

Abstract
Treatment of metmyoglobin with H2O2 is known to lead to the crosslinking of an active site tyrosine residue to the heme [Catalano, C. E., Y. S. Choe, and P. R. Ortiz de Montellano (1989) J. Biol. Chem. 264, 10534–10541]. We have found in this study that this reaction also leads to an altered heme product not covalently bound to the protein. This product was characterized by visible absorption, infrared absorption, and mass and NMR spectrometry as an iron chlorin product formed from the saturation of the double bond between carbon atoms at positions 17 and 18 of pyrrole ring D with concomitant addition of a hydroxyl group on the carbon atom at position 18 and lactonization of the propionic acid to the carbon atom at position 17. Studies with the use of 18O-labeled H2O2, O2, and H2O clearly indicate that the source of the added oxygen on the heme is water. Evidently, water adds regiospecifically to a cationic site formed on a carbon atom at position 18 after oxidation of the ferric heme prosthetic group with peroxide. Prolonged incubation of the reaction mixture containing the iron hydroxychlorin product led to the formation of an iron dihydroxychlorin product, presumably from a slow addition of water to the initial iron hydroxychlorin. The iron chlorin products characterized in this study are distinct from the meso-oxyheme species, which is thought to be formed during peroxide-mediated degradation of metmyoglobin, cytochrome P450, ferric heme, and model ferric hemes, and give further insight into the mechanism of H2O2-induced heme alterations.
The reactions of H2O2 with hemoproteins have been extensively investigated as models for the study of protein radicals (1–8), heme alteration (4, 6, 9–11), and proteolytic susceptibility (12–18). This reaction is thought to play a role in ischemia-reperfusion injury (9, 19–24), toxicity from hemoglobin-based red cell substitutes (10), regulation of arachidonic acid metabolism (25, 26), and the self-deactivation of cytochrome P450 (27). In the case of myoglobin, one of the best characterized of these hemoproteins, the reaction of H2O2 is known to lead to intermolecular crosslinking of myoglobin to form oligomers (5), or between myoglobin and other molecules (24), as well as to the crosslinking of the heme to the protein at tyrosine residue 103 (4). It has recently been shown with the use of radiolabeled heme that the protein-bound heme adduct accounts for 58% of the heme that is altered, whereas 29% of the altered heme is accounted for in a novel product not bound to the protein (28). This paper describes the complete structural elucidation of this product as an iron hydroxychlorin compound with saturation of ring D and reports the kinetics of the compound’s formation and subsequent hydrolysis to form a corresponding diol. Although oxidation of the heme at the meso position has been proposed to occur during peroxide-mediated alteration of metmyoglobin (11), cytochrome P450 (29), ferric heme (29, 30), and model ferric hemes (31), we found in the current study that iron hydroxychlorins are formed.
EXPERIMENTAL PROCEDURES
Materials.
Hydrogen peroxide (30%), stannous chloride, and metmyoglobin (horse heart) were purchased from Fisher, Aldrich, and Sigma, respectively. All experiments were performed with the use of buffers pretreated with Chelex 100, which was purchased from Bio-Rad. Pyridine-d5 (99.98 atom %) was purchased from Merck. Methanol-d4, deuterium oxide, and 18O2 were purchased from Cambridge Isotope Laboratories (Woburn, MA). H218O2 (2% solution, 90 atom %) and H218O (95 atom %) were obtained from ICON Isotopes (Summit, NJ).
Preparation of Altered Heme Products.
