

Abstract
Reactive oxygen species (ROS) contribute to the development of chronic ethanol-induced liver injury. Although ROS modulate the activity of many signal transduction pathways, the molecular targets of ROS during ethanol exposure are not well understood. Here, we investigated whether specific ROS-sensitive signal transduction pathways contribute to increased tumor necrosis factor α (TNF-α) production by Kupffer cells after chronic ethanol feeding to rats. Lipopolysaccharide (LPS) rapidly increased ROS production, measured by dihydrorhodamine fluorescence, in Kupffer cells from ethanol- and pair-fed rats, and ROS production was 2.5-fold greater in ethanol-fed compared with pair-fed. Pretreatment with diphenyleneiodonium (DPI), which inhibits reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, normalized ROS production in Kupffer cells from ethanol-fed rats. LPS rapidly increased Rac1-guanosinetriphosphatase (GTPase) activity and p67phox translocation to the plasma membrane in Kupffer cells from pair-fed rats. After ethanol feeding, Rac1-GTPase activity was already increased over pair-fed at baseline and remained elevated over pair-fed after LPS stimulation. Further, LPS-stimulated p67phox translocation to the plasma membrane was enhanced after chronic ethanol feeding. LPS-stimulated extracellular signal-regulated kinase (ERK)1/2 and p38 phosphorylation, two signaling pathways regulated by ROS, were increased twofold in Kupffer cells from ethanol-fed rats compared with pair-fed controls. However, only LPS-stimulated ERK1/2 phosphorylation was inhibited by DPI, which also reduced LPS-stimulated TNF-α production in Kupffer cells from pair- and ethanol-fed rats. These results demonstrate that chronic ethanol feeding increases LPS-stimulated NADPH oxidase-dependent production of ROS in Kupffer cells. Further, ERK1/2 is an important target of NADPH oxidase-derived ROS in Kupffer cells, contributing to enhanced LPS-stimulated TNF-α production by Kupffer cells after chronic ethanol feeding.
Keywords: macrophages, inflammation, signal transduction
INTRODUCTION
Production of tumor necrosis factor α (TNF-α) is one of the earliest responses of the liver to injury [1]. TNF-α acts as a principal mediator of the inflammatory response in mammals and has been implicated in the pathogenesis of a wide variety of chronic inflammatory diseases [2, 3], as well as in the progression of ethanol-induced liver injury [1, 4]. Kupffer cells, the resident macrophage in the liver, are important producers of TNF-α and other inflammatory cytokines in the liver. Lipopolysaccharide (LPS), present in the cell wall of gram-negative bacteria, is an important activator of TNF-α production by Kupffer cells [4]. Although enhanced expression of TNF-α after ethanol exposure is thought to be in part a result of an increased exposure to LPS [4], chronic ethanol also increases the susceptibility of animals to LPS-induced liver injury [5–7]. Further, after chronic ethanol feeding, LPS-stimulated TNF-α expression in isolated Kupffer cells is increased compared with expression in Kupffer cells from pair-fed control rats [8, 9].
Although the mechanisms for this increased sensitivity to LPS are not understood completely, a number of studies have identified the mitogen-activated protein kinase (MAPK) family member extracellular signal-regulated kinase (ERK)1/2 as a key target for chronic ethanol in Kupffer cells [8, 10]. ERK1/2 activity is required for increased LPS-stimulated TNF-α expression by Kupffer cells after chronic ethanol exposure [8, 10]. ERK1/2 contributes to LPS-stimulated TNF-α production, at least in part, by increasing expression of early growth response-1 protein [11, 12], a transcription factor required for increased LPS-stimulated TNF-α mRNA expression after chronic ethanol exposure in macrophages [13] and mice [14]. Taken together, these studies point to a critical role for increased ERK1/2 activity in mediating the chronic effects of ethanol in the liver. However, the mechanisms by which chronic ethanol increases LPS-stimulated ERK1/2 activity in Kupffer cells are not well understood.
Chronic ethanol exposure causes oxidative stress in the liver and enhances the formation of free radicals [15–17]. Although it is widely accepted that reactive oxygen species (ROS) play a critical role in the development of alcoholic liver injury [15–17], neither the sources nor the molecular targets of ROS produced during ethanol exposure are well understood. One important source of ROS during ethanol exposure is the production of highly reactive intermediates during ethanol oxidation by hepatocytes [16]. In addition, Kupffer cells are an important source of ROS during ethanol exposure [15] and in response to LPS [18]. Reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-dependent production of ROS is implicated in ethanol-induced liver injury, as p47phox −/− mice are resistant to chronic ethanol-induced injury [19]. Although ROS modulate the activity of a number of signal transduction pathways, including MAPKs and nuclear factor (NF)-κB [20], the molecular targets of ROS during ethanol exposure are not well understood. Many studies have focused on the role of ROS in activation of NF-κB during ethanol exposure (for example, see ref. [21, 22]. However, little information is available as to the role of ROS in mediating the chronic effects of ethanol on additional signaling pathways, such as MAPKs, known to be modulated by ROS.
Here, we have tested the hypothesis that increased ROS production by Kupffer cells via NADPH oxidase contributes to enhanced activation of ERK1/2 and TNF-α production after chronic ethanol exposure. We show that chronic ethanol exposure increased LPS-stimulated ROS production by Kupffer cells and have identified increased production of ROS via NADPH oxidase as an essential pathway in mediating increased activation of ERK1/2 and as a consequence, increased TNF-α production after chronic ethanol exposure.
MATERIALS AND METHODS
Materials
Adult male Wistar rats weighing 170–180 g were purchased from Harlan Sprague Dawley (Indianapolis, IN). Lieber-DeCarli ethanol diet was purchased from Dyets (Bethlehem, PA). Cell culture reagents were purchased from Gibco/Invitrogen (Carlsbad, CA). Antibodies were from the following sources: phospho-ERK1/2 (against Tyr-204), phospho-p38 (against Thr-180 and Tyr-182), and p38 (Santa Cruz Biotechnology, CA); ERK1/2 and Rac1 (Upstate Biotechnology, Lake Placid, NY); p67phox (BD Transduction, San Jose, CA); inhibitor of κB (IκB)-α (Cell Signaling, Beverly, MA); Na−-K−-adenosinetriphosphatase (NaK-ATPase; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City); transferrin receptor (Stressgen, Victoria, BC, Canada). Anti-rabbit and anti-mouse immunoglobulin G-peroxidase were purchased from Boehringer-Mannheim (Indianapolis, IN). Rac1 pull-down assay was from Pierce (Rockford, IL). LPS from Escherichia coli Serotype 026:B6 (cell culture-tested, purified by trichloroacetic acid extraction and gel filtration chromatography) was purchased from Sigma Chemical Co. (St. Louis, MO). Endotoxin contamination was monitored routinely in the laboratory using a kinetic chromogenic test based on the Limulus amebocyte lysate assay (Kinetic-QCL, BioWhittaker, Walkersville, MD).
Chronic ethanol feeding protocol
Rats were acclimatized for 3 days after arrival and then allowed free access to the Lieber-DeCarli liquid diet without ethanol for 2 days. Rats were then randomly assigned to pair- or ethanol-fed groups [23]. Ethanol-fed rats were allowed free access to a liquid diet containing 17% of calories as ethanol for 2 days, and then the ethanol content of the diet was increased to 35% of the calories for the duration of the 4-week feeding protocol. Controls were pair-fed with a liquid diet in which maltose dextrins were substituted isocalorically for ethanol. The Institutional Animal Care and Use Committee at Case Western Reserve University (Cleveland, OH) approved all procedures involving animals.
