

Summary
Preliminary studies suggested that age at onset (AAO) may help to define homogeneous bipolar affective disorder (BPAD) subtypes. This candidate symptom approach might be useful to identify vulnerability genes. Thus, the probability of detecting major disease-causing genes might be increased by focusing on families with early-onset BPAD type I probands. This study was conducted as part of the European Collaborative Study of Early Onset BPAD (France, Germany, Ireland, Scotland, Switzerland, England, Slovenia). We performed a genome-wide search with 384 microsatellite markers using non parametric linkage analysis in 87 sib-pairs ascertained through an early-onset BPAD type I proband (age at onset of 21 years or below). Non parametric multi-point analysis suggested eight regions of linkage with p-values <0.01 (2p21, 2q14.3, 3p14, 5q33, 7q36, 10q23, 16q23 and 20p12). The 3p14 region showed the most significant linkage (genome-wide p-value estimated over 10.000 simulated replicates of 0.015 [0.01–0.02]). After genome-wide search analysis, we performed additional linkage analyses with increase marker density using markers in four regions suggestive for linkage and having an information contents lower than 75% (3p14, 10q23, 16q23 and 20p12). For these regions, the information content improved by about 10%. In chromosome 3, the non parametric linkage score increased from 3.51 to 3.83. This study is the first to use early onset bipolar type I probands in an attempt to increase sample homogeneity. These preliminary findings require confirmation in independent panels of families.
Keywords: Adolescent; Adult; Age of Onset; Bipolar Disorder; classification; epidemiology; genetics; Child; Chromosome Mapping; Chromosomes, Human; genetics; Chromosomes, Human, Pair 3; genetics; Europe; Female; Genome, Human; Genomic Imprinting; genetics; Humans; Lod Score; Male; Microsatellite Repeats; Phenotype; Statistics, Nonparametric
Introduction
Bipolar affective disorder (BPAD) is a chronic psychiatric disorder with a lifetime cumulative risk of around one percent, affecting males and females equally (1). Family, twin and to a less extent adoption studies strongly support a genetic component of BPAD, buts its mode of inheritance remains unclear (1–3). Genome-wide searches have detected numerous chromosomal regions possibly linked to BPAD, leading to conflicting results. A recent meta-analysis of 18 BPAD genome scan data-sets (4) suggested that no chromosomal region achieved genome-wide statistical significance according to several simulation-based criteria. However, the most significant p values (<0.01) were observed for chromosomes 9p22.3–21.1, 10q11.21–22.1 and 14q24.1–32.12. The discrepancies between studies may be explained by differences in study design (including sample size, type of family structure, mode of ascertainment, phenotypic definitions and statistical methods used) and potential false-positive results. These conflicting results may also be due to the lack of a consensus definition for the affected phenotype and the possible clinical heterogeneity of BPAD (5).
Several studies have suggested that early-onset BPAD is a clinically and genetically homogeneous subtype of BPAD with greater heritability than late-onset BPAD. Clinical studies have demonstrated that early-onset BPAD is a more severe form of the disease, characterized by frequent psychotic features, more mixed episodes, greater psychiatric co-morbidity (with panic disorder and alcohol/drug abuse or dependence) and poorer response to prophylactic lithium (6–8). Familial aggregation is stronger in relatives of early-onset (15% to 30%) than of late-onset (3%) BPAD probands (6–10). Finally, several studies have suggested the existence of an intra-familial correlation for age at onset (AAO) among bipolar siblings (11, 12). A recent segregation analysis showed that BPAD is transmitted differently in early-and late-onset BPAD families (10). Several association studies investigating early-onset BPAD subgroups suggested the implication of candidate genes such as those encoding glycogen synthase kinase 3-beta (OMIM number 605004) (13) and the dopamine receptor D3 (OMIM number 126451) (14). Finally, polymorphisms in the serotonin transporter (SLC6A4 - OMIM number 182138) and apolipoprotein E (OMIM number 107741) genes seem to influence BPAD age at onset (15–17).
Thus, we hypothesized that focusing on families ascertained through an early-onset type I BPAD proband might reduce the heterogeneity of the BPAD phenotype, thus increasing the probability of detecting risk loci by linkage analysis. Such a “symptom candidate approach” (5) using age at onset has been already used in other complex diseases such as breast cancer or Alzheimer disease and has been useful in identifying susceptibility genes (18, 19). We recruited a large sample of sib-pairs including one sibling with early-onset type I BPAD as part of the European Collaborative Study of Early Onset BPAD (France, Germany, Ireland, Scotland, Switzerland, England and Slovenia). The aim of the study is to find genetic factors involved in BPAD in an homogeneous sample ascertained through an early onset type I BPAD proband. This is the first genome-wide search performed using both bipolar I disorder and AAO as inclusion criteria for ascertainment.
Materials and Methods
Subjects
Families were recruited within the European Collaborative Study of Early-Onset BPAD and were eligible for inclusion only if the proband met the DSM-IV criteria (20) for BPAD type I with an AAO of 21 years or below. BPAD type I is defined by the presence of manic episodes with or without depressive episodes. BPAD type II is defined by the presence of hypomanic episodes with depressive episodes (20). The age at onset threshold was chosen according to a previous admixture analysis (21) that has been replicated (22) in two large independent samples of BPAD I patients. These studies identified three AAO subgroups: early (AAO < 21 years old), intermediate and late onset (AAO > 37 years old). Here, AAO was defined for all centers as the age of the first thymic episode (depressive, manic or hypomanic) determined by reviewing medical case notes and information from semi-structured interviews. Affected sibling was diagnosed with BPAD (type I or II) or schizo-affective disorder with no specific requirements regarding AAO, or with major depressive episode (MDE, single or recurrent) with an AAO of 21 years or below. In this study, patients suffering from recurrent major depressive episodes with age at onset below 21 years old were considered as affected, because it was postulated that they belong to the bipolar spectrum disorder as defined by Ghaemi et al. (23). In addition, siblings of BPAD I early onset probands suffering from only one major depressive episode beginning before the age of 21 years old are known to be at high risk of progression to bipolar disorder (24, 25).