Metmyoglobin (10.0 μmol) was treated with H2O2 (10.0 μmol) in 100 ml of 50 mM potassium phosphate, pH 7.4, at 25°C. After 5 min, the reaction mixture was injected onto a Hi-Pore RP-304 (25 × 2.15 cm; Bio-Rad) reverse-phase column equilibrated with solvent A (0.1% trifluoroacetic acid) at a flow rate of 20 ml/min. A linear gradient was run to 30%, 35%, and 100% solvent B (0.1% trifluoroacetic acid in CH3CN) over 30, 50 and 60 min, respectively. Fractions corresponding to altered heme product II (retention time of 42 min) were collected. The purified heme product from seven such HPLC runs was pooled and loaded onto a C18 Sep-Pak cartridge (Millipore), which was then washed with water (100 ml). The purified heme product was eluted with approximately 10 ml of methanol and dried under a stream of nitrogen. The altered heme product I (retention time of 39 min) was prepared as described above except that the reaction was allowed to proceed for 72 h before purification.
The altered heme products from H218O2-treated samples were prepared as above except that only one reaction mixture was prepared. For studies with the use of 18O2 or H218O, the altered heme products were isolated as above except that the reaction mixture contained metmyoglobin (5.8 μmol) and H2O2 (5.8 μmol) in 2 ml of 50 mM potassium phosphate, pH 7.4. In studies with 18O2, the gas was added to the reaction mixture after evacuation by a vacuum pump. In studies with H218O, the 18O-enriched water was diluted with natural abundance water so that the final concentration was 47.5 atom % 18O.
Mass Spectral Analysis of the Heme Products.
Mass spectra were obtained on a tandem time-of-flight mass spectrometer built in-house and described previously (32). This instrument consists of two reflectron mass analyzers in tandem separated by a collision chamber for inducing fragmentation. However, in this work mass spectra were recorded in the “double reflectron” mode to obtain sufficient mass resolution to resolve completely the isotopic contributions to the molecular ion peak. Samples were diluted in a solution of methanol/0.1% trifluoroacetic acid in H2O (1:1), mixed with a solution of caffeic acid in water, and deposited on the probe tip of the mass spectrometer. The sample was ionized using a PTI PL2300 (Ontario, Canada) pulsed nitrogen laser, and spectra were recorded by a Tekronix TDS 540 digital oscilloscope, downloaded to a 486 PC, and averaged using tofware (ILYS Software, Pittsburgh).
NMR Analysis of the Heme Products.
Approximately 0.2–0.3 mg of the purified heme product was added to 2–3 mg of SnCl2 in 0.5 ml of pyridine-d5 as described (33). A 360 MHz Bruker (Billerica, MA) AMX with the sample maintained at 280°K was used. Typically, 256 free induction decays were collected with an accumulation time of 4 s for one-dimensional spectra. Phase-sensitive nuclear Overhauser effect (spectroscopy) [NOE(SY)] spectra were obtained by the method of States et al. (34) with the use of 1024 by 1024 data matrices and a mixing time of 0.8 s. This allowed collection of 256 t1 increments, each with 64 free induction decays. Carbon-13 peaks were detected by heteronuclear multiple bond correlation as described (35). The compounds were stable in the pyridine/SnCl2 solution for at least 6 h, and the spectra were recorded within this time. However, the compounds decomposed to unidentified products after more prolonged periods in solution.
Other Methods.
Visible absorption spectra were obtained with a Hewlett–Packard 8450A diode array spectrophotometer. HPLC was performed with the use of a Hewlett–Packard 1050 System with a 1040M diode array detector. Fourier transform infrared spectra were obtained on samples in KBr pellets with an FTS-45 Instrument from Bio-Rad Laboratories, Digilab Division.
RESULTS
Formation of Altered Heme Products.
Treatment of metmyoglobin with a stoichiometric amount of H2O2 led to covalent alteration of the heme to protein-bound products (Fig. 1B, peaks III and IV), which have been previously described (4, 9), as well as one major dissociable heme product (peak II), which we characterize in this report. The amount of product corresponding to peak II decreased in the reaction mixture after prolonged incubations at room temperature, whereas a product corresponding to peak I was formed (Fig. 1C). A more detailed kinetic analysis of this reaction over a 14-h period suggested that the decrease in peak II was concomitant with an increase in peak I (Fig. 2B). The other products (peak III and IV) appeared to be stable. In addition, we found that roughly one-half of the native heme was altered during the reaction (Fig. 2A).
