

Abstract
We studied the involvement of interferon-regulated, PKR on 2-ME–mediated actions in human osteosarcoma cells. Our results show that PKR is activated by 2-ME treatment and is necessary for 2-ME–mediated induction of osteosarcoma cell death.
Introduction
Osteosarcoma is the most common primary bone tumor and most frequently develops during adolescence. 2-Methoxyestradiol (2-ME), a metabolite of 17β-estradiol, induces interferon gene expression and apoptosis in human osteosarcoma cells. In this report, we studied the role of interferon-regulated double-stranded (ds)RNA-dependent protein kinase (PKR) protein on 2-ME–mediated cell death in human osteosarcoma cells.
Materials and Methods
Western blot analyses were used to measure PKR protein and phosphorylation levels. Cell survival and apoptosis assays were measured using trypan blue exclusion and Hoechst dye methods, respectively. A transient transfection protocol was used to express the dominant negative PKR mutants.
Results and Conclusions
PKR was increased in 2-ME–treated MG63 cells, whereas 17β-estradiol, 4-hydroxyestradiol, and 16α-hydroxyestradiol, which do not induce cell death, had no effect on PKR protein levels. Also, 2-ME treatment induced PKR kinase activity as indicated by increased autophosphorylation and phosphorylation of the endogenous substrate, eukaryotic initiation factor (eIF)-2α. dsRNA poly (I).poly (C), an activator of PKR protein, increased cell death when osteosarcoma cells were treated with a submaximal concentration of 2-ME. In contrast, a serine-threonine kinase inhibitor SB203580 and a specific PKR inhibitor 2-aminopurine (2-AP) blocked the 2-ME–induced cell death in MG63 cells. A dominant negative PKR mutant protein conferred resistance to 2-ME–induced cell death to MG63 osteosarcoma and 2-ME–mediated PKR regulation did not require interferon gene expression. PKR protein is activated in cell free extracts by 2-ME treatment, resulting in autophosphorylation and in the phosphorylation of the substrate eIF-2α. We conclude from these results that PKR is regulated by 2-ME independently of interferon and is essential for 2-ME–mediated cell death in MG63 osteosarcoma cells.
Keywords: estrogen metabolite, MG63 cells, interferon, protein kinase, double-stranded RNA-dependent protein kinase
INTRODUCTION
Osteosarcoma, although relatively uncommon, is the most common bone tumor and occurs most frequently during adolescence. A combination of surgery and chemotherapy has led to improved survival rate. However, a specific therapy is not yet available, and the mortality rate is still quite high, in part because 30% of patients diagnosed with osteosarcoma will develop metastatic diseases.(1,2)
2-Methoxyestradiol (2-ME) is a potential therapeutic agent for several types of cancer,(3–5) including bone cancer.(6) 2-ME induces apoptosis in osteosarcoma cells but not in normal osteoblasts.(6) 17β-Estradiol is metabolized by multiple hydroxylation pathways, resulting in the formation of 2-, 4-, and 16α-hydroxylated derivatives.(7) 2-Hydroxylation of 17β-estradiol is followed by methylation of the product and leads to the generation of 2-ME.(8) The anti-tumor activities of the 2-ME metabolite are not shared by 17β-estradiol or 4- and 16α-hydroxylated metabolites. Instead, the other hydroxylation pathways generate 4- and 16α-hydroxyestradiol (4-OHE and 16-OHE), which have been shown under some circumstances to exhibit tumor-promoting actions.(7)
The cytotoxic activity of 2-ME involves disruption of the cytoskeleton and induction of apoptosis. 2-ME inhibits tubulin formation that antagonizes mitosis.(9) Additionally, 2-ME stimulates expression of the p53 gene, which leads to induction of apoptosis.(5,10) 2-ME, however, can induce apoptosis in the absence of p53 protein in a pancreatic tumor cell line.(3,11) Taken together, these series of studies suggest that 2-ME activates multiple proapoptotic signaling pathways.
2-ME–induced apoptosis in osteosarcoma cells is preceded by an increase in interferon gene expression.(6) Interferon-regulated cell death and anti-cancer mechanisms have been well described and are often mediated by double-stranded (ds)RNA-dependent protein kinase (PKR), ribonuclease L, 2′5′-oligoadenylate synthetase, and Janus kinase (Jak)-signal transducer and activator of transcription (Stat) signaling.(12–20)
PKR is an interferon-induced, dsRNA-dependent serine-threonine protein kinase.(14,21) PKR is activated by viruses that produce dsRNA.(22–24) In the presence of dsRNA and ATP, PKR is autophosphorylated on several of its serine and threonine residues.(25–30) Once phosphorylated, PKR catalyzes the phosphorylation of target substrates. Protein synthesis initiation factor eIF-2α is the most completely characterized substrate for PKR. EIF-2α phosphorylation leads to an inhibition of cellular and viral protein synthesis, growth suppression, and apoptotic cell death. Other substrates (e.g., I-κB) have also been shown for the PKR protein.(31,32) In addition, PKR is involved in the regulation of several transcription factors including NF-κB,(31,32) interferon-regulating factor (IRF),(33) p53,(34) and Stat(35) proteins, which play a crucial role in inflammation, cell growth, differentiation, proliferation, and induction of apoptosis in a variety of different tumor cells.
Overexpression of dominant negative PKR protein carrying a loss of function mutation has been shown to protect cells from apoptosis induced by ds RNA, lipopolysaccharide, and TNF-α.(36) Expression of catalytically inactive mutant forms of the kinase can cause malignant transformation, suggesting that PKR is a tumor suppressor.(37,38) The purpose of this study in osteosarcoma cells was to investigate involvement of the interferon-regulated PKR pathway in 2-ME–mediated cell death.
MATERIALS AND METHODS
Metabolite treatment and cell growth assay
MG63 osteosarcoma cells were plated at 5 × 104 cells/well into 24-well plates containing 1 ml/well DMEM/F12 medium containing 10% charcoal-stripped FBS. After allowing the cells to attach overnight, the media in the wells were replaced with fresh 1 ml of medium containing 5 µM 2-ME, 4-OHE, 16α-OHE, 2-methoxyestrone (2-MEOE1), and 17β-estradiol (E2) (Sigma Chemical, St Louis, MO, USA). Cells were treated with PKR inhibitor 2-aminopurine (2-AP; Sigma; 25 µM), serine-threonine kinase inhibitor SB 203580 (Calbiochem, San Diego, CA, USA; 25 mM), and PKR activator poly (I).poly (C) (Sigma; 50 µg/ml). Viable cells were counted using the trypan blue method as described.(6) For transient transfection assays, cells were grown in 6-well plates at a plating density of 1.5 × 106 cells/well.