Each participant (proband, sibling and parents) was interviewed by a trained psychiatrist using one of the following instruments: Diagnostic Interview for Genetic Studies (DIGS) (26), Composite International Diagnostic Interview (CIDI) (27), Structured Clinical Interview for DSM-IV (SCID) (28) or Schedule for Affective Disorders and Schizophrenia (SADS) (29). These diagnostic questionnaires produce DSM-IV diagnoses. If parents were not evaluated, their phenotype was defined as unknown. Dizygotic and monozygotic twins were excluded. All subjects were of Caucasian origin. Seventy families were recruited as part of the European Collaborative Study of Early-Onset BPAD. Among these 70 families, 27 came from Germany (10 of these German families were included in a previous genome scan (30)), 16 from Ireland, 11 from France, 6 from Scotland, 6 from Switzerland and 4 from England. The protocol was approved by an ethics committee in each participating country and all subjects signed informed consent forms.
Disease Classification
All probands were diagnosed with early-onset type I BPAD and were thus considered to be affected. For the sib-pair analysis, we defined two phenotypes. For the “broad phenotype”, siblings were considered as affected if they had BPAD type I or II or bipolar-type schizo-affective disorder, regardless of AAO, or if they had a major depressive episode (single or recurrent) with an AAO of 21 years or below. For the “narrow phenotype”, siblings were considered as affected if they suffered from BPAD type I with an AAO of 21 years or less, as for probands. The sample consisted of 87 sib-pairs according to the broad phenotype. Among them, 29 sib-pairs met the criteria for the narrow phenotype definition.
Genotyping
Genomic DNA was isolated from peripheral blood leukocytes by the phenol/chloroform method. The genome-screen included 384 highly polymorphic microsatellite markers (Linkage Marker Set MD 10 (Applied Biosystems, Foster City, CA, USA)) and was performed at the Centre National de Génotypage (CNG - France). The average spacing between markers was 9.2 cM, with the largest gaps between D6S1610-D6S257 (23.8 cM) and D21S1911-D21S1914 (22.5 cM). The average heterozygosity was 75%. Automatic genotyping was performed by applying the following processes implemented in the Genetic Profiler software (version 1.1) to the raw MegaBACE data: trace processing, fragment sizing, allele calling and assigning genotype quality scores. A quality score (QS) was assigned to each genotype. This QS is based on the quality of the amplification, the morphology of the marker, the presence or the absence of spurious peaks and the allele favoritism (case of the heterozygous samples only) but also on the deviation of the observed value from the median value for the allele, weighted by the standard deviation of the distribution over all observations. Before statistical analysis, rigorous genotype quality assurance was performed to ensure accurate binning of alleles. The consistency of the data with mendelian inheritance and the lack of recombination between loci were evaluated using Pedcheck (31) and other purpose-written programs. Potentially incorrect genotypes were re-examined and retested if necessary. The mean success rate of genotyping is 95 per cent. Allele frequencies were determined using the FBAT software (32) and were compared to those available in databases (http://www.cephb.fr; http://www.genethon.fr; http://www.chg.duke.edu). We checked that the marker maps used in our analysis were consistent with published maps (http://research.marshfieldclinic.org). Marker locations (distance from p-ter) were extracted from the Marshfield database (http://research.marshfieldclinic.org). Cytogenetic location was extracted from the Human Genome Browser on the Sanger Institute website (http://www.ensembl.org/Homo_sapiens). DNA was available for all sib-pairs and for 76.9 % of parents.
Additional linkage analyses with increased marker density
After genome-wide search analysis, we performed additional linkage analyses with increased marker density using 28 additional markers. Eleven of them were located in regions suggestive for linkage at p=0.01 and having an average information contents lower than 75% (3p14, 10q23, 16q23 and 20p12). This led to an average spacing between markers around 4 cM for these regions. Seventeen markers were spread in other regions with low informativity.
Non Parametric Linkage analysis
The traditional LOD-score method is not directly applicable when the disease model is unknown, as is the case for many common heritable disorders. We thus performed linkage analyses using model-free methods that do not require specification of the underlying genetic model for the trait being investigated. Non parametric bi-point and multipoint linkage analyses were performed using the NPL (Non Parametric Lodscore) statistic of the GENEHUNTER 2.1 program (33). The strength of the linkage was expressed as NPL score and p-value. The NPL score was defined as the sum of NPL over the n pedigrees. For the ith pedigree, the NPL score was defined as NPL(i)=[S(v)-m]/s, where S(v) is the number of pairs of alleles identical by descent for the two sibs given the inheritance vector v, and m and s are respectively the mean and the standard deviation of S under the uniform distribution over the possible inheritance vectors. If allele sharing identical by descent in sib-pairs could not be directly determined (e.g. when DNA from one or both parents was missing), parental haplotypes were estimated based on the haplotypes of their offsprings. Under the hypothesis of no linkage, when there is complete parental information, the p-value can be calculated from the distribution of all the possible inheritance vectors. When parental haplotypes are estimated, we can either use an estimation of the complete information case or use the property that the NPL score tends towards a standard normal variable with a mean of 0 and a variance of 1. As few families in our sample included three affected sibs, all possible pairs were considered without using any weighting procedure. Indeed, when the number of sib-ships of size 3 or more is moderate, forming all possible pairs only slightly increases type I errors, whereas frequently used weighting procedures decrease the efficiency of the tests (34).
Test for parent-of-origin effect
Paternal and maternal inheritances were investigated using the ASPEX program (35). This method reports identity-by-descent information for all affected sib-pairs in nuclear families. At a given locus, for each parent, the program compares the corresponding alleles for each member of an affected sib-pair. If both alleles can uniquely be identified, the pair is scored as either identical or non-identical by descent from that parent, at that position. If the match is ambiguous, the position is scored as being uninformative. If data from additional siblings are available, they are used to reconstruct missing parents. To test for a parent-of-origin effect, we used a classical chi-square test to compare maternal and paternal allele sharing. To correct for multi-testing, we applied the classical Bonferroni correction.