Figure 1.

HPLC profile of untreated and H2O2-treated metmyoglobin. (A) Untreated metmyoglobin. (B) Reaction mixture containing metmyoglobin treated with H2O2 at pH 7.4 for 5 min. (C) Reaction mixture containing metmyoglobin treated with H2O2 at pH 7.4 for 21 h. Metmyoglobin (10 μM) was treated with H2O2 (10 μM) in 3.0 ml of 50 mM potassium phosphate, pH 7.4, at room temperature. Aliquots (100 μl) were taken at the indicated times and injected onto an HPLC column (Vydac C4, 10 μm, 0.46 × 25 cm, Hesperia, CA) equilibrated with solvent A (0.1% trifluoroacetic acid) at a flow rate of 1 ml/min. A linear gradient was run to 36%, 40%, 42%, 51%, and 100% solvent B (0.1% trifluoroacetic acid in CH3CN) over 5, 10, 5, 5, and 10 min, respectively. Absorbance at 210 and 400 nm was measured.
Figure 2.

Kinetics of heme loss and formation of altered heme products. (A) Amount of residual heme after treatment of metmyoglobin with H2O2. (B) Relative amount of heme products formed from the treatment of metmyoglobin with H2O2. The preparation and analysis of the samples are as described in Fig. 1. The relative amounts of heme metabolites were quantified by peak areas at 400 nm and expressed as a percentage of the peak area of heme at 400 nm of the untreated sample. The values are relative amounts since the absorptivities of the products are not known. The Roman numerals correspond to the peaks indicated in Fig. 1.
Characterization of Product Corresponding to Peaks II and I.
The compounds corresponding to peaks II and I were purified by the use of HPLC. The visible absorption spectrum of each of the purified products was found to be highly similar to that of iron chlorins, which have a maxima at 602 nm (ref. 36; Fig. 3 A and B). Thus, it appeared that a degree of saturation at a pyrrole group had occurred. The mass spectrum of product II showed a molecular ion at m/z 632.5 corresponding to the addition of oxygen to the heme, whereas that of product I showed a molecular ion at m/z 650.2 corresponding to the addition of two oxygens and two protons to the heme (Fig. 4 A and B). The one-dimensional proton NMR spectrum of product II clearly showed that all protons corresponding to those on four methyl, two propionic, two vinyl, and four meso groups were present (Fig. 5A). An upfield shift of one of the methyl groups was noted (1.96 ppm), which was consistent with an iron chlorin structure. A similar finding was observed for the proton NMR signals of product I (Fig. 5B).
Figure 3.

Absorption spectra of heme products I and II. (A) Heme product II. (B) Heme product I. Heme products were isolated as described in Experimental Procedures. Samples were dissolved in methanol (dashed line) or 0.1% trifluoroacetic acid in CH3CN (solid line).
Figure 4.

Mass spectra of heme products I and II. (A) Heme product II. (B) Heme product I. Heme products were prepared as described in Experimental Procedures.
Figure 5.

Proton NMR spectra of heme products I and II. (A) Heme product II. (B) Heme product I. Heme products were prepared and analyzed as described in Experimental Procedures.