Transient transfection
MG63 cells plated in 6-well plates were transfected at 60% confluence with 5 µg of pcDNA3-PKR-K296R or empty vector pcDNAneo using the transfection agent lipofectamine as described in the manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA). Twenty-four hours after transfection, the cells were treated with vehicle and 2-ME, and the cell viability was measured by trypan exclusion after 72 h.(6)
Hoechst staining of apoptotic cells
The apoptosis assay to detect chromatin condensation was performed using Hoechst staining as described.(39) Hoechst stain is a nuclear counterstain that emits blue fluorescence. It is membrane permeable and stains with low cytotoxicity. It intercalates in A-T regions and was used to distinguish condensed pyknotic nuclei in apoptotic cells. Twenty-four hours after transfection, the MG63 cells were treated with vehicle and 2-ME. After 72 h of treatment, the cells were fixed with 1% paraformaldehyde and stained for 60 minutes with Hoechst 33258 (Sigma) diluted to 5 µg/ml in PBS containing 0.01% Tween 20 as described.(39)
Preparation of cytoplasmic extract and protein analysis
Cytoplasmic extracts were prepared as described.(40) Briefly, cells harvested after 2-ME treatment were lysed by suspending in Nonidet P-40 lysis buffer containing 0.5% Nonidet P-40, 90 mM KCl, 1 mM magnesium acetate, 2 mM 2-mercaptoethanol, 10 mg/ml leupeptin, and 10 mM HEPES (pH 7.6). After centrifugation at 10,000g for 10 minutes, the resultant supernatant containing cytoplasmic extract (60 µg protein) was analyzed by Western blot hybridization using anti-PKR (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-actin (Sigma), anti-eIF-2α (Cell Signaling Technology, Danvers, MA, USA), anti-phospho-PKR (pT446), and anti-phospho-eIF-2α (Cell Signaling) antibodies.
In vitro PKR kinase assay
The cytoplasmic extract prepared from untreated MG63 cells was used for in vitro kinase assay. The assay mixture contained protein (60 µg) extract, 20 mM HEPES (pH 7.5), 80 mM KCl, 1 mM MgCl2, 1 mM MnCl2, 0.1 mM ATP, and 0.1 mM 2-ME in a final volume of 20 µl. The kinase assay mixture was incubated with vehicle or different concentrations of 2-ME for 30 minutes at 30°C. The proteins in the reaction mixture were analyzed by Western blot hybridization using anti-phospho-PKR and anti-phospho-eIF-2α antibodies.
Statistical analysis
All values are expressed as mean ± SE. Significant differences between groups were determined by Fisher’s protected least significant difference posthoc test for multiple group comparisons after detection of significance by one-way ANOVA. p < 0.05 was considered significant.
RESULTS
2-ME increases PKR protein levels and activity before cell death
Western blot hybridization using an anti-PKR antibody was used to determine whether 2-ME has an effect on PKR protein levels (Fig. 1). PKR protein was increased in 2-ME–treated cells after 24 h, whereas E2, 4-OHE, and 16α-OHE had no effect. Hybridization of the same blot using an anti-actin antibody showed that none of the test compounds had an effect on actin protein expression (Fig. 1A). Compared with solvent control, osteosarcoma cell survival in the presence of 2-ME, E2, 4-OHE, and 16α-OHE was 19%, 98%, 82%, and 99%, respectively (Fig. 1B).
FIG. 1.
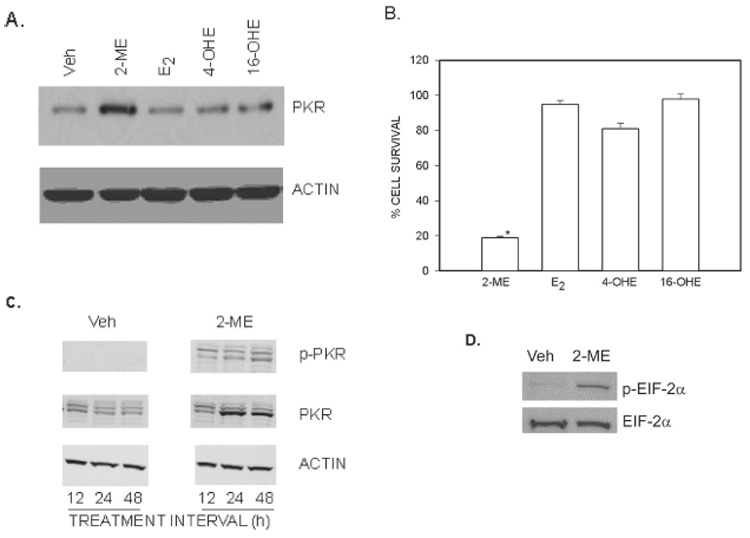
Association between an increase of PKR protein expression and cell survival in MG63 osteosarcoma cells. (A) PKR expression. Cytoplasmic extracts prepared from cells treated for 24 h with vehicle (Veh) or 10 µM of 2-ME, E2, 4-OHE, and 16-OHE was analyzed by Western blot hybridization using anti-PKR antibody and anti-actin antibody (Sigma). (B) Cell survival. Cells were treated with Veh or 5 µM of 2-ME, E2, 4-OHE, and 16-OHE for 72 h. The cells were harvested, and the viable cell counts were taken after staining with trypan blue. Values are mean ± SE (N = 3 replicate cultures). *p ≤ 0.05 vs. Veh. (C) Time-course of 2-ME effects on PKR. Cytoplasmic extracts prepared from cells treated for 12, 24, and 48 h with Veh or 10 µM of 2-ME were analyzed by Western blot hybridization using anti-phospho-PKR (pT446; Epitomics), anti-PKR (Santa Cruz Biotechnology), and anti-Actin (Sigma) antibodies. (D) EIF-2α phosphorylation. Cytoplasmic extracts prepared from cells treated with Veh or 10 µM of 2-ME for 24 h were analyzed by Western blot hybridization using anit-phospho-eIF-2α (pT446; Epitomics) and anti-eIF-2α (Cell Signaling Technology).
The effects of 2-ME treatment on autophosphorylation and expression of PKR protein were determined by Western blot analysis of extracts prepared from MG63 cells using phosphor-PKR (pT446) and anti-PKR antibodies (Fig. 1C). 2-ME treatment resulted in the phosphorylation of PKR protein at 12, 24, and 48 h after treatment, but the increases in PKR protein levels did not occur until 24 h after 2-ME treatment, suggesting that the phosphorylation and induction of PKR follow a different time-course. Hybridization of the same blot with an anti-actin antibody showed that 2-ME treatment did not affect actin protein levels at any of the time-points tested (Fig. 1C).
To determine whether 2-ME–mediated increases in PKR phosphorylation is followed by downstream phosphorylation of eIF-2α, we analyzed the cytoplasmic extracts from vehicle and 2-ME–treated cells by Western blot hybridization using anti-phospho-eIF-2α and anti-eIF-2α antibodies. 2-ME treatment increased the eIF-2α phosphorylation levels compared with vehicle but did not have any effect on total protein levels of eIF-2α (Fig. 1D).