Simulation study
As we conducted multiple analyses for multiple markers, we carried out a simulation study to estimate the genome-wide empirical p values. Given the small number of families, it is preferential to use simulations, rather than asymptotic theory (Holmans 2001). Marker allele frequencies and map distances were kept as in the original sample and genotypes were dropped through the 70 families, with the SIMULATE program (36). This program was modified to keep the original founder’s genotypes, under the hypothesis of no linkage between the disease and the markers. Our aim was to obtain the same information for replicates and the original set: the same parental genotypes, the same map distances, missing individuals for each marker and phenotypes for each individual. To estimate the genome-wide p values, we simulated 10.000 replicates for the 22 autosomes. The phenotypes were not simulated, and each individual was attributed his or her real set of phenotypes. These replicates were analyzed by genome-wide multipoint analyses. For each replicate i, the maximum NPL score (NPLMi) was recorded. For example, the genome-wide p value for a NPL of 3.51 was determined by the number of times NPLMi exceeded 3.51 divided by the number of replicates. This gave us the genome-wide p value accounting for multiple testing at all positions of the genome. We were also able to calculate the number of occurrences of a given NPL in each replicate NbT. This process was implemented in the FDB linkage and association management system (37).
Results
Description of the sample
We recruited 70 nuclear families ascertained through an early onset BPAD, including 87 sib-pairs according to the broad phenotype definition. Among them, 29 sib-pairs were considered as affected using the narrow phenotype definition. For the broad phenotype, mean AAO was 17.2 ± 2.4 years (range 11–21 years) for probands and 22.0 ± 6.8 years (range 9–46 years) for siblings. The sex (male/female) ratio was 0.66 for probands and 0.76 for siblings. The siblings were diagnosed with: BPAD type I (69.6%; N=55), BPAD type II (13.9%; N=11), bipolar-type schizo-affective disorder (10.1%; N=8) and major depressive episode (single or recurrent) (6.3%; N=5). For the narrow phenotype, mean AAO was 17.4 ± 2.5 years (range 12–21 years) for probands and 17.5 ± 2.9 years (range 9–21) for siblings. The sex ratio was 0.80 for probands and 0.87 for siblings.
Non parametric linkage analysis
The results of the non parametric bi-point linkage analysis are presented in table 1. When all p-values were non-significant over a region of more than 20 cM between two markers in favor of linkage (i.e. with a p<0.05), markers were assumed to detect different regions of linkage. Four regions had a p-value ≤ 0.01: the 3p14.1–14.3 region linked to the broad (p=0.002) and narrow (p=0.01) phenotypes respectively; the 10q23.33-q24.31 and 20p12.2 regions linked to the broad phenotype only (p=0.01 and p=0.002 respectively) and the 16q23.1 region linked to the narrow phenotype only (p=0.004). The 2p21-p23.2 and the 17q11.2-q22 regions had a p-value < 0.05 for adjacent markers (p=0.02 for the broad phenotype in 2p and p=0.03 for the narrow phenotype in 17q). None of the 13 chromosome X markers was suggestive for linkage.
Table 1.
Non parametric bi-point linkage analysis - Regions detected with a p value ≤0.05
| Cytogenetic location | Marker name | Dist. from p-ter (cM) | IC* | Narrow phenotype | Broad phenotype | ||
|---|---|---|---|---|---|---|---|
| NPL | p-value | NPL | p-value | ||||
| 2p23.2 | D2S165 | 47.43 | 0.70 | 0.59 | 0.17 | 1.67 | 0.05 |
| 2p22.3 | D2S367 | 54.96 | 0.74 | 0.9 | 0.08 | 1.7 | 0.04 |
| 2p21 | D2S2259 | 64.29 | 0.64 | 0.69 | 0.14 | 1.61 | 0.05 |
| 2p21 | D2S391 | 70.31 | 0.58 | 0.42 | 0.25 | 1.94 | 0.02 |
| # | |||||||
| 2q14.3 | D2S347 | 131.51 | 0.56 | 0.57 | 0.18 | 1.65 | 0.05 |
| # | |||||||
| 3p14.2 | D3S1300 | 80.32 | 0.68 | 1.47 | 0.01 | 2.88 | 0.002 |
| 3p14.1 | D3S1285 | 91.18 | 0.56 | 0.46 | 0.23 | 1.96 | 0.02 |
| # | |||||||
| 4q13.3 | D4S392 | 78.97 | 0.64 | 0.70 | 0.14 | 1.68 | 0.04 |
| # | |||||||
| 5q32 | D5S436 | 147.49 | 0.68 | 1.10 | 0.04 | 0.84 | 0.20 |
| # | |||||||
| 7q21.3 | D7S657 | 104.86 | 0.65 | 0.62 | 0.16 | 1.64 | 0.05 |
| # | |||||||
| 7q36 | D7S798 | 168.98 | 0.67 | 0.12 | 0.43 | 1.83 | 0.03 |
| # | |||||||
| 10p13 | D10S1653 | 40.36 | 0.7 | 1.07 | 0.04 | 0.51 | 0.30 |
| # | |||||||
| 10q23.33 | D10S185 | 116.34 | 0.66 | 0.50 | 0.22 | 2.25 | 0.01 |
| 10q24.31 | D10S192 | 124.27 | 0.66 | −0.06 | 0.54 | 1.88 | 0.03 |
| # | |||||||
| 16p13.2 | D16S404 | 18.07 | 0.69 | 1.26 | 0.02 | 1.14 | 0.12 |
| # | |||||||
| 16q23.1 | D16S516 | 100.39 | 0.6 | 1.64 | 0.004 | 1.36 | 0.08 |
| # | |||||||
| 17q11.2 | D17S798 | 53.41 | 0.58 | 1.17 | 0.03 | 0.33 | 0.37 |
| 17q21.32 | D17S1868 | 64.16 | 0.63 | 0.97 | 0.06 | 0.87 | 0.19 |
| 17q22 | D17S787 | 74.99 | 0.63 | 1.01 | 0.05 | 0.9 | 0.18 |
| # | |||||||
| 20p12.2 | D20S186 | 32.3 | 0.65 | 0.3 | 0.32 | 2.93 | 0.002 |
indicates two different regions (markers were assumed to detect different regions of linkage if all p-values were non-significant over a region of >20 cM between two markers with significant linkage).