A detailed, two-dimensional NOE(SY) NMR study was conducted on product II to determine the nature of the addition of the oxygen atom (Table 1). It was evident that the saturation of the pyrrole ring must have occurred by the addition of oxygen but not a concomitant proton. We therefore focused on the interactions of the upfield methyl, which must be at the site of saturation. The upfield methyl (1.96 ppm) showed NOE(SY) interactions with propionic acid protons at 3.24 and 3.54 ppm, clearly indicating the site as either ring C or D. Furthermore, the upfield methyls showed NOE(SY) interactions with a meso proton at 9.36 ppm, which was identified as the δ-meso by its interaction with another methyl group at 3.26 ppm (Table 1). The other meso positions did not show such an interaction. Thus, the saturated pyrrole was that of ring D. Three possible structures could be envisioned to account for saturation of ring D and addition of oxygen without addition of a proton: a carbonyl on position 17 with concomitant rearrangement of the propionic acid group to carbon 18; a heme epoxide across positions 17 and 18; or an alcohol at position 18 with concomitant formation of a lactone from the propionic acid group. The carbonyl product at position 17 is unlikely since NOE(SY) interactions between δ-meso proton and ring D methyl, and not propionic protons, were observed. To further differentiate between the remaining structures, the infrared and 13C NMR spectra were measured. The infrared spectrum of product II gave a band at 1773 cm−1 (Fig. 6), which is characteristic of the stretching frequency of a carbonyl on a 5-membered lactone (37) and consistent with that reported for a lactonized iron chlorin propionic acid (38). The broad band at 3500 cm−1 reflects the carboxylic acid functional groups, and the bands near 2900 cm−1 are consistent with C-H stretching frequencies of methyl groups (37). The bands at 1638 cm−1 and 1680 cm−1 are consistent with C = C stretching frequencies and those near 1200 cm−1 are consistent with C-O-C stretching frequencies of a lactone (37). The 13C chemical shift of the carbons coupled to the upfield methyl protons (1.96 ppm) were observed at 161, 83, and 96 ppm, by the use of heteronuclear multiple bond correlation (ref. 35; data not shown). Epoxides are known to exhibit chemical shifts near 40 ppm (37). The downfield 13C chemical shift at 161 ppm is consistent with an sp2 pyrrole carbon, and those at 83 and 96 ppm likely correspond to sp3 carbons with oxygen substituents (38, 39). Thus, the data show that the structure of product II is that of an iron hydroxychlorin product (Fig. 7A) with C19 corresponding to the carbon at 161 ppm and C17 and C18 corresponding to the upfield 13C resonances. In view of the highly similar results obtained for product I, the compound appeared to be due to the hydrolysis of product II (Fig. 7B).
Table 1.
NMR chemical shifts and NOE(SY) interactions of heme product II
| Proton | Chemical shift, ppm | Interactions |
|---|---|---|
| Meso α | 9.97 | 3.32 |
| Meso β | 9.95 | 3.54, 6.10 |
| Meso δ | 9.36 | 1.96, 3.26 |
| Meso γ | 9.32 | 4.11, 4.21 |
| Vinyl α ring A | 8.44 (d, d = 17.8, 11.2) | 5.94 |
| Vinyl α ring B | 8.25 (d, d = 17.8, 11.3) | 5.77 |
| Vinyl β-trans ring A | 6.28 (d, d = 17.8, 2.0) | |
| Vinyl β-trans ring B | 6.10 (d, d = 17.8, 1.9) | |
| Vinyl β-cis ring A | 5.94 (d, d = 11.6, 1.9) | |
| Vinyl β-cis ring B | 5.77 (d, d = 11.8, 1.9) | 3.54, 9.95, 8.25 |
| Propionics α ring C | 4.21 (m) | 3.30, 9.32 |
| Propionics α ring D | 4.11 (m) | 3.24, 3.54 |
| Propionics β ring C | 3.30 (m) | 4.21 |
| Propionics β ring D | 3.24, 3.54 (m) | 1.96 |
| Methyl ring A | 3.26 | 9.36 |
| Methyl ring B | 3.32 | 9.97 |
| Methyl ring C | 3.54 | 4.21, 9.95 |
| Methyl ring D | 1.96 | 9.36 |
Figure 6.

Fourier transform infrared spectrum of heme product II. Spectrum was obtained as described in Experimental Procedures.
Figure 7.

Structures of heme products I and II.
Origin of Added Oxygen Atoms.