Poly (I).poly(C) enhances the potency of 2-ME–mediated killing of osteosarcoma cells
The effects of poly (I).poly(C), a known activator of PKR on MG63 osteosarcoma cell survival, is shown in Fig. 2. 2-ME treatment resulted in a dose-related decrease in cell survival. Poly (I).poly(C) treatment resulted in a significant decrease in cell survival and further reduced cell survival in 2-ME–treated cultures.
FIG. 2.
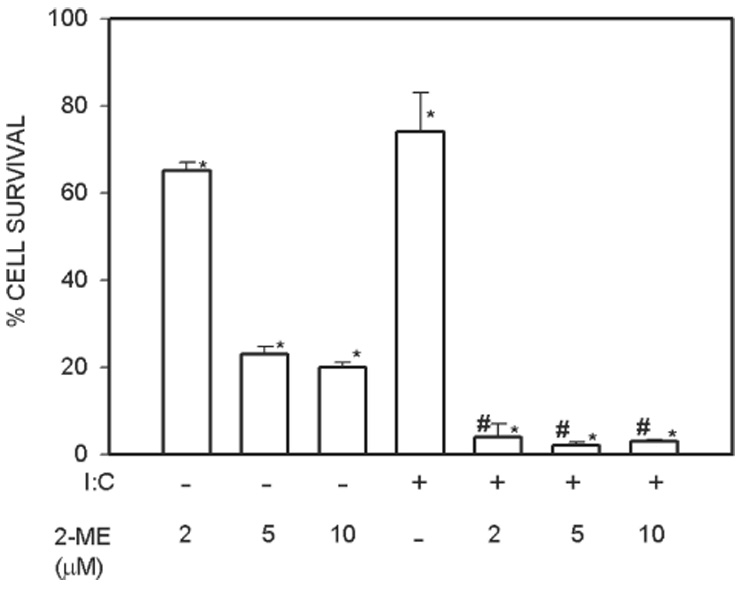
Poly (I).Poly(C) enhances 2-ME–mediated cell death. MG63 cells were treated with vehicle (Veh) and 5 µM of 2-ME in the presence and absence of 100 µg/ml Poly (I).Poly(C) for 72 h. Values are mean ± SE (N = 3 replicate cultures). *p ≤ 0.05 vs. Veh; #p ≤ 0.05 vs. 2-ME.
2-ME–induced cell death is antagonized by PKR inhibitors
The effects of 2-AP, a specific PKR inhibitor, and SB 203580, a general serine-threonine kinase inhibitor, were determined on 2-ME–treated MG63 cells. Cell survival in 2-ME–treated cultures increased in the presence of 2-AP (55%) and SB 203580 (70%; Fig. 3A). Neither 2-AP nor SB 203580 had an independent effect on cell survival. The respective effects of 2-ME and 2-AP on apoptosis was measured by Hoechst dye staining (Fig. 3B). Hoechst dye staining is used for detecting apoptosis, and it distinguishes condensed pyknotic nuclei in apoptotic cells.(39) The 2-ME–mediated increases in Hoechst dye–positive MG63 cells was reduced significantly in cultures treated with 2-AP. 2-AP had no independent effect on Hoechst dye staining.
FIG. 3.
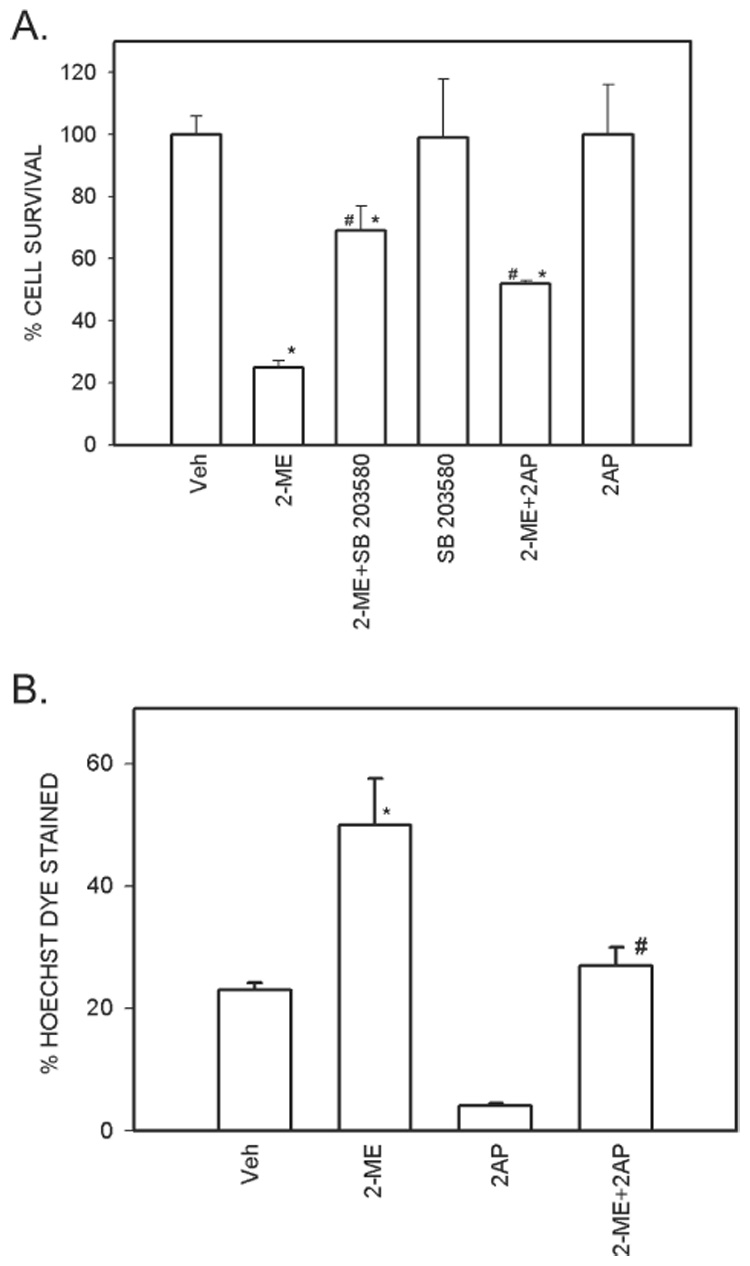
PKR inhibitors antagonize 2-ME effects in osteosarcoma cells. (A) Cell survival. MG63 cells were treated with vehicle (Veh) and 5 µM 2-ME in the presence and absence of 25 µM SB 203580 or 50 µM of 2-AP for 72 h. Values are mean ± SE (N = 3 replicate cultures). *p ≤ 0.05 vs. Veh; #p ≤ 0.05 vs. 2-ME. (B) Hoechst dye staining. MG63 cells were treated with vehicle (Veh) and 5 µM of 2-ME in the presence and absence of 50 µM of 2-AP for 72 h. Values are mean ± SE (N = 3 replicate cultures). *p ≤ 0.05 vs. Veh; #p ≤ 0.07 vs. 2-ME.