IC: Information Content
Non parametric multipoint linkage analysis plots are described in figure 1 for both phenotypes. Multipoint NPL value are reported in table 2 for each chromosomal regions identified in bi-point analysis. Most of the 13 regions reached NPL values at the maximum multipoint test statistic that were higher than those obtained by single point analysis. The only exceptions were found at chromosomes 7q21.3, 10p13 and 16q23.1, for which a lower or equal significance level in multipoint analysis was obtained comparing to single-point analysis. Non parametric multipoint linkage analysis of these regions revealed significant linkage (p≤0.01) for at least one phenotype in the following regions: 2p21 (NPL=2.8, p=0.003), 2q14.3 (NPL=2.24; p=0.01), 7q36 (NPIX2.43; p=0.007), 10q23 (NPL=2.45; p=0.007) and 20p12 (NPL=2.25; p=0.01) only for the broad phenotype, 3p14 for both the narrow (NPL=1.65; p=0.004) and broad phenotypes (NPL=3.51 p=0.0002), and 5q33 (NPL=1,4; p=0.01) and 16q23 (NPL=1.64; p=0.005) only for the narrow phenotype.
Figure 1.
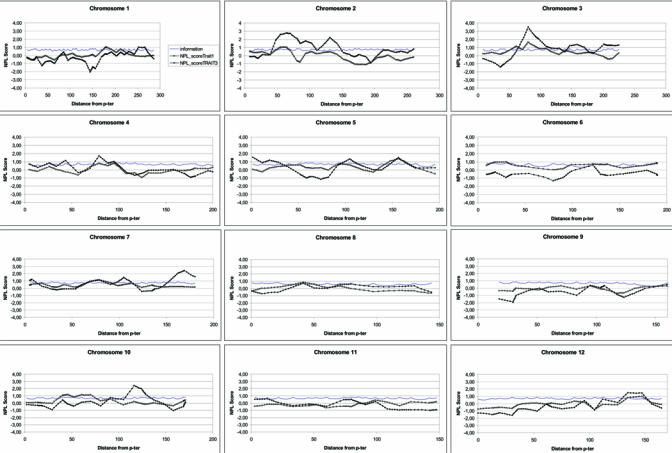
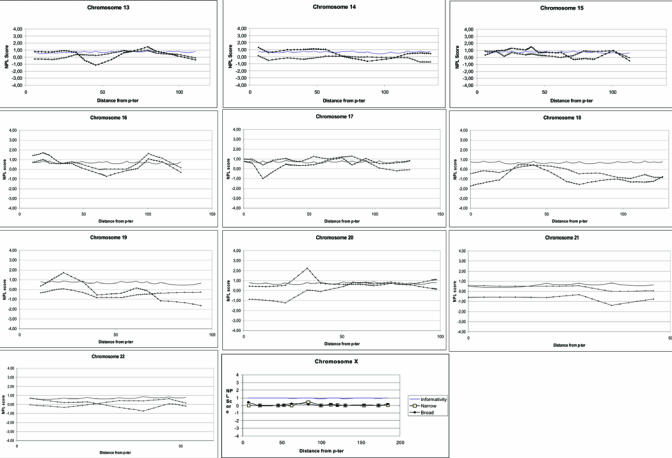
Non parametric multipoint linkage analysis plots
Table 2.
Non parametric multipoint linkage analysis
| Marker name | Distance of peak from p-ter | IC at peak | Narrow phenotype | Broad phenotype | ||
|---|---|---|---|---|---|---|
| NPL | p-value | NPL | p-value | |||
| D2S2259-D2S391 | 64.29 | 0.83 | 1.03 | 0.05 | 2.78 | 0.003 |
| D2S347 | 131.51 | 0.74 | 0.48 | ns | 2.24 | 0.01 |
| D3S1300 | 80.32 | 0.83 | 1.65 | 0.004 | 3.51 | 0.0002 |
| D4S392 | 78.97 | 0.78 | 0.81 | ns | 1.73 | 0.04 |
| D5S410 | 156.47 | 0.80 | 1.37 | 0.01 | 1.51 | 0.06 |
| D7S657 | 104.86 | 0.81 | 0.48 | ns | 1.49 | 0.07 |
| D7S798 | 168.98 | 0.82 | 0.19 | ns | 2.43 | 0.007 |
| D10S1653 | 40.36 | 0.85 | 1.07 | 0.04 | 0.43 | ns |
| D10S185 | 116.34 | 0.80 | 0.20 | ns | 2.45 | 0.007 |
| D16S404 | 18.07 | 0.86 | 0.99 | ns | 1.70 | 0.04 |
| D16S516 | 100.39 | 0.76 | 1.64 | 0.005 | 1.07 | 0.14 |
| D17S944 | 82.56 | 0.79 | 1.28 | 0.02 | 0.39 | ns |
| D20S186 | 32.30 | 0.81 | 0.05 | ns | 2.25 | 0.01 |
ns: non significant
Additional linkage analyses with increased marker density
Among the most significant regions (p<0.01 in multipoint analyses) and when information contents was lower than 75%, eleven additional markers were genotyped (in regions 3p14, 10q23, 16q23 and 20p12). For these regions, the location of the peak of linkage remained unchanged and the information content improved by about 10% in each region. In chromosome 3 region, the NPL score increased from 3.51 to 3.83 (broad phenotype, p=0.00005) and from 1.65 to 1.70 (narrow phenotype, p=0.003). In chromosome 10, the NPL score decreased from 2.45 to 2.25 (broad phenotype, p=0.01). In chromosome 16, the NPL score decreased from 1.64 to 1.33 (narrow phenotype, p=0.02). In chromosome 20, the NPL score increased from 2.25 to 2.5 (broad phenotype, p=0.006). We have also genotyped 17 markers spread in other regions with low informativity. Analyses of additional markers in these regions did not change the results of the multipoint linkage analysis (data not shown).
Simulation study
For the broad phenotype, a NPL of 3.51 corresponded to a genome-wide p-value of 0.015 [0.01–0.02] on the basis of simulations. According to simulations, a NPL score equal and higher than 3.23 would be needed to achieve empirical genome-wide significance at the 0.05 level in our data. Therefore, only the 3p14 region was still significant according to the genome-wide p-value.
Paternal and maternal contributions to linkage detection
Parent-of-origin effect was investigated in the eight regions linked to at least one phenotype with a p-value lower than 0.01 (see Table 3). An excess of maternal sharing compared to paternal sharing was suggested for 3p14 for the narrow phenotype (ORmaternal=9.3 [1.6–54.8]; p=0.008) and an excess of paternal sharing was suggested for 10q23 with both phenotypes (ORpaternal=5.0 [1.0–24.9]; p=0.03 for the narrow phenotype and ORpaternal=2.3 [1.0–5.5]; p=0.05 for the broad one). When Bonferroni’s correction for multi-testing was applied, no parent-of-origin effect remained significant.