The source of the added oxygen atoms found in both products was examined by use of 18O-enriched water, O2, and H2O2 and subsequent purification and mass spectral analysis of the heme products (Table 2). It was found that water, and not O2 or H2O2, was the source for the added oxygen for both products I and II. The molecular ion cluster for product II exhibited an isotopic distribution consistent with the presence of 16O and 18O in abundance of 52.5% and 47.5%, respectively, as expected from the 18O abundance of the water used (47.5%). The molecular ion cluster for product I exhibited an isotopic distribution consistent with the presence of two 16O, one 16O and one 18O, and two 18O compounds in abundance of 27.6%, 49.8%, and 22.6%, respectively, as expected from the statistical addition of water with 18O abundance of 47.5% to product II. The oxygen was apparently not exchangeable under the conditions of purification, which were performed with the use of natural abundance water. These results are consistent with the finding that product formation was independent of the concentration of O2 in the reaction mixtures (data not shown). Similar amounts of products were formed upon peroxide treatment of metmyoglobin in a reaction mixture that was degassed and made anaerobic by a previously described method (33) or in a reaction mixture that was bubbled with dioxygen for 10 min.
Table 2.
Masses of the heme products
| Product | 18O added to reaction mixture | Observed molecular ion(s), m/z | Predicted molecular formula | Theoretical value for predicted molecular formula |
|---|---|---|---|---|
| Heme | C34H32FeN4O4 | 616.5 | ||
| II | 632.5 | C34H32FeN4O5 | 632.2 | |
| II | H218O2 | 632.3 | C34H32FeN4O5 | 632.2 |
| II | H218O | 632.2* | C34H32FeN4O5 | 632.2 |
| II | 634.2* | C34H32FeN4O418O | 634.2 | |
| II | 18O2 | 632.3 | C34H32FeN4O5 | 632.2 |
| I | 650.2 | C34H34FeN4O6 | 650.2 | |
| I | H218O2 | 650.2 | C34H34FeN4O6 | 650.2 |
| I | H218O | 650.2† | C34H34FeN4O6 | 650.2 |
| I | 652.2† | C34H34FeN4O518O | 652.2 | |
| I | 654.2† | C34H34FeN4O418O2 | 654.2 | |
| I | 18O2 | 650.3 | C34H34FeN4O6 | 650.2 |
The isotopic distribution was consistent with normal abundance and 18O-enriched compounds in relative abundance of 52.5% and 47.5% of the total, respectively.
The isotopic distribution was consistent with the presence of two 16O, one 16O and one 18O, and two 18O compounds in relative abundance of 27.6%, 49.8%, and 22.6%, respectively.
DISCUSSION
We have shown that the peroxide-mediated alteration of the heme of metmyoglobin leads to formation of an iron chlorin product. This was unexpected as the initial oxidation of the meso carbon has been proposed to account for the peroxide-dependent degradation of the heme of myoglobin (11), heme of cytochrome P450 (29), as well as heme (29, 30) and model hemes (29, 31) in solution. Furthermore, the peroxide-mediated reaction is thought to mimic, in part, the initial oxidation of the meso carbon that occurs in the physiological heme degradation reaction catalyzed by heme oxygenase as well as in the coupled oxidation reaction of heme and hemoproteins (30, 31, 40). More recently, it has been shown that peroxide alone can support the heme oxygenase reaction to give verdoheme, strongly suggesting that a ferric iron-bound peroxide complex is responsible for meso-hydroxylation (41). It appears from our studies that the mechanisms of formation of the products from the reaction of H2O2 with metmyoglobin are quite different from that of the heme oxygenase reaction. The source of oxygen in the meso-modified heme, formed from the heme oxygenase, is dioxygen, whereas that for the iron chlorin product, formed from the peroxide reaction with metmyoglobin, is water. We propose the following cationic mechanism to account for the addition of water to the heme in the peroxide reaction with metmyoglobin.