Osteosarcoma cells expressing transdominant PKR mutants are resistant to 2-ME–mediated reduction in cell survival
The effect of 2-ME on MG63 cells that express trans-dominant negative PKR mutant K296R are shown in Fig. 4. It has been shown that overexpression of K296R that lacks an autophosphorylation site inhibits endogenous PKR protein. 2-ME was ineffective in reducing cell survival of osteosarcoma cells that express PKR mutants.
FIG. 4.
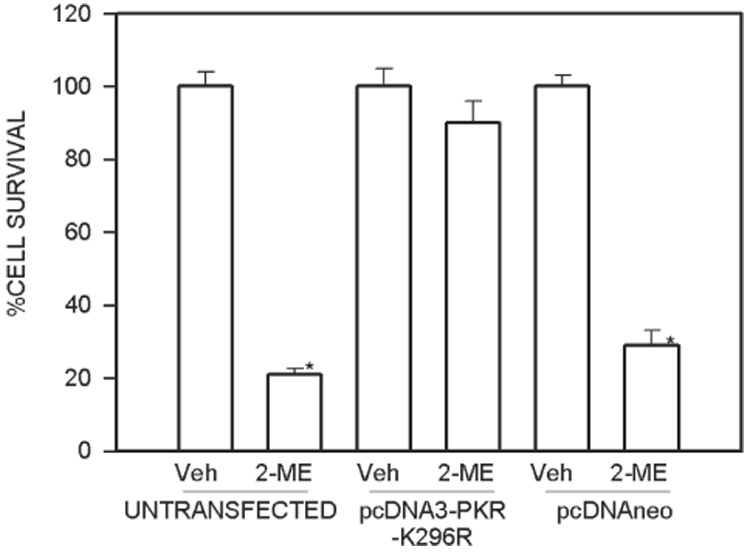
A dominant negative mutant PKR increases cell survival in 2-ME–treated cultures of osteosarcoma cells. MG63 cells were transfected with the plasmid pcDNA3-PKR-K296R or empty vector pcDNAneo and treated with vehicle (Veh) or 5 µM 2-ME for 72 h. The cells were harvested, and the viable cell counts were taken after staining with trypan blue. Values are mean ± SE (N = 3 replicate cultures). *p ≤ 0.05 vs. Veh.
2-ME–mediated cell death and PKR regulation are independent of interferon expression
To determine whether the interferon gene is involved in the action of PKR, 2-ME was administered in the presence and absence of anti-interferon β neutralizing antibodies. Neither the 2-ME–mediated cell death (Fig. 5A) nor the 2-ME–mediated PKR induction (Fig. 5B) was inhibited by anti-interferon β antibodies. The cell survival was decreased to 42% and 44%, respectively, in the absence and presence of neutralizing anti-interferon antibody. On its own, the anti-interferon β antibody did not affect cell death and PKR (Figs. 5A and 5B), but other studies reveal that the anti-interferon antibody blocks interferon-induced cell death in osteosarcoma cells (Fig. 6).
FIG. 5.
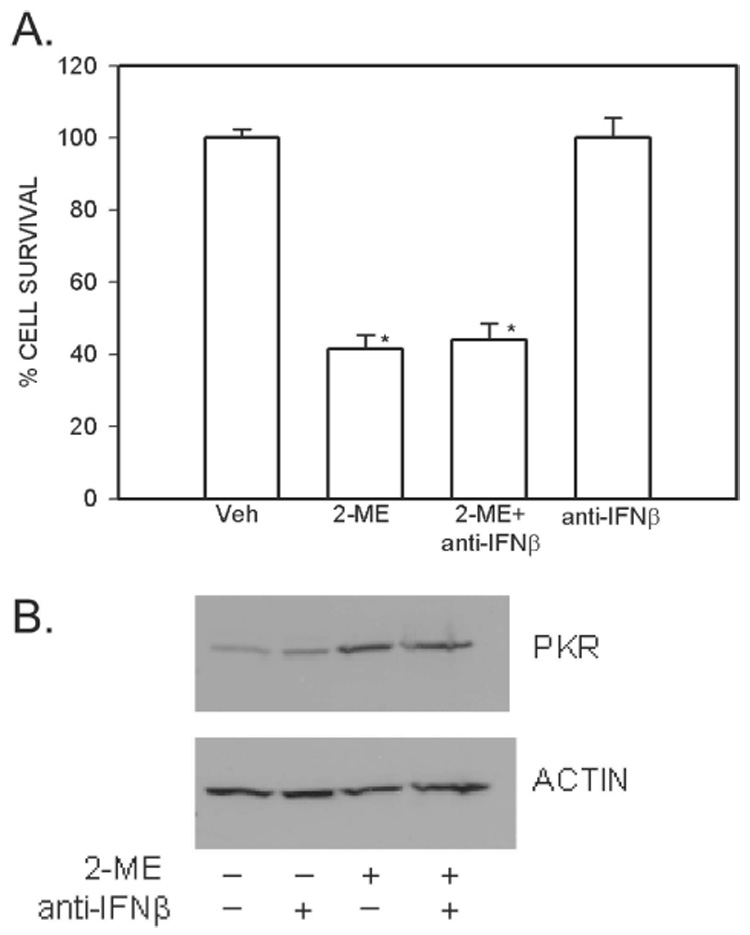
2-ME–mediated induction of cell death and PKR expression are not blocked by anti-IFNβ antibody. (A) Cell survival. MG63 cells were treated with vehicle (Veh) and 5 µM 2-ME in the presence and absence of 1 µg/ml of anti-IFNβ antibody for 72 h. The cells were harvested, and the viable cell counts were taken after staining with trypan blue. Values are mean ± SE (N = 3 replicate cultures). *p ≤ 0.05 vs. Veh. (B) Protein expression. Cytoplasmic extracts were prepared from cells treated for 24 h with Veh or 10 µM 2-ME in the presence or absence of 1 µg/ml of anti-IFNβ antibody and analyzed by Western blot hybridization using anti-PKR antibody and anti-actin antibody (Sigma).
FIG. 6.