Isolation and culture of Kupffer cells
Kupffer cells were isolated and cultured as described previously [24] except that Connaught’s Medical Research Labs (CMRL) media were used to isolate and culture Kupffer cells. Briefly, livers were perfused with 0.05% collagenase, and the resulting suspension of liver cells was treated with 0.02% pronase for 15 min at 12°C. The resulting cell suspension from two rats per treatment group was pooled and then centrifuged three times at 50 g for 2 min, and the supernatant was collected after each centrifugation. The pooled supernatant was then purified by centrifugal elutriation. Isolated Kupffer cells were suspended in CMRL media with 10% fetal bovine serum and penicillin-streptomycin (CMRL/10% FBS) at a concentration of 2 × 106 cells/ml and plated onto 96-well [0.2 × 106/well for TNF-α enzyme-linked immunosorbent assays (ELISAs) or 0.03 × 106/well for dihydrorhodamine (DHR) fluorescence], 6-well (6 × 106/well), and 24-well (1.5 × 106/well) culture plates. After 1 h, nonadherent cells were removed by aspiration, and fresh media were added. The purity of the Kupffer cell preparations has been described previously [24] and was monitored routinely by staining with antibody against ED2 (CD163; Serotec, Raleigh, NC), a rat macrophage marker. Assays were started after 16–18 h in culture. In some experiments, inhibitors were added to the Kupffer cell culture prior to the LPS treatment. Culture media were replaced with fresh CMRL/10% FBS, with or without inhibitors 2 h prior to LPS treatment.
Measurement of ROS
Kupffer cells were cultured for 16–18 h and then pretreated with antioxidants or enzymatic inhibitors. Cells were then stimulated with 100 ng/ml LPS for 0–15 min at 37°C in a 5% CO2 atmosphere. Media were then replaced with 100 μl DHR (10 μM, Molecular Probes, Eugene OR) and diluted in CMRL/10% FBS, and cells were incubated for 15 min at 37°C in a 5% CO2 in the dark. Fluorescence was measured in a fluorescence plate reader (Perkin Elmer, Wellesley, MA) using an excitation wavelength of 505 nm and emission detection wavelength of 530 nm.
Phosphorylation/dephosphorylation of MAPKs
To measure phosphorylation of ERK1/2 and p38, cultured Kupffer cells were treated with antioxidants and enzyme inhibitors and then stimulated or not with 100 ng/ml LPS for 0–60 min. At the end of the treatment, cells were moved to ice, washed with 2 ml ice-cold phosphate-buffered saline (PBS) containing 1 mM sodium orthovanadate, and lysed in Laemmli sample buffer [30 mM Tris, pH 6.8, 5% glycerol, 1% sodium dodecyl sulfate (SDS), 2.5% β-mercaptoethanol, and 0.125 mg/ml bromophenol blue] containing 1 mM sodium orthovanadate.
The rate of ERK1/2 dephosphorylation after LPS stimulation was assessed using a technique adapted from Yaglom et al. [25]. After 16–18 h culture, Kupffer cells were stimulated with 100 ng/ml LPS for 30 min and then treated with 10 mM 2-deoxyglucose and 5 μM rotenone for 0–10 min to inhibit further phosphorylation events. Cells were then moved to ice and washed with 2 ml ice-cold PBS buffer with 1 mM sodium orthovanadate and lysed in 1% Triton-lysis buffer (50 mM Tris, 6.4 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM sodium pyrophosphate, 1 mM activated sodium vanadate, 10 mM NaF, and protease inhibitor cocktail, Complete-EDTA free™, Roche Molecular Biochemicals, Indianapolis, IN). Protein was normalized, and samples were prepared by boiling in Laemmli sample buffer. Proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE), probed by Western blot with monoclonal antibody (mAb) against phospho-ERK1/2, then probed with polyclonal antibody to total ERK1/2 or with antibody to phospho-p38, and then stripped and probed again with antibody against total p38 protein.
IκB-α degradation
Kupffer cells were cultured for 16–18 h and then pretreated with or without diphenyleneiodonium chloride (DPI). Cells were then stimulated with or without 100 ng/ml LPS for 30 min. Cells were then moved to ice and washed with 2 ml ice-cold PBS buffer and lysed in 1% Triton-lysis buffer. Protein was normalized, and samples were prepared by boiling in Laemmli sample buffer. Proteins were separated by SDS-PAGE and probed by Western blot with antibody against IκB-α. Equal loading was assessed by probing with antibody to total ERK1/2, which is not affected by chronic ethanol feeding or LPS exposure [10].
Translocation of p67phox to the membrane
After 16–18 h in culture, Kupffer cells were stimulated or not with 100 ng/ml LPS for 0–10 min. Cells were then washed with cold PBS with 1 mM sodium orthovanadate and homogenized in 20 mM Tris-HCl (pH 7.4), 1 mM EDTA, and 250 mM sucrose with protease inhibitor cocktail, 1 mg/ml bacitracin, and 1 mg/ml benzamidine in a glass-on-glass dounce homogenizer with a loose-fitting pestle and centrifuged at 200 g for 15 min to remove nuclei and unbroken cells. The resulting supernatant was then centrifuged at 15,000 g for 15 min at 4°C. Pellets were washed with 1 mL homogenization buffer, and the resulting plasma membrane-enriched fraction was resuspended in radioimmunoprecipitation buffer [50 mM Tris, pH 7.4, 1% Nonidet P-40 (NP-40), 150 mM NaCl, 1 mM EDTA, with protease inhibitor cocktail]. Protein concentration was measured and normalized in Laemmli sample buffer. Samples were separated by SDS-PAGE and probed by Western blotting with antibody specific for p67phox. Western blots were probed with antibody to NaK-ATPase to ensure equal loading of plasma membrane proteins between samples.
Rac1 activation assay
Activation of Rac1 in response to LPS was measured using a Rac1 pull-down assay (Pierce). After culture for 16–18 h, Kupffer cells were stimulated or not with 100 ng/ml LPS for 0–2.5 min at 37°C. Cells were then washed with cold PBS and lysed in lysis/binding buffer (25 mM Tris HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1% NP-40, 1 mM dithiothreitol, 5% glycerol, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mM phenylmethylsulfonyl fluoride). Lysates were centrifuged for 15 min at 16,000 g at 4°C, and supernatants were incubated with a glutathione S-transferase-p21-activated protein kinase 1-p21 binding domain fusion protein for 60 min at 4°C. Complexes were collected on SwellGel immobilized protein discs (Pierce), washed three times with lysis/binding buffer, resuspended in 40 μl 2× Laemmli sample buffer, and boiled for 5 min. Proteins were resolved by 12% SDS-PAGE, and the quantity of Rac1 pulled down was assessed by Western blot.
Western blotting
Bound antibodies were detected using an enhanced chemiluminescence reagent. Immunoreactive protein quantity analysis was performed on a Macintosh computer using the public domain NIH Image program [developed at the National Institutes of Health (NIH), Bethesda, MD]. Film exposure times were in the linear range of detection.