Table 3.
Test for parent-of-origin effect
| Narrow phenotype | Broad phenotype | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Maternal meiosis | Paternal meiosis | Maternal meiosis | Paternal meiosis | |||||||
| Locus | %ibd | χ2 | %ibd | χ2 | Test for parental contribution χ2 (p-value) | %ibd | χ2 | %ibd | χ2 | Test for parental contribution χ2 (p-value) |
| D2S2259 | 53.3 | 0.07 | 57.9 | 0.47 | 0.07 (ns) | 65.1 | 3.93 | 57.1 | 1.00 | 0.6 (ns) |
| D2S391 | 57.1 | 0.29 | 53.3 | 0.07 | 0.04 (ns) | 71.8 | 7.41 | 57.1 | 0.86 | 1.9(ns) |
| D2S347 | 77.8 | 2.78 | 55.6 | 0.11 | 1.0 (ns) | 69.7 | 5.12 | 60.6 | 1.48 | 0.6 (ns) |
| D3S1300 | 41.2 | 0.53 | 86.7 | 8.07 | 7.0 (0.008) | 52.1 | 0.08 | 70.2 | 7.68 | 3.3 (0.07) |
| D5S410 | 56.2 | 0.25 | 72.2 | 3.56 | 0.9 (ns) | 50.0 | 0.00 | 53.1 | 0.18 | 0.01 (ns) |
| D7S798 | 44.4 | 0.22 | 47.8 | 0.04 | 0.05 (ns) | 59.6 | 1.72 | 52.6 | 0.16 | 0.5 (ns) |
| D10S185 | 81.2 | 6.25 | 46.7 | 0.07 | 4.0 (0.04) | 70.8 | 8.33 | 51.1 | 0.02 | 3.8 (0.05) |
| D16S516 | 73.3 | 3.27 | 80.0 | 5.40 | 0.2 (ns) | 61.8 | 1.88 | 60.0 | 1.40 | 0.02 (ns) |
| D20S186 | 61.5 | 0.69 | 50.0 | 0.00 | 0.4 (ns) | 69.6 | 7.04 | 71.1 | 8.02 | 0.03 (ns) |
ns: nonsignificant
Discussion
The aim of our study was to search for genes implicated in bipolar affective disorder. We hypothesized that ascertaining families through an early onset bipolar I patient led to select an homogeneous sample of patients. As the traditional LOD score method cannot be applied directly when the disease model is unknown, we used model-free methods to search for chromosome regions linked with BPAD in a sample of 70 European families ascertained through an early-onset BPAD type I proband. Non parametric multi-point analysis identified eight regions of linkage with p-values ≤0.01 (2p21, 2q14.3, 3p14, 5q33, 7q36, 10q23, 16q23, 20p12). The genome-wide p-value at 3p14 was 0.01, estimated by our simulation study of 10.000 replicates. Interestingly, an excess of maternal sharing for the 3p14 region and an excess of paternal sharing for 10q23 were suggested but these results did not remain significant after Bonferroni’s correction for multi-testing.
In this multicentric study, diagnoses for affective disorders were generated using the following instruments : DIGS, CIDI, SCID or SADS. All these questionnaires generated DSM-IV criteria based diagnoses. Differences in sensitivity and specificity of these instruments may have introduced some clinical heterogeneity. However, inclusion criteria were strictly based on DSM-IV affective disorders definitions and all centers used the same stringent definition for age at onset. Using these strict inclusion criteria may have restricted the heterogeneity inherent in our multicentric study design.
As expected, there is a high concordance between probands and siblings diagnoses as 70% of siblings (N=55) were diagnosed with BPAD type I, 14% with BPAD type II (N=11), 10% with schizo-affective disorder bipolar type (N=8) and 6% of major depressive episode (single or recurrent; N=5). We determined a strict threshold to define age at onset for proband inclusion criteria (equal or lower than 21 years). Using such a criteria led us to analyse a small sample of sib-pairs for the narrow phenotype. With a less stringent AAO criteria (equal or lower than 25 years), 74 of 87 sib-pairs (85%) would have been concordant for age at onset, whatever there primary affective diagnoses were.
Regions 2p21, 3p14, 5q33 and 20p12 appeared to be linked to both phenotypes, whereas 2p14.3, 7q36 and 10q23 showed linkage only to the broad one. These results might be interpreted as a gain of power due to sample size (29 sib-pairs for the narrow phenotype versus 87 for the broad one).
Examination of (i) the number of studies revealing positive linkage, (ii) the degree of overlap between the detected regions and (iii) similarities in phenotypic definition may shed some light on these results. Seven of these regions have previously been reported to be linked to BPAD. Regions 3p14, 7q36, 10q23 and 20p12 are particularly interesting as they were detected in our sample and in at least two of the previously published scans in BPAD using a significance threshold of p<0.01 (see table 4). Chromosomal regions 2p21 (38, 39), 3p14 (40–42), 7q36 (43, 44) and 20p12 (45, 46) were previously identified in three independent studies, reinforcing the hypothesis of linkage. Some of these regions (2p21, 7q36, 20p12) were also detected in a previous genome scan (30). However, since some families were analyzed in this paper and in our study, our results cannot be considered to be an independent replication. The 16q24 region, which was significant only with our narrow definition, has not been described in any BPAD genome scan. This result could be a false positive due to multi-testing. Our approach, using both BPAD type I and AAO as inclusion criteria for probands, allowed us to select only seven regions among numerous regions suggested by published genome-wide searches of BPAD and to suggest the implication of the 3p14 region.
Table 4.