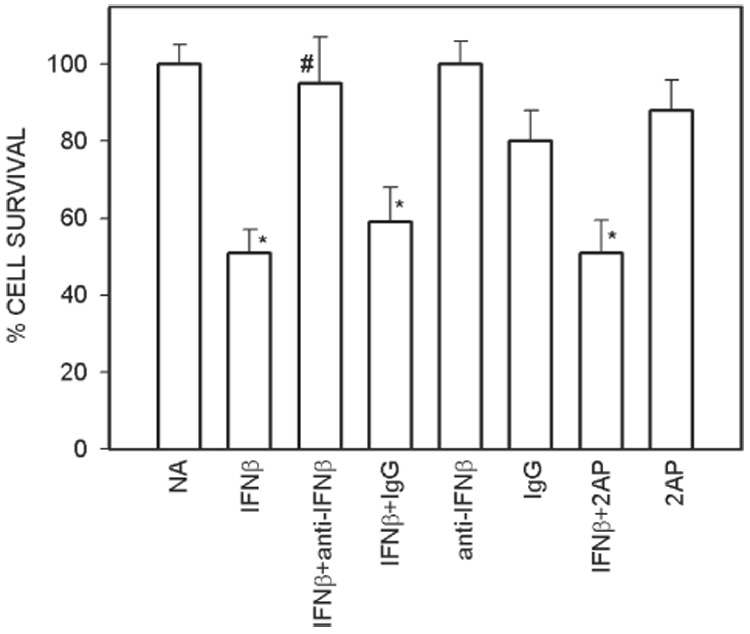
Interferon treatment induces cell death in osteosarcoma cells. MG63 cells were exposed to no addition (NA) or 2000 units/ml of IFNβ in the presence and absence of 1 µg/ml of anti-IFNβ antibody or 1 µg/ml of control IgG or 50 µM 2-AP. Values are mean ± SE (N = 3 replicate cultures). *p ≤ 0.05 vs. NA; #p ≤ 0.05 vs. IFNβ.
Direct effects of interferon in osteosarcoma cells
Interferon induces anti-growth and anti-proliferative effects that are mediated through interferon-regulated genes. To study whether PKR gene expression is involved in direct anti-osteosarcoma effects of interferon, we studied the cell survival in interferon-treated cells in the presence of the PKR inhibitor, 2-AP. Interferon decreased the cell survival to 45% of the control value, and addition of anti-interferon antibody blocked cell death (Fig. 6). The control IgG had no effect on its own or on the interferon-mediated decrease in cell survival. Finally, the interferon-mediated decrease in cell survival was not blocked by the PKR inhibitor 2-AP (Fig. 6).
Activation of PKR in a cell-free system
To understand the mechanism of PKR activation, we examined the effect of vehicle and different concentrations of 2-ME in a cell-free system (Fig. 7). The Western blot hybridization experiments show that PKR protein is activated in the presence of 2-ME. At 2 µM, the effect was small, whereas PKR phosphorylation increased in the presence 2-ME at 10 and 50 µM. 2-ME treatment also resulted in the phosphorylation of eIF-2α.
FIG. 7.
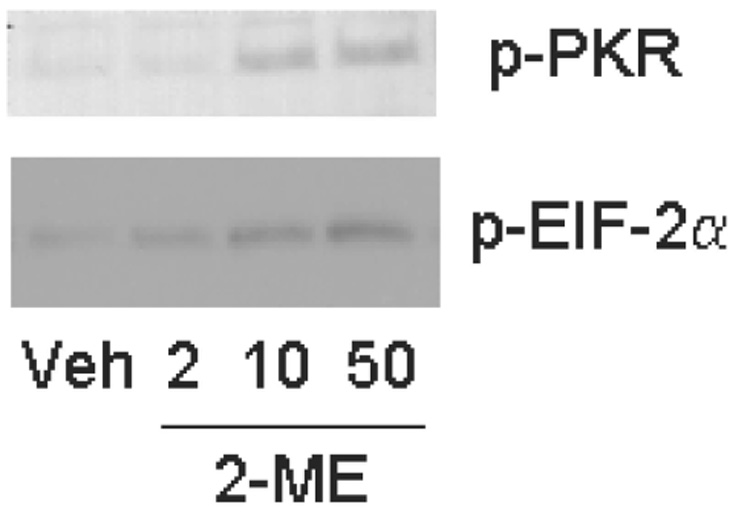
2-ME activates PKR in a cell-free system. Cytoplasmic extracts prepared from untreated MG63 cells were incubated in kinase assay mixture with vehicle (Veh) or 2, 10, and 50 µM 2-ME and assayed for PKR activity in vitro. The kinase assay mixture was analyzed by Western blot hybridization using anit-phospho-PKR (pT446; Epitomics) and anti-eIF-2α (Cell Signaling Technology).
DISCUSSION
In addition to its well-known function in mediating the antiviral and anti-proliferative effects of interferon, PKR is implicated in apoptosis, signal transduction, transcriptional regulation, translational control, cell growth, differentiation, and tumor suppression.(14–17,19,21,31,32,37,38) These results provide evidence that PKR mediates 2-ME–induced osteosarcoma cell apoptosis. PKR activity and gene expression are increased in 2-ME–treated MG63 osteosarcoma cells. In contrast, structurally similar compounds (E2, 4-OHE, and 16-OHE) that do not affect cell death did not increase the level of PKR protein. Furthermore, an activator of PKR enhanced the potency of 2-ME, whereas the chemical inhibitors and the transdominant mutant inhibitors of PKR increased osteosarcoma cell survival in 2-ME–treated cultures.
PKR is found in a latent form in most cells and requires dsRNA for its activation. PKR is activated in virus-infected cells where dsRNA is generated because of symmetrical transcription, and in other cells, where RNA forms hairpin structures.(22,24) The synthetic dsRNA poly (I).poly (C) is a well-known activator of PKR protein and has been shown to induce PKR activity in a number of cell types.(22,32,41) In this study, poly(I).poly(C) treatment enhanced the effects of 2-ME, indicating that PKR is activated to a greater extent in cells co-treated with 2-ME and poly (I).poly(C). The dsRNA poly (I).poly(C) induced cell death directly, suggesting that activation of PKR is sufficient to initiate cell death in osteosarcoma cells. This possibility needs to be verified because dsRNA poly (I).poly (C) has been shown to independently regulate several genes.(42)
PKR mediates the apoptotic actions of interleukins, interferons, heparin, TNF-α, platelet-derived growth factor, lipopolysaccharides, and ceramides. The PKR enzyme phosphorylates its substrates on serine/threonine. Our work shows that a serine/threonine kinase inhibitor SB 203580 antagonized 2-ME, providing evidence for the involvement of PKR in 2-ME–mediated osteosarcoma cell death. This involvement is further supported by the observation that 2-AP antagonizes 2-ME–mediated cell killing in MG63 cells; 2-AP is a well-established inhibitor of PKR kinase activity and PKR-mediated activation of signaling cascades.(41,43-46)
The dominant negative PKR mutant K296R has a mutation at a lysine residue within the ATP binding site and is completely inactive for autophosphorylation and substrate phosphorylation.(37,38,47) The dominant negative phenotype observed by mutant PKR overexpression results from competition for binding to potential dsRNA activators.(37,38,47) MG63 osteosarcoma cell lines become partially resistant to the proapoptotic actions of 2-ME, when K296R is transiently expressed. However, the incomplete reduction in 2-ME–mediated apoptosis could mean that other 2-ME–dependent pathways might be involved.