Real-time PCR
After 16–18 h in culture, Kupffer cells were pretreated for 2 h with or without 10 μM DPI and then stimulated or not with 100 ng/ml LPS for 60 min. Total RNA was isolated from Kupffer cells using the RNeasy Micro kit (Qiagen, Valencia, CA) with on-column DNA digestion using the RNase-free DNase set (Qiagen) according to the manufacturer’s instructions. Total RNA (200–300 ng) was reverse-transcribed using the RETROscript kit (Ambion, Austin, TX) with random decamers as primers. Real-time PCR amplification was performed in an iCycler (Bio-Rad, Hercules, CA) using SYBR Green PCR core reagents (Applied Biosystems, Warrington, UK). The relative amount of target mRNA was determined using the comparative threshold (Ct) method by normalizing target mRNA Ct values to those of β-actin (ΔCt). The primer sequences are as follows: TNF-α forward, 5′-GAA CAA CCC TAC GAG CAC CT-3′; TNF-α reverse, 5′-GGG TAG TTT GGC TGG GAT AA-3′; β-actin forward, 5′-CGG TCA GGT CAT CAC TAT CG-3′, β-actin reverse, 5′-TTC CAT ACC CAG GAA GGA AG-3′. All primers used for real-time PCR analysis were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA). Statistical analysis of real-time PCR results was performed on ΔCt values.
ELISA for TNF-α
After treatment with DPI, Kupffer cells were stimulated with or without 100 ng/ml LPS for 4 h at 37°C in a 5% CO2. Cell culture media were removed and stored at −20°C for TNF-α assay using ELISA (R&D Systems, Minneapolis, MN). TNF-α concentration was measured in the cell culture media. Total TNF-α per well was calculated and then normalized to the number of Kupffer cells per well.
Statistical analysis
Because of the limited number of Kupffer cells available from each animal, data from several feeding trials are presented. Values reported are mean ± sem. Data were analyzed by Student’s t-test or general linear models procedure followed by least square means analysis of differences between groups (SAS, Carey, IN), blocking for trial effects if data from more than one trial were used. Data were log-transformed if needed to obtain a normal distribution.
RESULTS
Chronic ethanol feeding increases the sensitivity of isolated Kupffer cells to LPS stimulation, leading to increased production of TNF-α [10, 24]. This increased TNF-α production is mediated, at least in part, via ERK1/2 activation [8, 10]. Stimulation of Kupffer cells with LPS increased phosphorylation of ERK1/2 in Kupffer cells from pair-fed and ethanol-fed rats (Fig. 1A). However, LPS stimulation of ERK1/2 phosphorylation was higher in Kupffer cells from ethanol-fed rats compared with pair-fed rats (Fig. 1A) [10]. Increased phosphorylation of ERK1/2 after ethanol feeding was observed as early as 10 min after treatment with LPS and was maintained over 60 min (Fig. 1A). Maximum phosphorylation of ERK1/2, observed between 15 and 30 min, was twofold higher in Kupffer cells from ethanol-fed rats compared with pair-fed rats (Fig. 1A).
Fig. 1.
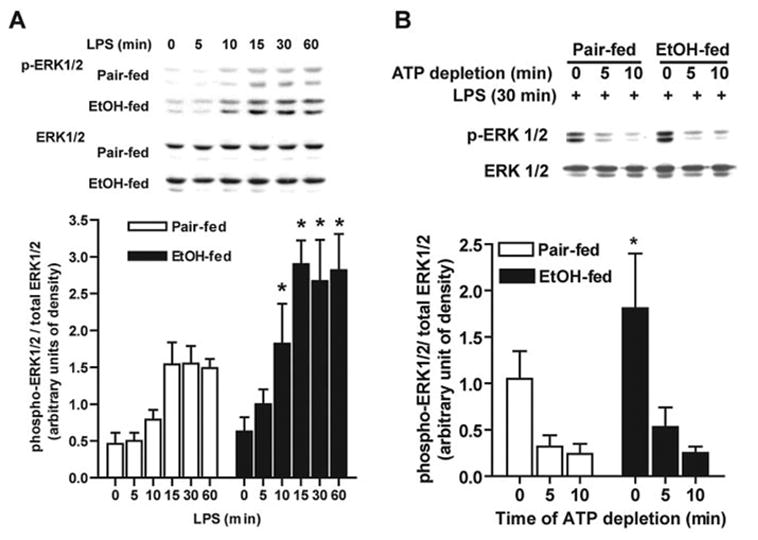
Chronic ethanol feeding increases LPS-stimulated ERK1/2 phosphorylation in Kupffer cells. (A) LPS-stimulated ERK1/2 phosphorylation (p-ERK1/2) was measured in Kupffer cells from pair- and ethanol (EtOH)-fed rats after culture for 16–18 h. Cells were then treated with or without 100 ng/ml LPS for 0–60 min, and ERK1/2 phosphorylation was assessed by Western blot. (B) Dephosphorylation of ERK1/2 after LPS treatment was measured in Kupffer cells from pair- and ethanol-fed rats. After culture for 16–18 h, cells were stimulated with 100 ng/ml LPS for 30 min and then treated or not with 10 mM 2-deoxy glucose and 5 μM rotenone to deplete adenosine 5′-triphosphate (ATP) and inhibit further phosphorylation. Phospho-ERK1/2 relative to total ERK1/2 was measured by Western blotting. Values represent mean ± sem, n = 4 (A) and n = 7 (B); *, P < 0.05, compared with pair-fed at the same time-point.
To investigate the mechanism of increased LPS-stimulated phosphorylation of ERK1/2 after chronic ethanol feeding, we hypothesized that chronic ethanol decreased ERK1/2 dephosphorylation and/or increased ERK1/2 phosphorylation. To investigate whether chronic ethanol feeding changed the rate of ERK1/2 dephosphorylation, Kupffer cells from pair- and ethanol-fed rats were stimulated with or without 100 ng/ml LPS for 30 min. Cells were then treated with 2-deoxyglucose and rotenone to rapidly deplete ATP and prevent further phosphorylation events [25]. The rate of dephosphorylation of ERK1/2 was followed over 10 min (Fig. 1B). Five minutes after ATP depletion, ERK1/2 phosphorylation was reduced to 30% in pair-fed and ethanol-fed rats and further reduced to 22% and 14% after 10 min in Kupffer cells from pair-fed and ethanol-fed rats, respectively (Fig. 1B). There was no difference in the rate of ERK1/2 dephosphorylation between Kupffer cells from pair- and ethanol-fed rats, demonstrating that inhibition of dephosphorylation of phospho-ERK1/2 did not contribute to increased LPS-stimulated ERK1/2 after chronic ethanol feeding.
Kupffer cells isolated from rats treated with LPS exhibit an increased rate of ROS production [18]. As ROS can enhance ERK1/2 phosphorylation in other cell types [26], we hypothesized that increased ROS production may contribute to increased LPS-stimulated ERK1/2 phosphorylation after chronic ethanol feeding. To test this hypothesis, we assessed the effect of chronic ethanol on production of ROS measured by DHR 123 fluorescence [27]. No differences in DHR fluorescence were observed between Kupffer cells from ethanol- and pair-fed rats at baseline (2185±307 arbitrary units of fluorescence in pair-fed and 2135±290 in ethanol-fed; n=5). LPS treatment rapidly increased DHR fluorescence in Kupffer cells from ethanol- and pair-fed rats (Fig. 2A). DHR fluorescence was 2.5-fold higher after chronic ethanol feeding after 5–15 min of stimulation with LPS (Fig. 2A).