Regions detected in published genome screens and replicated in the European families for BPAD phenotypes (p<0.01)
| Region | Distance from p-ter Marschfield (cM) | Early onset BPAD Peak of linkage | Previous genome wide searches of BPAD |
|---|---|---|---|
| Phenotype (p-value) | Authors (Year) | ||
| 2p22-p25 | 28–55 | Detera-Wadleigh (1999), Cichon (2001) | |
| 2p21 | 64 | Broad (p=0.003) | |
| 2p14 | 87 | Liu (2003) | |
| # | |||
| 2q14 | 131 | Broad (p=0.01) | |
| 2q21 | 142–147 | Cichon (2001); Ewald (2003) (47) | |
| # | |||
| 3p14 | 62–79 | Fallin (2004), Kelsoe (2001) | |
| 3p14 | 80 | Narrow (p=0.004) Broad (p=0.0002) | |
| 3p13-p14 | 90–103 | Me Innes (1996), Kelsoe (2001), Cichon (2001), Radhakrishna (2001) | |
| # | |||
| 5q33 | 156 | Narrow (p=0.01) | |
| 5q33 | 160 | Edenberg (1997) (48) | |
| 5q35 | 184 | Detera-Wadleigh (1999) | |
| # | |||
| 7q34-q36 | 147–161 | Cichon (2001), Detera-Wadleigh (1997), Curtis (2003) | |
| 7q36 | 168 | Broad (p=0.007) | |
| # | |||
| 10q23 | 116 | Broad (p= 0.007) | |
| 10q24-q25 | 124–134 | Cichon (2001), Kelsoe (2001) | |
| # | |||
| 16q24 | 100 | Narrow (p=0.005) | |
| 16q24 | 129 | Me Innes (1996) | |
| # | |||
| 20p12 | 25 | Morissette (1999) | |
| 20p12 | 32 | Broad (p=0.01) | |
| 20p12 | 33–39 | Radhakrishna (2001), Morissette (1999), Cichon (2001) |
Markers were assumed to detect different regions of linkage if all p-values were nonsignificant over a region of > 20 cM between two markers with significant linkage.
We investigated if any candidate genes reported as associated with early-onset BPAD could be located near regions of linkage. Our study showed a bi-point NPL of 1.2 (p=0.03) for the narrow phenotype at marker D17S798, which is about 3 cM from the serotonin transporter gene (SLC6A4). We did not find any significant linkage in regions containing other candidate genes (i.e. glycogen synthase kinase 3-beta and dopamine receptor D3 located in 3q13 and apoliproprotein E located in 19q13). These genes might have only a weak effect on BPAD or this result may be due to the peculiarities of our sample, such as mode of ascertainment or false positive results (13,14,16).
As there is large confidence interval for linkage in these regions (i.e 38 cM for 2p21, 25 cM for 2q14.3, 18 cM for 3p14, 33 cM for 5q33, 18 cM for 7q36, 17 cM for 10q23 and 12 cM for the 20p12 regions), we are currently undergoing the fine mapping of the regions of interest. Our preliminary findings require confirmation in independent panels of families.
Acknowledgments
We thank all the families who participated to this study. We thank Magalie Duroux (Laboratory INSERM U513 - Créteil - France) for preparing DNA samples for genotyping. We thank Catherine Bonaïti-Pellié for helpful comments about this work.
Footnotes
This work was supported by:
The French study was supported by grants from Assistance Publique-Hôpitaux de Paris, Ministère de la Recherche (PHRC, AOM98152) and the National Alliance for Research on Schizophrenia and Depression (NARSAD). B. Etain received a grant from Institut National de la Santé et de la Recherche Médicale (Poste d’Accueil INSERM).
The German study received the following support: the National Genomic Network (NGFN) of the German Ministry of Education and Research, the Deutsche Forschungsgememschaft (SFB 400 subprojects Dl and D3, Graduiertenkolleg GRK 246, FOR 423 subproject D1), the Alfried Krupp von Bohlen und Halbach-Stiftung, the Interuniversity Attraction Poles program P5/19 of the Belgian Federal Science Policy Office, and the National Alliance for Research on Schizophrenia and Depression (NARSAD). Research at the District Hospital Haar was supported by grants from the German Research Society (DFG), AL 230-1/2/3 – 230-5/1/2 and the SFB 400.
D. Blackwood and W. Muir were supported by grants from The Health Foundation, London. The Medical Research Council, UK; The Chief Scientist Office of the Scottish Executive. The Irish study was supported by Aware, the Irish charity helping to defeat depression, the Health Research Board (H01069 HRB RP153/2000), and the Friends of St. Patrick’s Hospital. The Swiss study was supported by five grants from the Swiss National Foundation (#32-40677.94 to F. Ferrero, #32-47315.96 and # 32-061974.00 to M. Preisig, #32-66793.01 and #32-102168.03 to A. Malafosse).
References
- 1.Taylor L, Faraone SV, Tsuang MT. Family, twin, and adoption studies of bipolar disease. Curr Psychiatry Rep. 2002;4(2):130–3. doi: 10.1007/s11920-002-0046-1. [DOI] [PubMed] [Google Scholar]
- 2.McGuffm P, Rijsdijk F, Andrew M, Sham P, Katz R, Cardno A. The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch Gen Psychiatry. 2003;60(5):497–502. doi: 10.1001/archpsyc.60.5.497. [DOI] [PubMed] [Google Scholar]
- 3.Smoller JW, Finn CT. Family, twin, and adoption studies of bipolar disorder. Am J Med Genet. 2003;123C(1):48–58. doi: 10.1002/ajmg.c.20013. [DOI] [PubMed] [Google Scholar]
- 4.Segurado R, Detera-Wadleigh SD, Levinson DF, Lewis CM, Gill M, Nurnberger JI, Jr, et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part III: Bipolar disorder. Am J Hum Genet. 2003;73(1):49–62. doi: 10.1086/376547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leboyer M, Bellivier F, Nosten-Bertrand M, Jouvent R, Pauls D, Mallet J. Psychiatric genetics: search for phenotypes. Trends Neurosci. 1998;21(3):102–5. doi: 10.1016/s0166-2236(97)01187-9. [DOI] [PubMed] [Google Scholar]
- 6.Weissman MM, Gershon ES, Kidd KK, Prusoff BA, Leckman JF, Dibble E, et al. Psychiatric disorders in the relatives of probands with affective disorders. The Yale University-National Institute of Mental Health Collaborative Study. Arch Gen Psychiatry. 1984;41(1):13–21. doi: 10.1001/archpsyc.1984.01790120015003. [DOI] [PubMed] [Google Scholar]
- 7.Schurhoff F, Bellivier F, Jouvent R, Mouren-Simeoni MC, Bouvard M, Allilaire JF, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215–21. doi: 10.1016/s0165-0327(99)00111-1. [DOI] [PubMed] [Google Scholar]
- 8.Strober M, Morrell W, Burroughs J, Lampert C, Danforth H, Freeman R. A family study of bipolar I disorder in adolescence. Early onset of symptoms linked to increased familial loading and lithium resistance. J Affect Disord. 1988;15(3):255–68. doi: 10.1016/0165-0327(88)90023-7. [DOI] [PubMed] [Google Scholar]
- 9.Faraone SV, Glatt SJ, Tsuang MT. The genetics of pediatric-onset bipolar disorder. Biol Psychiatry. 2003;53(11):970–7. doi: 10.1016/s0006-3223(02)01893-0. [DOI] [PubMed] [Google Scholar]