The initial steps in the reaction of H2O2 with metmyoglobin have been extensively investigated (42–44) and appear to involve the formation of a Fe+4OH heme species and a hydroxyl radical equivalent, which could be localized on the protein or the heme (Scheme I). 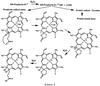 When the radical is localized on the protein, an aromatic amino acid radical is formed, most likely involving tyrosine residues. The protein radical can lead to crosslinking reactions with other proteins (5, 24), other organic molecules (24), or with the heme (4). The crosslinking of the heme to the protein has been reported to occur by the attack of a tyrosine radical (residue 103) to the heme, possibly at a meso position (4). When the radical is localized on the heme, as is common for cytochrome c peroxidases, a heme cation radical is formed (Scheme I). Although we have shown the cation radical on ring D, this is but one of many resonance stabilized forms. Attack by the carboxyl oxygen of the propionic acid side chain at the cationic site would lead to a lactone heme radical species, which could delocalize the lone electron to the heme iron to give ferric iron and porphyrin cation. This cation can be attacked by water to give the iron hydroxychlorin product isolated in our study (product II). Further reaction with water, presumably in the active site of myoglobin, can give rise to the iron dihydroxychlorin product isolated in this study (product I).
When the radical is localized on the protein, an aromatic amino acid radical is formed, most likely involving tyrosine residues. The protein radical can lead to crosslinking reactions with other proteins (5, 24), other organic molecules (24), or with the heme (4). The crosslinking of the heme to the protein has been reported to occur by the attack of a tyrosine radical (residue 103) to the heme, possibly at a meso position (4). When the radical is localized on the heme, as is common for cytochrome c peroxidases, a heme cation radical is formed (Scheme I). Although we have shown the cation radical on ring D, this is but one of many resonance stabilized forms. Attack by the carboxyl oxygen of the propionic acid side chain at the cationic site would lead to a lactone heme radical species, which could delocalize the lone electron to the heme iron to give ferric iron and porphyrin cation. This cation can be attacked by water to give the iron hydroxychlorin product isolated in our study (product II). Further reaction with water, presumably in the active site of myoglobin, can give rise to the iron dihydroxychlorin product isolated in this study (product I).
Although we have depicted the lactonization of the propionic acid preceding the addition of water in the formation of product II (Scheme I), it is also possible that water initially adds to the carbon atom at position 18, and then the lactonization of the propionic acid to the carbon atom at position 17 subsequently occurs. In either case, the regiospecificity of the water addition is presumably dictated in part by the accessibility of water to the heme as rings A and B were not attacked. The reason for the specificity for saturation of ring D over that of ring C, even though both are apparently exposed to water, remains to be examined. The sharpness of the proton signal corresponding to the D-ring methyl group of product II suggests that the addition of water was stereospecific. However, we do not know the absolute stereochemistry of the carbon atoms at positions 17 or 18, and the reaction mechanism does not preclude any stereoisomer.
Iron hydroxychlorins with saturation of a double bond on ring C, but not on ring D, have been isolated and characterized from catalase HPII from Escherichia coli (45) and bacterial terminal oxidase (38). Since the origin of the added oxygen atoms in the bacterial products has not been defined, it is not clear if the mechanism proposed here or an enzymatic hydroxylation, as proposed by others (38, 45), is responsible for the synthesis of heme d in bacteria. The iron chlorins isolated in our study, however, appear to be novel heme products with the potential of being biomarkers of oxidative damage to metmyoglobin.
Acknowledgments
We are grateful to Drs. Lance R. Pohl, James R. Gillette, and Minor J. Coon for critically reviewing this manuscript. We thank Drs. T. Spande and N. Whittaker for measurement of Fourier transform infrared spectra. This work was supported in part by a Pharmaceutical Research and Manufacturers of America Foundation Research Starter Grant to Y.O.
Footnotes
Abbreviation: NOE(SY), nuclear Overhauser effect (spectroscopy).