Fig. 2.
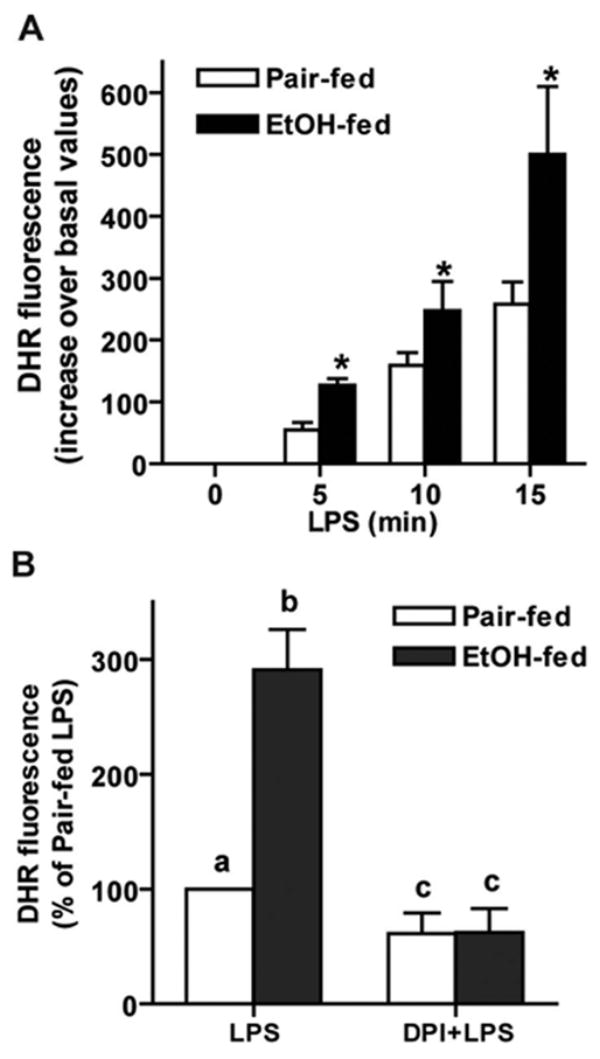
Chronic ethanol feeding increases LPS-stimulated DHR fluorescence in isolated Kupffer cells, which, isolated from pair- and ethanol-fed rats, were cultured 16–18 h and then stimulated with or without 100 ng/ml LPS for 0–15 min (A). Media were then replaced with 10 μM DHR, and cells were incubated for 15 min. Fluorescence was measured at an excitation wavelength of 505 nm and emission detection wavelength of 530 nm. Values represent mean ± sem, corrected for DHR fluorescence at zero time; n = 5; *, P < 0.05, compared with pair-fed. DHR fluorescence at zero time did not differ between pair-fed (2185± 307 arbitrary units of fluorescence) and ethanol-fed (2135± 290). (B) Pretreatment with DPI chloride decreased LPS-stimulated ROS production. Kupffer cells were treated with or without 10 μM DPI for 2 h and then stimulated with LPS for 5 min, followed by incubation with DHR as described above; n = 13, values with different superscripts; a, P < 0.05 compared with pair-fed cells not treated with LPS; b, P < 0.05, compared with pair-fed cells treated with LPS; c, P < 0.05 compared with cells treated with LPS but not treated with DPI. DHR fluorescence is expressed as percent of pair-fed LPS treated (177±87), as several different lots of DHR were used in these experiments, and each lot had a different relative fluorescence.
LPS is a potent activator of NADPH oxidase activity in macrophages [28–30]. To test whether LPS-stimulated NADPH oxidase activity contributed to increased ROS accumulation, Kupffer cells were pretreated with DPI, an inhibitor of flavoenzymes, which blocks NADPH oxidase activity. DPI reduced LPS-stimulated DHR fluorescence by 40% in Kupffer cells from pair-fed and 80% in ethanol-fed (Fig. 2B). After DPI treatment, there was no longer any difference in LPS-stimulated ROS production by Kupffer cells between pair- and ethanol-fed rats (Fig. 2B).
NADPH oxidase is a multisubunit enzyme. Although two subunits, p22phox and gp91phox, are localized to the membrane of macrophages and neutrophils, translocation of p40phox, p47phox, and p67phox from the cytosol to the membrane is required for activation of NADPH oxidase activity [31]. Here, we monitored p67phox as a representative marker for translocation of NADPH oxidase subunits. Chronic ethanol feeding had no effect on quantity of total p67phox protein in homogenates of Kupffer cells (Fig. 3A). In response to LPS, p67phox rapidly translocated to a plasma membrane-enriched fraction of Kupffer cells from pair-fed rats (Fig. 3A). After chronic ethanol feeding, p67phox translocation to the plasma membrane in response to LPS was increased (Fig. 3A). In addition to the translocation of cytosolic components, the small GTP-binding protein Rac1 is an essential activator of NADPH oxidase catalytic activity [31]. In Kupffer cells from pair-fed rats, the quantity of GTP-bound Rac1 was low at baseline and increased rapidly in response to LPS treatment (Fig. 3B). After chronic ethanol feeding, the quantity of GTP-bound Rac1 was already elevated at baseline and did not increase further in response to LPS treatment (Fig. 3B). Taken together, these data indicate that chronic ethanol feeding resulted in a dysregulation of NADPH oxidase activation. A constitutive activation of Rac1 as well as an enhanced translocation of the NAPDH oxidase subunit p67phox to the plasma membrane were associated with increased LPS-stimulated ROS production.
Fig. 3.
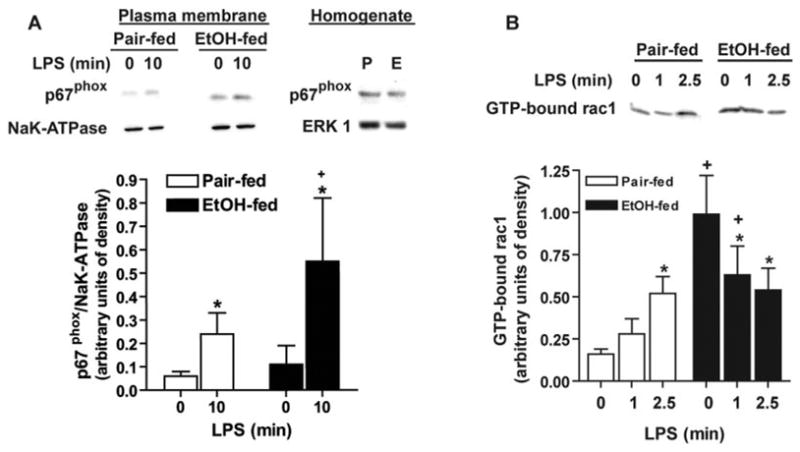
(A) Chronic ethanol feeding increases LPS-stimulated p67phox translocation. Kupffer cells isolated from pair (P)- and ethanol (E)-fed rats were cultured 16–18 h and then stimulated with or without 100 ng/ml LPS for 10 min. Kupffer cells were lysed, and plasma membrane-enriched fractions and homogenates were probed for p67phox by Western blot. NaK-ATPase was used as a control for equal loading of plasma membrane fractions. Values represent means ± sem; n = 5; +, P < 0.05, compared with pair-fed at each time-point; *, P < 0.05, compared with cells not treated with LPS. (B) Chronic ethanol feeding increases guanosine 5′-triphosphate (GTP)-bound Rac1. Kupffer cells isolated from pair- and ethanol-fed rats were cultured 16–18 h and then stimulated with or without 100 ng/ml LPS for up to 2.5 min. Kupffer cells were lysed, and GTP-bound Rac1 was measured using a Rac1 pull-down assay. Values represent means ± sem; n = 6; +, P < 0.05, compared with pair-fed at each time-point; *, P < 0.05, compared with cells not treated with LPS.