Interferon is a well-known inducer of PKR. Interferon in the absence of 2-ME acts directly and induces cell death in osteosarcoma cells. However, our studies show that regulation of PKR and induction of cell death by 2-ME do not require interferon. Whereas it is not surprising that interferon has direct effects, it is somewhat surprising that addition of a PKR inhibitor to interferon-treated cells has minimal effect and does not reverse 2-ME actions. It is possible that interferon-mediated cell killing may be mediated by other pathways like Jak-Stat or 2-5A system. Further work is necessary to delineate the specific pathways associated with interferon-induced cell death in osteosarcoma cells.
Activation of PKR leads to transcriptional and translational increases in cell death pathway–associated genes.(15,16,48) The translational regulation is triggered by the phosphorylation of the α subunit of eIF-2 by the activated (autophosphorylated) PKR protein.(22,25–27) This PKR-mediated phosphorylation of eIF-2α results in the inhibition of initiation of mRNA translation and represents an important mechanism of interferon action. PKR protein levels and activity increased in 2-ME–treated cells, but by different time-courses. The increase in PKR activity is accompanied by an increase in eIF-2α phosphorylation. Increases in PKR activities and expression has been shown in other cells to cause growth suppression and cell death,(15) but further studies are required to determine whether eIF-2α inhibition or other PKR-regulated mechanisms contribute to 2-ME–mediated anti-osteosarcoma effects.
PKR can also be activated directly or indirectly by calcium depletion, reactive oxygen intermediates, and the endoplasmic reticulum overload response through the activator protein protein activator of interferon-induced protein kinase (PACT).(49) 2-ME directly activated PKR in cell free extracts, leading to autophosphorylation and substrate phosphorylation. PKR is activated by nucleic acids and polysaccharides, but this is the first example of PKR activation by a steroid. Caspases and mitogen-activated protein kinase (MAP) kinases play a role in signal transducing events associated with PKR.(21,50) Studies carried out in the presence of several inhibitors to understand the upstream events in 2-ME–mediated PKR activation revealed that PKR activation is not dependent on caspases and MAP kinases (AM and KS, unpublished observations).
Studies in the recent past have focused on the involvement of translational processes and factors in control of cell proliferation, indicating that protein synthesis can be an additional target for anti-cancer strategies. Eukaryotic protein synthesis is a complex biochemical process, involving more than a hundred different polypeptides(51); seven of them have been identified as targets for regulatory pathways.(51) PKR was previously thought to be a translational inhibitor. Subsequently, it was shown to have in vivo substrates in addition to eIF-2α and is now considered to regulate gene transcription and signal transduction pathways.(52) PKR may also have a tumor suppressor function.(37,38) Expression of a functionally defective mutant of human PKR in NIH3T3 cells resulted in malignant transformation, suggesting that PKR may function as a suppressor of cell proliferation and tumorigenesis.(53) Moreover, mutant forms of eIF-2α also caused malignant transformation of NIH3T3 cells.(53) The role of PKR in cancer has been well documented. A copy of the gene for PKR protein is rearranged in a murine lymphocytic leukemia, although it is not clear whether this mutation contributes to the phenotype of the tumor.(54) Also, it has been shown that patients bearing tumors with a higher PKR content have a more favorable prognosis. PKR activation and subsequent apoptosis could be a mechanism by which 2-ME exerts its anti-osteosarcoma effects. Other studies have identified a cellular inhibitor of PKR, a p58 protein, that functions as an oncogene through the downregulation of PKR.(55)
Although many studies show a role in the suppression of tumorigenesis, higher levels of PKR have been observed in breast carcinoma.(56,57) The PKR activity in breast cancer is inhibited by the transdominant inhibitors present in the cells.(57)
This study shows that PKR contributes to pro-apoptotic actions of 2-ME in osteosarcoma cells and this process involves the phosphorylation of translational control protein eIF-2α. The transfection of dominant negative mutants of various translational factors can inhibit cell proliferation and several of them are being studied for delivery by viral-mediated gene therapy. Further studies to determine whether translational control factors other than eIF-2α potentiate the 2-ME–mediated anti-osteosarcoma effects are warranted.
The cross-talk between signal transduction and protein synthesis and the availability of different molecular tools interfering with both processes suggest the design of new multimodal therapeutic strategies for osteosarcoma and other cancers. Therefore, the use of 2-ME along with strategies to modify PKR response could increase the combined antitumor effects and offers strong promises in the treatment of osteosarcoma.
ACKNOWLEDGMENTS
These studies were supported by NIH Grants CA 10287801A1, AR 48033, and AA 011140, NASA Grant NAG9-1458, and the Mayo Clinic. The authors thank Dr Michael Mathews (University of Medicine and Dentistry of New Jersey, Newark, NJ, USA) for pcDNA3-PKR-K296R plasmid.
Footnotes
The authors state that they have no conflicts of interest.