References
- 1.Kelman D J, DeGray J A, Mason R P. J Biol Chem. 1994;269:7458–7463. [PubMed] [Google Scholar]
- 2.King N K, Looney F D, Winfield M E. Biochim Biophys Acta. 1967;133:65–82. doi: 10.1016/0926-6577(64)90179-2. [DOI] [PubMed] [Google Scholar]
- 3.Ruf H H, Raab-Brill U, Blau C. Biochem Soc Trans. 1993;21:739–744. doi: 10.1042/bst0210739. [DOI] [PubMed] [Google Scholar]
- 4.Catalano C E, Choe Y S, Ortiz de Montellano P R. J Biol Chem. 1989;264:10534–10541. [PubMed] [Google Scholar]
- 5.Tew D, Ortiz de Montellano P R. J Biol Chem. 1988;263:17880–17886. [PubMed] [Google Scholar]
- 6.King N K, Looney F D, Winfield M E. Biochim Biophys Acta. 1967;133:65–82. doi: 10.1016/0926-6577(64)90179-2. [DOI] [PubMed] [Google Scholar]
- 7.Smith W L, Eling T E, Kulmacz R J, Marnett L J, Tsai A. Biochemistry. 1992;31:3–7. doi: 10.1021/bi00116a001. [DOI] [PubMed] [Google Scholar]
- 8.Davies M J. Free Radical Res Commun. 1990;10:361–370. doi: 10.3109/10715769009149905. [DOI] [PubMed] [Google Scholar]
- 9.Osawa Y, Korzekwa K. Proc Natl Acad Sci USA. 1991;88:7081–7085. doi: 10.1073/pnas.88.16.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osawa Y, Darbyshire J F, Meyer C M, Alayash A I. Biochem Pharmacol. 1993;12:2299–2305. doi: 10.1016/0006-2952(93)90621-3. [DOI] [PubMed] [Google Scholar]
- 11.Cantoni L, Gibbs A H, De Matteis F. Int J Biochem. 1981;13:823–830. doi: 10.1016/0020-711x(81)90102-6. [DOI] [PubMed] [Google Scholar]
- 12.Fligiel S E G, Lee E C, McCoy J P, Johnson K J, Varani J. Am J Pathol. 1984;115:418–425. [PMC free article] [PubMed] [Google Scholar]
- 13.Fagan J M, Waxman L, Goldberg A L. J Biol Chem. 1986;261:5705–5713. [PubMed] [Google Scholar]
- 14.Davies K J A, Golberg A L. J Biol Chem. 1987;262:8220–8226. [PubMed] [Google Scholar]
- 15.Davies K J A. J Biol Chem. 1987;262:9895–9901. [PubMed] [Google Scholar]
- 16.Davies K J A, Delsignore M E. J Biol Chem. 1987;262:9908–9913. [PubMed] [Google Scholar]
- 17.Davies K J A, Lin S W, Pacifici R E. J Biol Chem. 1987;262:9914–9920. [PubMed] [Google Scholar]
- 18.Davies K J A, Lin S W. Free Radical Biol Med. 1988;5:225–236. doi: 10.1016/0891-5849(88)90016-0. [DOI] [PubMed] [Google Scholar]
- 19.Mitsos S E, Kim D, Lucchesi B R, Fantone J C. Lab Invest. 1988;59:824–830. [PubMed] [Google Scholar]
- 20.Arduini A, Eddy L, Hochstein P. Free Radical Biol Med. 1990;9:511–513. doi: 10.1016/0891-5849(90)90130-b. [DOI] [PubMed] [Google Scholar]
- 21.Arduini A, Mancinelli G, Radatti G L, Damonti W, Hochstein P, Cadenas E. Free Radical Biol Med. 1992;13:449–454. doi: 10.1016/0891-5849(92)90185-j. [DOI] [PubMed] [Google Scholar]
- 22.Galaris D, Cadenas E, Hochstein P. Arch Biochem Biophys. 1989;273:497–504. doi: 10.1016/0003-9861(89)90509-2. [DOI] [PubMed] [Google Scholar]