- 10.Grigoroiu-Serbanescu M, Martinez M, Nothen MM, Grinberg M, Sima D, Propping P, et al. Different familial transmission patterns in bipolar I disorder with onset before and after age 25. Am J Med Genet. 2001;105(8):765–73. doi: 10.1002/ajmg.10047. [DOI] [PubMed] [Google Scholar]
- 11.Baron M, Risch N, Mendlewicz J. Age at onset in bipolar-related major affective illness: clinical and genetic implications. J Psychiatr Res. 1982;17(1):5–18. doi: 10.1016/0022-3956(82)90030-9. [DOI] [PubMed] [Google Scholar]
- 12.Leboyer M, Bellivier F, McKeon P, Albus M, Borrman M, Perez-Diaz F, et al. Age at onset and gender resemblance in bipolar siblings. Psychiatry Res. 1998;81(2):125–31. doi: 10.1016/s0165-1781(98)00087-0. [DOI] [PubMed] [Google Scholar]
- 13.Benedetti F, Bernasconi A, Lorenzi C, Pontiggia A, Serretti A, Colombo C, et al. A single nucleotide polymorphism in glycogen synthase kinase 3-beta promoter gene influences onset of illness in patients affected by bipolar disorder. Neurosci Lett. 2004;355(1–2):37–40. doi: 10.1016/j.neulet.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 14.Chiaroni P, Azorin JM, Dassa D, Henry JM, Giudicelli S, Malthiery Y, et al. Possible involvement of the dopamine D3 receptor locus in subtypes of bipolar affective disorder. Psychiatr Genet. 2000;10(1):43–9. doi: 10.1097/00041444-200010010-00008. [DOI] [PubMed] [Google Scholar]
- 15.Bellivier F, Leroux M, Henry C, Rayah F, Rouillon F, Laplanche JL, et al. Serotonin transporter gene polymorphism influences age at onset in patients with bipolar affective disorder. Neurosci Lett. 2002;334(1):17–20. doi: 10.1016/s0304-3940(02)01029-7. [DOI] [PubMed] [Google Scholar]
- 16.Bellivier F, Laplanche JL, Schurhoff F, Feingold J, Feline A, Jouvent R, et al. Apolipoprotein E gene polymorphism in early and late onset bipolar patients. Neurosci Lett. 1997;233(1):45–8. doi: 10.1016/s0304-3940(97)00624-1. [DOI] [PubMed] [Google Scholar]
- 17.Ospina-Duque J, Duque C, Carvajal-Carmona L, Ortiz-Barrientos D, Soto I, Pineda N, et al. An association study of bipolar mood disorder (type I) with the 5-HTTLPR serotonin transporter polymorphism in a human population isolate from Colombia. Neurosci Lett. 2000;292(3):199–202. doi: 10.1016/s0304-3940(00)01464-6. [DOI] [PubMed] [Google Scholar]
- 18.Rosen EM, Fan S, Pestell RG, Goldberg ID. BRCA1 gene in breast cancer. J Cell Physiol. 2003;196(1):19–41. doi: 10.1002/jcp.10257. [DOI] [PubMed] [Google Scholar]
- 19.Rocchi A, Pellegrini S, Siciliano G, Murri L. Causative and susceptibility genes for Alzheimer’s disease: a review. Brain Res Bull. 2003;61(1):1–24. doi: 10.1016/s0361-9230(03)00067-4. [DOI] [PubMed] [Google Scholar]
- 20.American Psyciatric Association. Diagnostic and statistical manual of mental disorders, (DSM-IV) 4. Washington, DC: 1994. [Google Scholar]
- 21.Bellivier F, Golmard JL, Henry C, Leboyer M, Schurhoff F. Admixture analysis of age at onset in bipolar I affective disorder. Arch Gen Psychiatry. 2001;58(5):510–2. doi: 10.1001/archpsyc.58.5.510. [DOI] [PubMed] [Google Scholar]
- 22.Bellivier F, Golmard JL, Rietschel M, Schulze TG, Malafosse A, Preisig M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999–1001. doi: 10.1176/appi.ajp.160.5.999. [DOI] [PubMed] [Google Scholar]
- 23.Ghaemi SN, Ko JY, Goodwin FK. Cade’s disease and beyond : misdiagnosis, antidepressant use, and a proposed definition for bipolar spectrum disorder. Can J Psychiatry. 2002;47:125–134. doi: 10.1177/070674370204700202. [DOI] [PubMed] [Google Scholar]
- 24.Angst J, Sellaro R, Stassen HH, Gamma A. Diagnostic conversion from depression to bipolar disorders: results of a long-term prospective study of hospital admissions. Journal of Affective Disorders. 2005 doi: 10.1016/S0165-0327(03)00195-2. In Press. [DOI] [PubMed] [Google Scholar]
- 25.Smith DJ, Harrison N, Muir W, Blackwood DHR. The high prevalence of bipolar spectrum disorders in young adults with recurrent depression: toward an innovative diagnostic framework. Journal of Affective Disorders. 2005 doi: 10.1016/j.jad.2004.02.005. In Press. [DOI] [PubMed] [Google Scholar]
- 26.Nurnberger JJ, Blehar M, Kaufmann C, York-Cooler C, Simpson S, Harkavy-Friedman J, et al. Diagnostic interview for genetic studies. Rationale, unique features, and training. NIMH Genetics Initiative. Arch Gen Psychiatry. 1994;51(11):849–59. doi: 10.1001/archpsyc.1994.03950110009002. discussion 863–4. [DOI] [PubMed] [Google Scholar]
- 27.Wittchen HU, Robins LN, Cottier LB, Sartorius N, et al. Cross-cultural feasibility, reliability and sources of variance of the Composite International Diagnostic Interview (CIDI) Crit J Psychiatry. 1991;159:645–653. doi: 10.1192/bjp.159.5.645. [DOI] [PubMed] [Google Scholar]
- 28.Spitzer First MB, Gibbon Miriam Robert L, Williams Janet BW. Biometrics Research. New York State Psychiatric Institute; New York: 2002. Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Patient Edition. (SCID-I/P) [Google Scholar]
- 29.Endicott J, Spitzer RL. A diagnostic interview. The schedule for affective disorders and schizophrenia. Arch Gen Psychiatry. 1978;35:837–844. doi: 10.1001/archpsyc.1978.01770310043002. [DOI] [PubMed] [Google Scholar]
- 30.Cichon S, Schumacher J, Muller DJ, Hurter M, Windemuth C, Strauch K, et al. A genome screen for genes predisposing to bipolar affective disorder detects a new susceptibility locus on 8q. Hum Mol Genet. 2001;10(25):2933–44. doi: 10.1093/hmg/10.25.2933. [DOI] [PubMed] [Google Scholar]
- 31.O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63(1):259–66. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horvath S, Xu X, Laird NM. The family based association test method: strategies for studying general genotype—phenotype associations. Eur J Hum Genet. 2001;9(4):301–6. doi: 10.1038/sj.ejhg.5200625. [DOI] [PubMed] [Google Scholar]
- 33.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996;58(6):1347–63. [PMC free article] [PubMed] [Google Scholar]
- 34.Meunier F, Philippi A, Martinez M, Demenais F. Affected sib-pair tests for linkage: type I errors with dependent sib-pairs. Genet Epidemiol. 1997;14(6):1107–11. doi: 10.1002/(SICI)1098-2272(1997)14:6<1107::AID-GEPI91>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 35.Hinds DA, Risch N. The ASPEX package: affected sib-pair exclusion mapping. http://aspex.sourceforge.net/1996.