REFERENCES
- 1.O’Reilly R, Cheung NK, Bowman L, Castle V, Hoffer F, Kapoor N, Kletzel M, Lindsley K, Shamberger R, Tubergen D. NCCN pediatric neuroblastoma practice guidelines. The National Comprehensive Cancer Network. Oncology. 1996;10:1813–1822. [PubMed] [Google Scholar]
- 2.Ward WG, Mikaelian K, Dorey F, Mirra JM, Sassoon A, Holmes EC, Eilber FR, Eckardt JJ. Pulmonary metastases of stage IIB extremity osteosarcoma and subsequent pulmonary metastases. J Clin Oncol. 1994;12:1849–1858. doi: 10.1200/JCO.1994.12.9.1849. [DOI] [PubMed] [Google Scholar]
- 3.Schumacher G, Kataoka M, Roth JA, Mukhopadhyay T. Potent antitumor activity of 2-methoxyestradiol in human pancreatic cancer cell lines. Clin Cancer Res. 1999;5:493–499. [PubMed] [Google Scholar]
- 4.Seegers JC, Lottering ML, Grobler CJ, van Papendorp DH, Habbersett RC, Shou Y, Lehnert BE. The mammalian metabolite, 2-methoxyestradiol, affects P53 levels and apoptosis induction in transformed cells but not in normal cells. J Steroid Biochem Mol Biol. 1997;62:253–267. doi: 10.1016/s0960-0760(97)00043-5. [DOI] [PubMed] [Google Scholar]
- 5.Mukhopadhyay T, Roth JA. Induction of apoptosis in human lung cancer cells after wild-type p53 activation by methoxyestradiol. Oncogene. 1997;14:379–384. doi: 10.1038/sj.onc.1200835. [DOI] [PubMed] [Google Scholar]
- 6.Maran A, Zhang M, Kennedy AM, Sibonga JD, Rickard DJ, Spelsberg TC, Turner RT. 2-Methoxyestradiol induces interferon gene expression and apoptosis in osteosarcoma cells. Bone. 2002;30:393–398. doi: 10.1016/s8756-3282(01)00681-0. [DOI] [PubMed] [Google Scholar]
- 7.Zhu BT, Conney AH. Functional role of estrogen metabolism in target cells: Review and perspectives. Carcinogenesis. 1998;19:1–27. doi: 10.1093/carcin/19.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Gelbke HP, Knuppen R. The excretion of five different 2-hydroxyoestrogen monomethyl ethers in human pregnancy urine. J Steroid Biochem. 1976;7:457–463. doi: 10.1016/0022-4731(76)90112-6. [DOI] [PubMed] [Google Scholar]
- 9.D’Amato RJ, Lin CM, Flynn E, Folkman J, Hamel E. 2-Methoxyestradiol, an endogenous mammalian metabolite, inhibits tubulin polymerization by interacting at the colchicines site. Proc Natl Acad Sci USA. 1994;91:3964–3968. doi: 10.1073/pnas.91.9.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukhopadhyay T, Roth JA. Superinduction of wild-type p53 protein after 2-methoxyestradiol treatment of Ad5p53-transduced cells induces tumor cell apoptosis. Oncogene. 1998;17:241–246. doi: 10.1038/sj.onc.1201909. [DOI] [PubMed] [Google Scholar]
- 11.Pribluda VS, Gubish ER, Jr, Lavallee TM, Treston A, Swartz GM, Green SJ. 2-Methoxyestradiol: An endogenous antiangiogenic and antiproliferative drug candidate. Cancer Metastasis Rev. 2000;19:173–179. doi: 10.1023/a:1026543018478. [DOI] [PubMed] [Google Scholar]
- 12.Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC. Apoptosis and interferons: Role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237–249. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- 13.Castelli JC, Hassel BA, Maran A, Paranjape J, Hewitt JA, Li XL, Hsu YT, Silverman RH, Youle RJ. The role of 2′–5′ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ. 1998;5:313–320. doi: 10.1038/sj.cdd.4400352. [DOI] [PubMed] [Google Scholar]
- 14.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 15.Barber GN. The dsRNA-dependent protein kinase, PKR and cell death. Cell Death Differ. 2005;12:563–570. doi: 10.1038/sj.cdd.4401643. [DOI] [PubMed] [Google Scholar]
- 16.Der SD, Yang YL, Weissmann C, Williams BR. A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:3279–3283. doi: 10.1073/pnas.94.7.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaufman RJ. Double-stranded RNA-activated protein kinase mediates virus-induced apoptosis: A new role for an old actor. Proc Natl Acad Sci USA. 1999;96:11693–11695. doi: 10.1073/pnas.96.21.11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clemens MJ. Interferons and apoptosis. J Interferon Cytokine Res. 2003;23:277–292. doi: 10.1089/107999003766628124. [DOI] [PubMed] [Google Scholar]
- 19.Sen GC. Novel functions of interferon-induced proteins. Semin Cancer Biol. 2000;10:93–101. doi: 10.1006/scbi.2000.0312. [DOI] [PubMed] [Google Scholar]
- 20.Silverman RH. Implications for RNase L in prostate cancer biology. Biochemistry. 2003;42:1805–1812. doi: 10.1021/bi027147i. [DOI] [PubMed] [Google Scholar]
- 21.Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 22.Maran A, Mathews MB. Characterization of the double-stranded RNA implicated in the inhibition of protein synthesis in cells infected with a mutant adenovirus defective for VA RNA. Virology. 1988;164:106–113. doi: 10.1016/0042-6822(88)90625-3. [DOI] [PubMed] [Google Scholar]
- 23.Marques JT, Rebouillat D, Ramana CV, Murakami J, Hill JE, Gudkov A, Silverman RH, Stark GR, Williams BR. Down-regulation of p53 by double-stranded RNA modulates the antiviral response. J Virol. 2005;79:11105–11114. doi: 10.1128/JVI.79.17.11105-11114.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edery I, Petryshyn R, Sonenberg N. Activation of double-stranded RNA-dependent kinase (dsl) by the TAR region of HIV-1 mRNA: A novel translational control mechanism. Cell. 1989;56:303–312. doi: 10.1016/0092-8674(89)90904-5. [DOI] [PubMed] [Google Scholar]
- 25.Farrell PJ, Balkow K, Hunt T, Jackson RJ, Trachsel H. Phosphorylation of initiation factor elF-2 and the control of reticulocyte protein synthesis. Cell. 1977;11:187–200. doi: 10.1016/0092-8674(77)90330-0. [DOI] [PubMed] [Google Scholar]
- 26.Sen GC, Taira H, Lengyel P. Interferon, double-stranded RNA, and protein phosphorylation. Characteristics of a double-stranded RNA-activated protein kinase system partially purified from interferon treated Ehrlich ascites tumor cells. J Biol Chem. 1978;253:5915–5921. [PubMed] [Google Scholar]
- 27.Levin D, London IM. Regulation of protein synthesis: Activation by double-stranded RNA of a protein kinase that phosphorylates eukaryotic initiation factor 2. Proc Natl Acad Sci USA. 1978;75:1121–1125. doi: 10.1073/pnas.75.3.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galabru J, Hovanessian A. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J Biol Chem. 1987;262:15538–15544. [PubMed] [Google Scholar]
- 29.Kostura M, Mathews MB. Purification and activation of the double-stranded RNA-dependent eIF-2 kinase DAI. Mol Cell Biol. 1989;9:1576–1586. doi: 10.1128/mcb.9.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berry MJ, Knutson GS, Lasky SR, Munemitsu SM, Samuel CE. Mechanism of interferon action. Purification and substrate specificities of the double-stranded RNA-dependent protein kinase from untreated and interferon-treated mouse fibroblasts. J Biol Chem. 1985;260:11240–11247. [PubMed] [Google Scholar]