To determine if NADPH oxidase-dependent production of ROS contributed to enhanced LPS-stimulated ERK1/2 phosphorylation after chronic ethanol feeding, Kupffer cells were pretreated with DPI, and then LPS-stimulated ERK1/2 phosphorylation was assessed by Western blot. DPI reduced LPS-stimulated phosphorylation of ERK1/2 in Kupffer cells from pair- and ethanol-fed rats (Fig. 4). In DPI-treated Kupffer cells, there was no longer a difference in LPS-stimulated ERK1/2 phosphorylation between ethanol- and pair-fed rats (Fig. 4).
Fig. 4.
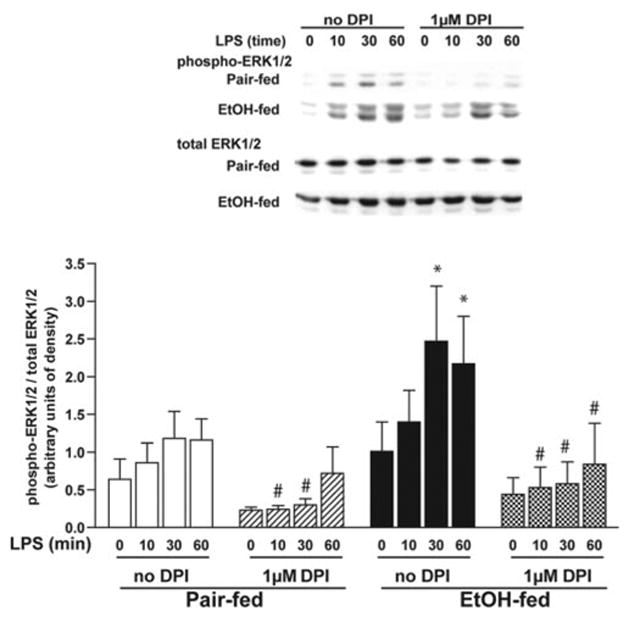
DPI chloride suppresses LPS-stimulated ERK1/2 phosphorylation. Kupffer cells from pair- and ethanol-fed rats were cultured 16–18 h. Cells were then preincubated with or without 1 μM DPI for 2 h and then treated with 100 ng/ml LPS for 0–60 min. Phosphorylation of ERK1/2 was assessed by Western blot. Values represent means ± sem; n = 8; *, P < 0.05, compared with pair-fed; #, P < 0.05, compared with cells not treated with DPI (within a diet group).
Although these data suggest that NADPH oxidase is an important contributor to increased ROS production by Kupffer cells in response to LPS, additional sources of ROS in Kupffer cells include production of ROS via cytochrome P450 (CYP2E1) or xanthine oxidase. Chronic ethanol exposure increases CYP2E1 expression in isolated Kupffer cells [8] (Fig. 5), and CYP2E1 overexpression in RAW 264.7 macrophages results in increased TNF-α secretion [32]. However, preincubation of Kupffer cells with 5 mM 4-methylpyrazole or 5 μM chlormethiazole, inhibitors of CYP2E1, or 300 μM allopurinol to inhibit xanthine oxidase had no effect on LPS-stimulated ERK1/2 phosphorylation in Kupffer cells from pair- or ethanol-fed rats (Fig. 5). Collectively, these data suggest that NADPH oxidase activity was a major determinant of increased LPS-stimulated ERK1/2 phosphorylation after chronic ethanol feeding.
Fig. 5.
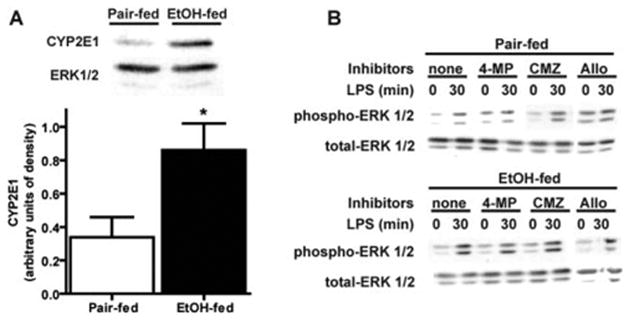
(A) Chronic ethanol feeding increases CYP2E1 protein expression in Kupffer cells, which were isolated from pair- and ethanol-fed rats and cultured for 16–18 h. Homogenates were prepared, and CYP2E1 protein was measured by Western blot. Values represent means ± sem; n = 9; *, P < 0.05, compared with pair-fed. (B) LPS-stimulated ERK1/2 phosphorylation in Kupffer cells treated with inhibitors of CYP2E1 or xanthine oxidase. Kupffer cells from pair- and ethanol-fed rats were cultured 16–18 h. Cells were then preincubated with or without 5 mM 4-methylpyrazole (4-MP), 5 μM chlor-methiazole (CMZ), or allopurinol (Allo) for 2 h and then treated with 100 ng/ml LPS for 0–60 min. Phosphorylation of ERK1/2 was assessed by Western blot. Blots are representative of three (for chlormethiazole and allopurinol) or seven (for 4-methylpyrazole) independent experiments.
After chronic ethanol feeding, LPS-stimulated p38 phosphorylation is also increased in Kupffer cells [8, 9] (Fig. 6A). As p38 activation is also subject to regulation in response to oxidants, we asked whether NADPH oxidase activity also contributed to the increase in LPS-stimulated p38 phosphorylation observed in Kupffer cells after chronic ethanol feeding. Preincubation with DPI decreased LPS-stimulated p38 phosphorylation in Kupffer cells from pair-fed rats (Fig. 6A). However, DPI was not able to suppress LPS-stimulated p38 phosphorylation in Kupffer cells after ethanol feeding (Fig. 6A). LPS also induces NF-κB dissociation from IκB-α, translocation of p65 to the nucleus, and stimulation of its DNA-binding activity in Kupffer cells. Although chronic ethanol feeding does not increase LPS-stimulated NF-κB DNA-binding activity in the model of cultured Kupffer cells studied here [10], we investigated the impact of DPI on LPS-stimulated IκB-α degradation as an indirect marker of NF-κB activation. Pretreatment with DPI prevented LPS-stimulated degradation of IκB-α in Kupffer cells from pair-fed rats but did not affect the decay of IκB-α in cells from ethanol-fed rats (Fig. 6B). These data suggest that the increase in NADPH oxidase-derived ROS generated after chronic ethanol feeding may specifically regulate the activation of ERK1/2, rather than multiple LPS-stimulated signaling cascades.
Fig. 6.