- 36.Terwilliger J, Speer M, Ott J. Chromosome-based method for rapid computer simulation in human genetic linkage analysis. Genet Epidemiol. 1993;10:217–224. doi: 10.1002/gepi.1370100402. [DOI] [PubMed] [Google Scholar]
- 37.Gallina S, Lecoeur C, Delannoy V, Dina C. European Mathematical Genetics Meeting. Lille; France: 2001. Intranet software environment for linkage analysis studies. [Google Scholar]
- 38.Detera-Wadleigh SD, Badner JA, Berrettini WH, Yoshikawa T, Goldin LR, Turner G, et al. A high-density genome scan detects evidence for a bipolar-disorder susceptibility locus on 13q32 and other potential loci on 1q32 and 18p11.2. Proc Natl Acad Sci U S A. 1999;96(10):5604–9. doi: 10.1073/pnas.96.10.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Juo SH, Dewan A, Grunn A, Tong X, Brito M, et al. Evidence for a putative bipolar disorder locus on 2p13–16 and other potential loci on 4q31, 7q34, 8q13, 9q31, 10q21–24, 13q32, 14q21 and 17q11–12. MolPsychiatry. 2003;8(3):333–42. doi: 10.1038/sj.mp.4001254. [DOI] [PubMed] [Google Scholar]
- 40.Fallin MD, Lasseter VK, Wolyniec PS, McGrath JA, Nestadt G, Valle D, et al. Genomewide Linkage Scan for Bipolar-Disorder Susceptibility Loci among Ashkenazi Jewish Families. Am J Hum Genet. 2004;75(2) doi: 10.1086/422474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kelsoe JR, Spence MA, Loetscher E, Foguet M, Sadovnick AD, Remick RA, et al. A genome survey indicates a possible susceptibility locus for bipolar disorder on chromosome 22. Proc Natl Acad Sci U S A. 2001;98(2):585–90. doi: 10.1073/pnas.011358498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McInnes LA, Escamilla MA, Service SK, Reus VI, Leon P, Silva S, et al. A complete genome screen for genes predisposing to severe bipolar disorder in two Costa Rican pedigrees. Proc Natl Acad Sci U S A. 1996;93(23):13060–5. doi: 10.1073/pnas.93.23.13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Detera-Wadleigh SD, Badner JA, Yoshikawa T, Sanders AR, Goldin LR, Turner G, et al. Initial genome scan of the NIMH genetics initiative bipolar pedigrees: chromosomes 4, 7, 9, 18, 19, 20, and 21q. Am J Med Genet. 1997;74(3):254–62. doi: 10.1002/(sici)1096-8628(19970531)74:3<254::aid-ajmg4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 44.Curtis D, Kalsi G, Brynjolfsson J, Mclnnis M, O’Neill J, Smyth C, et al. Genome scan of pedigrees multiply affected with bipolar disorder provides further support for the presence of a susceptibility locus on chromosome 12q23-q24, and suggests the presence of additional loci on 1p and 1q. Psychiatr Genet. 2003;13(2):77–84. doi: 10.1097/01.ypg.0000056684.89558.d2. [DOI] [PubMed] [Google Scholar]
- 45.Morissette J, Villeneuve A, Bordeleau L, Rochette D, Laberge C, Gagne B, et al. Genome-wide search for linkage of bipolar affective disorders in a very large pedigree derived from a homogeneous population in Quebec points to a locus of major effect on chromosome 12q23-q24. Am J Med Genet. 1999;88(5):567–87. doi: 10.1002/(sici)1096-8628(19991015)88:5<567::aid-ajmg24>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 46.Radhakrishna U, Senol S, Herken H, Gucuyener K, Gehrig C, Blouin JL, et al. An apparently dominant bipolar affective disorder (BPAD) locus on chromosome 20p11.2-q11.2 in a large Turkish pedigree. Eur J Hum Genet. 2001;9(1):39–44. doi: 10.1038/sj.ejhg.5200584. [DOI] [PubMed] [Google Scholar]
- 47.Ewald H, Kruse TA, Mors O. Genome wide scan using homozygosity mapping and linkage analyses of a single pedigree with affective disorder suggests oligogenic inheritance. Am J Med Genet. 2003;120B(1):63–71. doi: 10.1002/ajmg.b.20039. [DOI] [PubMed] [Google Scholar]
- 48.Edenberg HJ, Foroud T, Conneally PM, Sorbel JJ, Carr K, Crose C, et al. Initial genomic scan of the NIMH genetics initiative bipolar pedigrees: chromosomes 3, 5, 15, 16, 17, and 22. Am J Med Genet. 1997;74(3):238–46. [PubMed] [Google Scholar]