb.20.19.7121-7131.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Croft M, Carter L, Swain SL, Dutton RW. Generation of polarized antigen-specific CD8 effector populations: reciprocal action of interleukin (IL)-4 and IL-12 in promoting type 2 versus type 1 cytokine profiles. J Exp Med. 1994;180:1715–1728. doi: 10.1084/jem.180.5.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haring JS, Badovinac VP, Harty JT. Inflaming the CD8+ T cell response. Immunity. 2006;25:19–29. doi: 10.1016/j.immuni.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 41.Badovinac VP, Tvinnereim AR, Harty JT. Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science. 2000;290:1354–1358. doi: 10.1126/science.290.5495.1354. [DOI] [PubMed] [Google Scholar]
- 42.Whitmire JK, Tan JT, Whitton JL. IFN-gamma acts directly on CD8+ T cells to increase their abundance during virus infection. J Exp Med. 2005;201:1053–1059. doi: 10.1084/jem.20041463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sercan O, Hammerling GJ, Arnold B, Schuler T. Innate immune cells contribute to the IFN-gamma-dependent regulation of antigen-specific CD8+ T cell homeostasis. J Immunol. 2006;176:735–739. doi: 10.4049/jimmunol.176.2.735. [DOI] [PubMed] [Google Scholar]
- 44.Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, Johnston J, Madden K, Xu W, West J, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000;408(6808):57–63. doi: 10.1038/35040504. [DOI] [PubMed] [Google Scholar]
- 45.Valenzuela J, Hammerbeck CD, Mescher MF. Cutting Edge: Bcl-3 up-regulation by signal 3 cytokine (IL-12) prolongs survival of antigen-activated CD8 T cells. J Immunol. 2005;174:600–604. doi: 10.4049/jimmunol.174.2.600. [DOI] [PubMed] [Google Scholar]
- 46.Rocha B, Grandien A, Freitas A. Anergy and exhaustion are independent mechanisms of peripheral T cell tolerance. J Exp Med. 1995;181(3):993–1003. doi: 10.1084/jem.181.3.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJD, Suresh M, Altman JD, Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1999;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deeths MJ, Kedl RM, Mescher MF. CD8+ T cells become nonresponsive (anergic) following activation in the presence of costimulation. J Immunol. 1999;163(1):102–10. [PubMed] [Google Scholar]
- 49.Jenkins M, Schwartz R. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med. 1987;165:302–319. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mueller D, Jenkins M. Molecular mechanisms underlying function T-cell unresponsiveness. Current Opinion in Immunol. 1995;7:375–81. doi: 10.1016/0952-7915(95)80113-8. [DOI] [PubMed] [Google Scholar]
- 51.Tham EL, Shrikant P, Mescher MF. Activation-induced nonresponsiveness: A Th-dependent regulatory checkpoint in the CTL response. J Immunol. 2002;168:1190–1197. doi: 10.4049/jimmunol.168.3.1190. [DOI] [PubMed] [Google Scholar]
- 52.Otten G, Germain R. Split anergy in a CD8+ T cell: Receptor-dependent cytolysis in the absence of interleukin-2 production. Science. 1991;251:1228–1231. doi: 10.1126/science.1900952. [DOI] [PubMed] [Google Scholar]
- 53.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and Functional Profiling of Memory CD8 T Cell Differentiation. Cell. 2002;111(6):837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 54.Shrikant P, Mescher MF. Control of syngeneic tumor growth by activation of CD8+ T cells: efficacy is limited by migration away from the site and induction of nonresponsiveness. J Immunol. 1999;162:2858–2866. [PubMed] [Google Scholar]
- 55.Wherry EJ, Barber DL, Kaech SM, Blattman JN, Ahmed R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. PNAS. 2004;101(45):16004–16009. doi: 10.1073/pnas.0407192101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shrikant P, Mescher MF. Opposing effects of interleukin-2 in tumor immunotherapy: promoting CD8 T cell growth and inducing apoptosis. J Immunol. 2002;169:1753–1759. doi: 10.4049/jimmunol.169.4.1753. [DOI] [PubMed] [Google Scholar]
- 57.Blattman JN, Grayson JM, Wherry EJ, Kaech SM, Smith KA, Ahmed R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat Med. 2003;9(5):540–547. doi: 10.1038/nm866. [DOI] [PubMed] [Google Scholar]