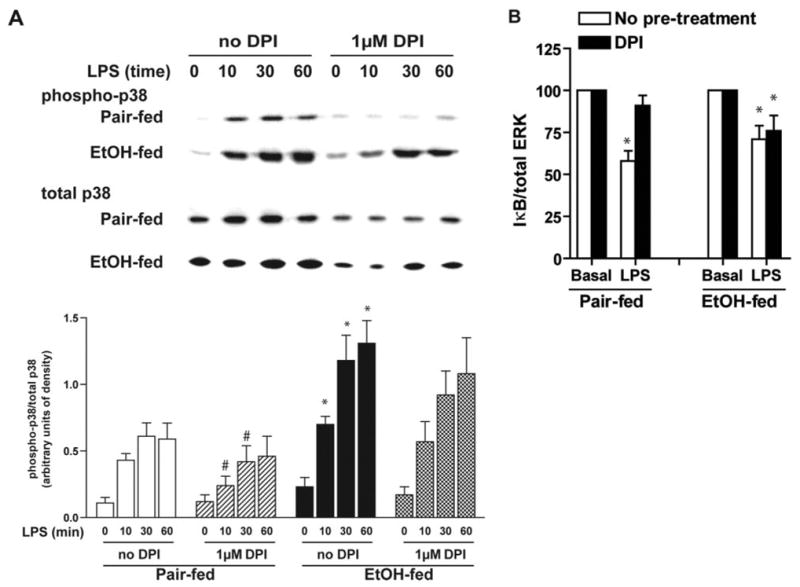
DPI chloride does not suppress LPS-stimulated p38 phosphorylation (A) or IκBα degradation (B) in Kupffer cells from ethanol-fed rats. Kupffer cells from pair- and ethanol-fed rats were cultured 16–18 h. Cells were then preincubated with or without 1 μM DPI for 2 h. (A) Phosphorylation of p38 was measured in response to treatment with 100 ng/ml LPS for 0–60 min. (B) Quantity of IκB-α was assessed by Western blot after 30 min stimulation with or without 100 ng/ml LPS. Values represent means ± sem; n = 7 for p38, and n = 6 for IκB-α. (A) *, P < 0.05, compared with pair-fed; #, P < 0.05, compared with cells not treated with DPI (within a diet group). (B) *, P < 0.05, compared with basal values within each diet group.
Increased activation of ERK1/2 by LPS in Kupffer cells after chronic ethanol feeding contributes to increased LPS-stimulated TNF-α secretion [10]. If ROS generated by NADPH oxidase contribute to increased ERK1/2 activation after chronic ethanol, then inhibition of NADPH oxidase with DPI should reduce LPS-stimulated TNF-α production by Kupffer cells after chronic ethanol feeding. Consistent with this hypothesis, pretreatment of Kupffer cells with DPI normalized LPS-stimulated TNF-α mRNA accumulation (Fig. 7A) and decreased the concentration of TNF-α in the media (Fig. 7B) to control levels.
Fig. 7.
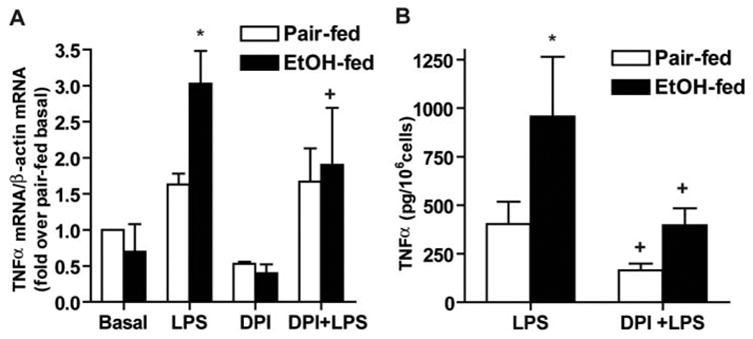
DPI chloride inhibits LPS-stimulated TNF-α production after chronic ethanol feeding. Kupffer cells from pair- and ethanol-fed rats were cultured 16–18 h. Cells were then preincubated with or without 1 μM DPI for 2 h and then treated with 100 ng/ml LPS. (A) After 60 min LPS treatment, total RNA was isolated, and TNF-α mRNA accumulation was measured by real-time PCR and normalized to β-actin mRNA. Values represent means ± sem; n = 3; *, P < 0.05, compared with pair-fed; +, P < 0.05, compared with LPS-treated cells not treated with DPI. (B) After 4 h, cell culture media were removed, and TNF-α concentrations were measured by ELISA. Values represent means ± sem; n = 7–8; *, P < 0.05 compared with pair-fed; +, P < 0.05 compared with LPS-treated cells not treated with DPI.
DISCUSSION
Increased production of ROS has been implicated in the development of alcoholic liver injury [15–17]. However, neither the source nor targets of the ROS produced during ethanol consumption are well defined. NADPH oxidase activity has been implicated as an important source of ROS during chronic ethanol exposure in rodent models of chronic ethanol exposure [15]. Proof of principle studies using p47phox−/− mice as well treatment of rats with DPI during chronic ethanol exposure indicate that NADPH oxidase activity is required for the development of chronic ethanol-induced liver injury [19, 33]. However, the mechanisms by which NADPH oxidase activity contributes to liver injury are not well understood. Here, we have demonstrated that chronic ethanol feeding enhances the ability of LPS to increase ROS production in Kupffer cells. Although baseline rates of ROS production were not affected by chronic ethanol feeding, LPS-stimulated ROS production was increased by 2.5-fold in Kupffer cells from ethanol-fed rats compared with cells from pair-fed rats. This increased ROS production was suppressed effectively by treatment with DPI, an inhibitor of NADPH oxidase. As a consequence of increased NADPH oxidase-derived ROS production, LPS-stimulated ERK1/2 phosphorylation increased in Kupffer cells after chronic ethanol feeding. Increased activation of this key MAPK signaling pathway after chronic ethanol feeding contributes to increased TNF-α production. These studies thus identify increased NADPH oxidase as an important site for ROS production and ERK1/2 signaling as an important target for ROS during LPS-mediated signal transduction by Kupffer cells after chronic ethanol feeding.
There is a growing appreciation of the specific role of ROS in the modulation/regulation of a number of signal transduction cascades [20, 34]. ROS contribute to cellular responses to a variety of hormones, neurotransmitters, and cytokines [20, 34]. Evidence also indicates that ROS contribute to LPS-stimulated signaling pathways in cells of the innate immune system (i.e., monocytes/macrophages, neutrophils) as well as nonimmune cells [35, 36]. In macrophages, several specific signaling pathways regulated by ROS have been identified. For example, Hsu and Wen [28] demonstrated that LPS-stimulated ROS production acts via p38 and ERK1/2 MAPKs to enhance interleukin-1 gene expression. Here, we find that NADPH oxidase-derived ROS contribute to LPS-mediated activation of the ERK1/2, p38, and NF-κB pathways in hepatic macrophages from pair-fed control rats (Figs. 4 and 6).