- 23.Galaris D, Eddy L, Arduini A, Cadenas E, Hochstein P. Biochem Biophys Res Commun. 1989;160:1162–1168. doi: 10.1016/s0006-291x(89)80125-1. [DOI] [PubMed] [Google Scholar]
- 24.Rice R H, Lee Y M, Brown W D. Arch Biochem Biophys. 1983;221:417–427. doi: 10.1016/0003-9861(83)90160-1. [DOI] [PubMed] [Google Scholar]
- 25.Smith W L, Meade E A, DeWitt D L. Ann NY Acad Sci. 1994;714:136–142. doi: 10.1111/j.1749-6632.1994.tb12037.x. [DOI] [PubMed] [Google Scholar]
- 26.Varfolomeyev S D, Mevkh A T. Biotechnol Appl Biochem. 1993;17:291–304. [PubMed] [Google Scholar]
- 27.Karuzina I I, Archakov A I. Free Radical Biol Med. 1994;16:73–97. doi: 10.1016/0891-5849(94)90245-3. [DOI] [PubMed] [Google Scholar]
- 28.Osawa Y, Williams M W. Free Radical Biol Med. 1996;21:35–41. doi: 10.1016/0891-5849(95)02215-5. [DOI] [PubMed] [Google Scholar]
- 29.Schaefer W H, Harris T M, Guengerich F P. Biochemistry. 1985;24:3254–3263. doi: 10.1021/bi00334a027. [DOI] [PubMed] [Google Scholar]
- 30.Brown S B, Hatzikonstantinou H, Herries D G. Biochem J. 1978;174:901–907. doi: 10.1042/bj1740901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones P, Prudhoe K, Robson T. Biochem J. 1973;135:361–365. doi: 10.1042/bj1350361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cornish T, Cotter R J. Rapid Commun Mass Spectrom. 1992;6:242–248. doi: 10.1002/rcm.1290060404. [DOI] [PubMed] [Google Scholar]
- 33.Osawa Y, Highet R J, Murphy C M, Cotter R J, Pohl L R. J Am Chem Soc. 1989;111:4462–4467. [Google Scholar]
- 34.States D J, Haberkorn R A, Ruben D J. J Magn Reson. 1982;48:286–292. [Google Scholar]
- 35.Bax A, Subramanian S. J Magn Reson. 1986;67:565. [Google Scholar]
- 36.Vavra M R, Timkovich R, Yap F, Gennis R B. Arch Biochem Biophys. 1986;250:461–468. doi: 10.1016/0003-9861(86)90750-2. [DOI] [PubMed] [Google Scholar]
- 37.Gordon A J, Ford R A. The Chemist’s Companion: A Handbook of Practical Data, Techniques, and References. New York: Wiley; 1972. p. 438. [Google Scholar]
- 38.Timkovich R, Cork M S, Gennis R B, Johnson P Y. J Am Chem Soc. 1985;107:6069–6075. [Google Scholar]
- 39.Fuhrhop J H, Smith K M. In: Porphyrins and Metalloporphyrins. Smith K M, editor. New York: Elsevier; 1975. pp. 757–869. [Google Scholar]
- 40.Falk J E. Porphyrins and Metalloporphyrins. New York: Elsevier; 1964. pp. 20–21. [Google Scholar]
- 41.Wilks A, Torpey J, Ortiz de Montellano P R. J Biol Chem. 1994;269:29553–29556. [PubMed] [Google Scholar]
- 42.King N K, Winfield M E. J Biol Chem. 1963;238:1520–1528. [PubMed] [Google Scholar]
- 43.King N K, Winfield M E. Aust J Biol Sci. 1966;19:211–217. [PubMed] [Google Scholar]
- 44.George P, Irvine D H. Biochem J. 1952;52:511–517. doi: 10.1042/bj0520511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiu J T, Loewen P C, Switala J, Gennis R B, Timkovich R. J Am Chem Soc. 1989;111:7046–7050. [Google Scholar]