- 31.Maran A, Maitra RK, Kumar A, Dong B, Xiao W, Li G, Williams BR, Torrence PF, Silverman RH. Blockage of NF-kappa B signaling by selective ablation of an mRNA target by 2-5A antisense chimeras. Science. 1994;265:789–792. doi: 10.1126/science.7914032. [DOI] [PubMed] [Google Scholar]
- 32.Kumar A, Haque J, Lacoste J, Hiscott J, Williams BR. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Mol Cell Biol. 1994;91:6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar A, Yang YL, Flati V, Der S, Kadereit S, Deb A, Haque J, Reis L, Weissmann C, Williams BR. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: Role of IRF-1 and NF-kappaB. EMBO J. 1997;16:406–416. doi: 10.1093/emboj/16.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cuddihy AR, Wong AH, Tam NW, Li S, Koromilas AE. The double-stranded RNA activated protein kinase PKR physically associates with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro. Oncogene. 1999;18:2690–2702. doi: 10.1038/sj.onc.1202620. [DOI] [PubMed] [Google Scholar]
- 35.Wong AH, Durbin JE, Li S, Dever TE, Decker T, Koromilas AE. Enhanced antiviral and antiproliferative properties of a STAT1 mutant unable to interact with the protein kinase PKR. J Biol Chem. 2001;276:13727–13737. doi: 10.1074/jbc.M011240200. [DOI] [PubMed] [Google Scholar]
- 36.Vorburger SA, Pataer A, Swisher SG, Hunt KK. Genetically targeted cancer therapy: Tumor destruction by PKR activation. Am J Pharmacogenomics. 2004;4:189–198. doi: 10.2165/00129785-200404030-00006. [DOI] [PubMed] [Google Scholar]
- 37.Koromilas AE, Roy S, Barber GN, Katze MG, Sonenberg N. Malignant transformation by a mutant of the IFN-inducible dsRNA-dependent protein kinase. Science. 1992;257:1685–1689. doi: 10.1126/science.1382315. [DOI] [PubMed] [Google Scholar]
- 38.Meurs EF, Galabru J, Barber GN, Katze MG, Hovanessian AG. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA. 1993;90:232–236. doi: 10.1073/pnas.90.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gingery A, Bradley E, Shaw A, Oursler MJ. Phosphatidylinositol 3-kinase coordinately activates the MEK/ERK and AKT/NFkappaB pathways to maintain osteoclast survival. J Cell Biochem. 2003;89:165–179. doi: 10.1002/jcb.10503. [DOI] [PubMed] [Google Scholar]
- 40.Kennedy AM, Shogren KL, Zhang M, Turner RT, Spelsberg TC, Maran A. 17beta-estradiol-dependent activation of signal transducer and activator of transcription-1 in human fetal osteoblasts is dependent on Src kinase activity. Endocrinology. 2005;146:201–207. doi: 10.1210/en.2004-0486. [DOI] [PubMed] [Google Scholar]
- 41.Hu Y, Conway TW. 2-Aminopurine inhibits the double-stranded RNA-dependent protein kinase both in vitro and in vivo. J Interferon Res. 1993;13:323–328. doi: 10.1089/jir.1993.13.323. [DOI] [PubMed] [Google Scholar]
- 42.Geiss G, Jin G, Guo J, Bumgarner R, Katze MG, Sen GC. A comprehensive view of regulation of gene expression by double-stranded RNA-mediated cell signaling. J Biol Chem. 2001;276:30178–30182. doi: 10.1074/jbc.c100137200. [DOI] [PubMed] [Google Scholar]
- 43.Jarrous N, Osman F, Kaempfer R. 2-Aminopurine selectively inhibits splicing of tumor necrosis factor alpha mRNA. Mol Cell Biol. 1996;16:2814–2822. doi: 10.1128/mcb.16.6.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugiyama T, Fujita M, Koide N, Mori I, Yoshida T, Mori H, Yokochi T. 2-aminopurine inhibits lipopolysaccharide-induced nitric oxide production by preventing IFN-beta production. Microbiol Immunol. 2004;48:957–963. doi: 10.1111/j.1348-0421.2004.tb03625.x. [DOI] [PubMed] [Google Scholar]
- 45.Thomis DC, Samuel CE. Mechanism of interferon action: Evidence for intermolecular autophosphorylation and autoactivation of the interferon-induced, RNA-dependent protein kinase PKR. J Virol. 1993;67:7695–7700. doi: 10.1128/jvi.67.12.7695-7700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ben-Asouli Y, Banai Y, Pel-Or Y, Shir A, Kaempfer R. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell. 2002;108:221–232. doi: 10.1016/s0092-8674(02)00616-5. [DOI] [PubMed] [Google Scholar]
- 47.Barber GN, Jagus R, Meurs EF, Hovanessian AG, Katze MG. Molecular mechanisms responsible for malignant transformation by regulatory and catalytic domain variants of the interferon-induced enzyme RNA-dependent protein kinase. J Biol Chem. 1995;270:17423–17428. doi: 10.1074/jbc.270.29.17423. [DOI] [PubMed] [Google Scholar]
- 48.Lee SB, Esteban M. The interferon-induced double-stranded RNA-activated protein kinase induces apoptosis. Virology. 1994;199:491–496. doi: 10.1006/viro.1994.1151. [DOI] [PubMed] [Google Scholar]
- 49.Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998;17:4379–4390. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saelens X, Kalai M, Vandenabeele P. Translation inhibition in apoptosis: Caspase-dependent PKR activation and eIF2-alpha phosphorylation. J Biol Chem. 2001;276:41620–41628. doi: 10.1074/jbc.M103674200. [DOI] [PubMed] [Google Scholar]
- 51.Rhoads RE. Signal transduction pathways that regulate eukaryotic protein synthesis. J Biol Chem. 1999;274:30337–30340. doi: 10.1074/jbc.274.43.30337. [DOI] [PubMed] [Google Scholar]
- 52.Jagus R, Joshi B, Barber GN. PKR, apoptosis and cancer. Int J Biochem Cell Biol. 1999;31:123–138. doi: 10.1016/s1357-2725(98)00136-8. [DOI] [PubMed] [Google Scholar]
- 53.Donze O, Jagus R, Koromilas AE, Hershey JW, Sonenberg N. Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J. 1995;14:3828–3834. doi: 10.1002/j.1460-2075.1995.tb00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abraham N, Jaramillo ML, Duncan PI, Methot N, Icely PL, Stojdl DF, Barber GN, Bell JC. The murine PKR tumor suppressor gene is rearranged in a lymphocytic leukemia. Exp Cell Res. 1998;244:394–404. doi: 10.1006/excr.1998.4201. [DOI] [PubMed] [Google Scholar]
- 55.Barber GN, Thompson S, Lee TG, Strom T, Jagus R, Darveau A, Katze MG. The 58-kilodalton inhibitor of the interferon-induced double-stranded RNA-activated protein kinase is a tetratricopeptide repeat protein with oncogenic properties. Proc Natl Acad Sci USA. 1994;91:4278–4282. doi: 10.1073/pnas.91.10.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim SH, Forman AP, Mathews MB, Gunnery S. Human breast cancer cells contain elevated levels and activity of the protein kinase, PKR. Oncogene. 2000;19:3086–3094. doi: 10.1038/sj.onc.1203632. [DOI] [PubMed] [Google Scholar]
- 57.Savinova O, Joshi B, Jagus R. Abnormal levels and minimal activity of the dsRNA-activated protein kinase, PKR, in breast carcinoma cells. Int J Biochem Cell Biol. 1999;31:175–189. doi: 10.1016/s1357-2725(98)00140-x. [DOI] [PubMed] [Google Scholar]