After chronic ethanol exposure, the few studies published have suggested a general role for increased ROS production in mediating the chronic effect of ethanol on LPS-mediated signaling in macrophages. For example, treatment of MonoMac6 macrophages or Kupffer cells with general antioxidant compounds, such as N-acetyl cysteine or dilinoleoylphosphatidylcholine (DLPC), abrogates the increase in LPS-stimulated TNF-α expression observed in these cells after chronic ethanol exposure during culture or in response to ethanol feeding [8, 22, 37]. In particular, Cao et al. [8] identified LPS-stimulated NF-κB, as well as ERK1/2 and p38 MAPKs, as ROS-sensitive targets for the inhibitory effects of DLPC in Kupffer cells isolated from rats fed chronic ethanol. Here, we have specifically identified NADPH oxidase-derived ROS as an important contributor to LPS-stimulated ERK1/2 phoshorylation in rat Kupffer cells. These results from Kupffer cells implicate a critical role of ROS in activation of the ERK1/2 pathway and are consistent with previous work in other cell types, demonstrating that ERK1/2 can be activated by ROS [20]. It is interesting that chronic ethanol feeding abrogated the ability of DPI to inhibit LPS-stimulated p38 phosphorylation and IκB-α degradation in Kupffer cells (Fig. 6). These data suggest that additional mechanisms for p38 and NF-κB activation must come into play after chronic ethanol exposure. These additional mechanisms could include contributions from CYP2E1-derived ROS and/or additional changes in LPS-mediated signaling in response to chronic ethanol feeding.
Although ROS production by Kupffer cells after chronic ethanol feeding may be mediated by a number of pathways, such as CYP2E1, xanthine oxidase, or NADPH oxidase, only inhibitors of NADPH oxidase were able to prevent chronic, ethanol-induced increases in LPS-stimulated ERK1/2 phosphorylation in Kupffer cells (Fig. 5). Although NADPH oxidase-derived ROS contributed to increased LPS-stimulated ERK1/2 phosphorylation after chronic ethanol, they did not contribute to enhanced LPS-stimulated p38 MAPK phosphorylation or degradation of IκB-α, an indirect indicator of NF-κB activation, after chronic ethanol feeding (Fig. 6). These data suggest a compartmentalization of ROS production within hepatic macrophages, and the LPS-stimulated ERK1/2 signaling pathway was more closely linked with NADPH oxidase-derived ROS compared with other LPS-activated signaling cascades. Studies using other model systems suggest that specific signaling cascades, including ERK1/2, may be linked with NADPH oxidase-dependent ROS production. However, such specificity in ROS-mediated signaling varies between different cell types and/or activation signals (for example, see refs. [38–40]), and further studies are required to define such an association in Kupffer cells after ethanol exposure.
LPS-mediated activation of NADPH oxidase involves the activation and recruitment of several cytosolic subunits to the plasma membrane [20, 35]. The mechanisms by which chronic ethanol feeding increased NADPH oxidase-dependent ROS production in response to LPS are not known. Although chronic ethanol feeding did not increase the total quantity of p67phox protein in Kupffer cells, LPS-stimulated translocation of p67phox to the plasma membrane was increased compared with pair-fed rats. Translocation of p47phox and p67phox is dependent on protein kinase C (PKC)δ-mediated phosphorylation in monocytes [41, 42]. As PKCδ is a target of ethanol action in a number of cell types [43], changes in PKCδ-mediated regulation of NADPH oxidase after chronic ethanol are currently under investigation. Further, chronic ethanol exposure resulted in a constitutive activation of Rac1, the small GTP-binding protein required for activation of NADPH oxidase. Disruption of the normal GTP/guanosine 5′-diphosphate cycling of Rac1 may contribute to enhanced LPS-stimulated NADPH oxidase activity after chronic ethanol feeding. A number of studies using other cells/tissues have identified the activation of small GTP-binding proteins as a target of ethanol action, consistent with the increase in the active form of Rac1 after chronic ethanol feeding in the current study. For example, ethanol exposure increases the relative proportion of the GTP-bound forms of several guanosinetriphosphatases (GTPases) including ras in mouse liver homogenates [44], RhoA in fetal rat astrocytes [45], cdc42 in SVEC4-10 cells [46], TC10 in rat adipocytes [47], as well as RhoA and cdc42 in vitro [48]. Potential mechanisms by which chronic ethanol targets small GTP-binding proteins include changes in the post-translational processing, such as impaired lipidation reactions [49], impaired regulation of GTPase cycling activity [50], and/or redox-dependent activation analagous to that observed in other ras and Rho family GTPases [51, 52].
Circulating TNF-α is increased in the blood of alcoholics and in animals chronically exposed to ethanol [4, 53]. Studies in transgenic mice lacking the TNF receptor I as well as treatment of mice and rats with antibodies to TNF-α during chronic ethanol exposure have demonstrated an essential role for TNF-α in the progression of chronic, ethanol-induced liver injury [1]. We have previously demonstrated the critical role of ERK1/2 in mediating increased LPS-stimulated TNF-α production by Kupffer cells after chronic ethanol feeding [10, 13]. Together with our previous results, the current data suggest that chronic, ethanol-induced increases in the ERK1/2 pathway are mediated, at least in part, via increased LPS-stimulated, NADPH oxidase-derived ROS production. These data thus identify ERK1/2 signaling as a molecular target of increased ROS production in Kupffer cells, which likely contributes to increased TNF-α production and therefore liver injury during chronic ethanol feeding.
Acknowledgments
This work was supported by NIH Grants AA013868 and AA11975. The mAb against α1 subunit of NaK-ATPase (α6F), developed by D. M. Fambrough, was obtained from the Developmental Studies Hybridoma Bank, maintained by the University of Iowa, Department of Biological Sciences, under Contract NO1-HD-7-3263.