- 58.Shrikant P, Khoruts A, Mescher MF. CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell and IL-2-dependent mechanism. Immunity. 1999;11:483–493. doi: 10.1016/s1074-7613(00)80123-5. [DOI] [PubMed] [Google Scholar]
- 59.Buentke E, Mathiot M, Tolaini M, Di Santo J, Zamoyska R, Seddon B. Do CD8 effector T cells need IL-7R expression to become resting memory cells? Blood. 2006;108:1949–1956. doi: 10.1182/blood-2006-04-016857. [DOI] [PubMed] [Google Scholar]
- 60.Prlic M, Lefrancois L, Jameson SC. Multiple choices: regulation of memory CD8 T cell generation and homeostasis by interleukin (IL)-7 and IL-15. J Exp Med. 2002;195:F49–F52. doi: 10.1084/jem.20020767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4(12):1191–8. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 62.Lacombe MH, Hardy MP, Rooney J, Labrecque N. IL-7 receptor expression levels do not identify CD8 memory T lymphocyte precursors following peptide immunization. J Immunol. 2005;175:4400–4407. doi: 10.4049/jimmunol.175.7.4400. [DOI] [PubMed] [Google Scholar]
- 63.Fields PE, Gajewski TF, Fitch FW. Blocked Ras activation in anergic CD4+ T cells. Science. 1996;271:1276–1278. doi: 10.1126/science.271.5253.1276. [DOI] [PubMed] [Google Scholar]
- 64.DeSilva DR, Feeser WS, Tancula EJ, Scherle PA. Anergic T cells are defective in both jun NH2-terminal kinase and mitogen-activated protein kinase signaling pathways. J Exp Med. 1996;183(5):2017–23. doi: 10.1084/jem.183.5.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li W, Whaley CD, Mondino A, Mueller DL. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+ T cells. Science. 1996;271:1272–1276. doi: 10.1126/science.271.5253.1272. [DOI] [PubMed] [Google Scholar]
- 66.Kang SM, Beverly B, Tran AC, Brorson K, Schwartz RH, Lenardo MJ. Transactivation by AP-1 is a molecular target of T cell clonal anergy. Science. 1992;21:1134–1138. doi: 10.1126/science.257.5073.1134. [DOI] [PubMed] [Google Scholar]
- 67.Zhang J, Salojin KV, Gao JX, Cameron MJ, Bergerot I, Delovitch TL. p38 mitogen-activated protein kinase mediates signal integration of TCR/CD28 costimulation in primary murine T cells. J Immunol. 1999;162:3819–3829. [PubMed] [Google Scholar]
- 68.Tham EL, Mescher MF. Signaling alterations in activation-induced non-responsive (AINR) CD8 T cells. J Immunol. 2001;167:2040–2048. doi: 10.4049/jimmunol.167.4.2040. [DOI] [PubMed] [Google Scholar]
- 69.Su B, Jacinto E, Hibi M, Kallunki T, Karin M, Ben-Neriah Y. JNK is involved in signal integration during costimulation of T lymphocytes. Cell. 1994;77:727–736. doi: 10.1016/0092-8674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 70.Uhlin M, Masucci MG, Levitsky V. Regulation of lck degradation and refractory state in CD8+ cytotoxic T lymphocytes. Proc Natl Acad Sci U S A. 2005;102:9264–9269. doi: 10.1073/pnas.0406333102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mueller DL. E3 ubiquitin ligases as T cell anergy factors. Nat Immunol. 2004;5:883–890. doi: 10.1038/ni1106. [DOI] [PubMed] [Google Scholar]
- 72.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Ann Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 73.Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006;27:195–201. doi: 10.1016/j.it.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 74.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 75.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 76.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 77.Carter LL, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, Collins M, Honjo T, Freeman GJ, Carreno BM. PD-1:PD-L inhibitory pathway affects both CD4+ and CD8+ T cells and is overcome by IL-2. Eur J Immunol. 2002;32:634–643. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 78.Lactchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Banchereau J, Pascual V. Type I interferon in Systemic Lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 80.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Ann Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 81.O’Shea JJ, Park H, Pesu M, Borie D, Changelian P. New strategies for immunosuppression: interfering with cytokines by targeting the Jak/Stat pathway. Curr Op Rheumatol. 2005;17:305–311. doi: 10.1097/01.bor.0000160781.07174.db. [DOI] [PubMed] [Google Scholar]