References
- 1.Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343:1467–1476. doi: 10.1056/NEJM200011163432007. [DOI] [PubMed] [Google Scholar]
- 2.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumor necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reimund JM, Wittersheim C, Dumont S, Muller CD, Baumann R, Poindron P, Duclos B. Mucosal inflammatory cytokine production by intestinal biopsies in patients with ulcerative colitis and Crohn’s disease. J Clin Immunol. 1996;16:144–150. doi: 10.1007/BF01540912. [DOI] [PubMed] [Google Scholar]
- 4.Thurman RG. Mechanisms of hepatic toxicity II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol. 1998;275:G605–G611. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 5.Honchel R, Ray M, Marsano L, Cohen D, Lee E, Shedlofsky S, McClain CJ. Tumor necrosis factor in alcohol enhanced endotoxin liver injury. Alcohol Clin Exp Res. 1992;16:665–669. doi: 10.1111/j.1530-0277.1992.tb00656.x. [DOI] [PubMed] [Google Scholar]
- 6.Koteish A, Yang S, Lin H, Huang X, Diehl AM. Chronic ethanol exposure potentiates lipopolysaccharide liver injury despite inhibiting Jun N-terminal kinase and caspase 3 activation. J Biol Chem. 2002;277:13037–13044. doi: 10.1074/jbc.M101632200. [DOI] [PubMed] [Google Scholar]
- 7.Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology. 2000;32:1008–1017. doi: 10.1053/jhep.2000.19621. [DOI] [PubMed] [Google Scholar]
- 8.Cao Q, Mak KM, Lieber CS. Dilinoleoylphosphatidylcholine decreases LPS-induced TNF-α generation in Kupffer cells of ethanol-fed rats: respective roles of MAPKs and NF-κB. Biochem Biophys Res Commun. 2002;294:849–853. doi: 10.1016/S0006-291X(02)00586-7. [DOI] [PubMed] [Google Scholar]
- 9.Kishore R, McMullen MR, Nagy LE. Stabilization of TNF-α mRNA by chronic ethanol: role of A+U rich elements and p38 mitogen-activated protein kinase signaling pathway. J Biol Chem. 2001;276:41930–41937. doi: 10.1074/jbc.M107181200. [DOI] [PubMed] [Google Scholar]
- 10.Kishore R, Hill JR, McMullen MR, Frenkel J, Nagy LE. ERK1/2 and Egr-1 contribute to increased TNF-α production in rat Kupffer cells after chronic ethanol feeding. Am J Physiol. 2002;282:G6–G15. doi: 10.1152/ajpgi.00328.2001. [DOI] [PubMed] [Google Scholar]
- 11.Guha M, O’Connell MA, Pawlinski R, Hollis A, McGovern P, Yan SF, Stern D, Mackman N. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor α expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood. 2001;98:1429–1439. doi: 10.1182/blood.v98.5.1429. [DOI] [PubMed] [Google Scholar]
- 12.Shi L, Kishore R, McMullen M, Nagy LE. Lipopolysaccharide stimulation of ERK1/2 increases TNF-α production via Egr-1. Am J Physiol Cell Physiol. 2002;282:C1205–C1211. doi: 10.1152/ajpcell.00511.2001. [DOI] [PubMed] [Google Scholar]
- 13.Shi L, Kishore R, McMullen M, Nagy LE. Chronic ethanol increases LPS-stimulated Egr-1 expression in RAW 264.7 macrophages: contribution to enhanced TNF-α production. J Biol Chem. 2002;277:14777–14785. doi: 10.1074/jbc.M108967200. [DOI] [PubMed] [Google Scholar]
- 14.McMullen MR, Pritchard MT, Wang Q, Millward CA, Croniger CM, Nagy LE. Early growth response-1 transcription factor is essential for ethanol-induced fatty liver injury in mice. Gastroenterology. 2005;128:2066–2076. doi: 10.1053/j.gastro.2005.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, Gabele E, Rusyn I, Yamashina S, Froh M, Adachi Y, Iimuro Y, Bradford BU, Smutney OM, Connor HD, Mason RP, Goyert SM, Peters JM, Gonzalez FJ, Samulski RJ, Thurman RG. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med. 2001;31:1544–1549. doi: 10.1016/s0891-5849(01)00748-1. [DOI] [PubMed] [Google Scholar]
- 16.Arteel GE. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology. 2003;124:778–790. doi: 10.1053/gast.2003.50087. [DOI] [PubMed] [Google Scholar]
- 17.Hoek JB, Pastorino JG. Ethanol, oxidative stress, and cytokine-induced liver cell injury. Alcohol. 2002;27:63–68. doi: 10.1016/s0741-8329(02)00215-x. [DOI] [PubMed] [Google Scholar]
- 18.Spolarics Z. Endotoxemia, pentose cycle, and the oxidant/antioxidant balance in the hepatic sinusoid. J Leukoc Biol. 1998;63:534–541. doi: 10.1002/jlb.63.5.534. [DOI] [PubMed] [Google Scholar]
- 19.Kono H, Rusyn I, Yin M, Gäbele E, Yamashina S, Dikalova A, Kadiiska MB, Connor HD, Mason RP, Segal BH, Bradford BU, Holland SM, Thurman RG. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106:867–872. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 21.Nanji AA, Jokelainen K, Rahemtulla A, Miao L, Fogt F, Matsumoto H, Tahan SR, Su GL. Activation of nuclear factor κ B and cytokine imbalance in experimental alcoholic liver disease in the rat. Hepatology. 1999;30:934–943. doi: 10.1002/hep.510300402. [DOI] [PubMed] [Google Scholar]
- 22.Hill DB, Devalaraja R, Joshi-Barve S, Barve S, McClain CJ. Antioxidants attenuate nuclear factor-κ B activation and tumor necrosis factor-α production in alcoholic hepatitis patient monocytes and rat Kupffer cells, in vitro. Clin Biochem. 1999;32:563–570. doi: 10.1016/s0009-9120(99)00056-9. [DOI] [PubMed] [Google Scholar]
- 23.Lieber CS, DeCarli LM. The feeding of alcohol in liquid diets: two decades of application and 1982 update. Alcohol Clin Exp Res. 1982;6:523–531. doi: 10.1111/j.1530-0277.1982.tb05017.x. [DOI] [PubMed] [Google Scholar]
- 24.Aldred A, Nagy LE. Ethanol dissociates hormone-stimulated cAMP production from inhibition of TNF-α production in rat Kupffer cells. Am J Physiol. 1999;276:G98–G106. doi: 10.1152/ajpgi.1999.276.1.G98. [DOI] [PubMed] [Google Scholar]
- 25.Yaglom J, O’Callaghan-Sunol C, Gabai V, Sherman MY. Inactivation of dual-specificity phosphatases is involved in the regulation of extracellular signal-regulated kinases by heat shock and hsp72. Mol Cell Biol. 2003;23:3813–3824. doi: 10.1128/MCB.23.11.3813-3824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torres M. Mitogen-activated protein kinase pathways in redox signaling. Front Biosci. 2003;8:d369–d391. doi: 10.2741/999. [DOI] [PubMed] [Google Scholar]
- 27.Wan CP, Myung E, Lau BH. An automated micro-fluorometric assay for monitoring oxidative burst activity of phagocytes. J Immunol Methods. 1993;159:131–138. doi: 10.1016/0022-1759(93)90150-6. [DOI] [PubMed] [Google Scholar]
- 28.Hsu HY, Wen MH. Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J Biol Chem. 2002;277:22131–22139. doi: 10.1074/jbc.M111883200. [DOI] [PubMed] [Google Scholar]
- 29.Landmann R, Scherer F, Schumann R, Link S, Sansano S, Zimmerli W. LPS directly induces oxygen radical production in human monocytes via LPS binding protein and CD14. J Leukoc Biol. 1995;57:440–449. doi: 10.1002/jlb.57.3.440. [DOI] [PubMed] [Google Scholar]
- 30.Uchikura K, Wada T, Hoshino S, Nagakawa Y, Aiko T, Bulkley GB, Klein AS, Sun Z. Lipopolysaccharides induced increases in Fas ligand expression by Kupffer cells via mechanisms dependent on reactive oxygen species. Am J Physiol Gastrointest Liver Physiol. 2004;287:G620–G626. doi: 10.1152/ajpgi.00314.2003. [DOI] [PubMed] [Google Scholar]
- 31.Diebold BA, Bokoch GM. Rho GTPases and the control of the oxidative burst in polymorphonuclear leukocytes. Curr Top Microbiol Immunol. 2005;291:91–111. doi: 10.1007/3-540-27511-8_6. [DOI] [PubMed] [Google Scholar]
- 32.Cao Q, Mak KM, Lieber CS. Cytochrome P4502E1 primes macrophages to increase TNF-α production in response to lipopolysaccharide. Am J Physiol Gastrointest Liver Physiol. 2005;289:G95–107. doi: 10.1152/ajpgi.00383.2004. [DOI] [PubMed] [Google Scholar]
- 33.Kono H, Rusyn I, Uesugi T, Yamashina S, Connor HD, Dikalova A, Mason RP, Thurman RG. Diphenyleneiodonium sulfate, an NADPH oxidase inhibitor, prevents early alcohol-induced liver injury in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1005–G1012. doi: 10.1152/ajpgi.2001.280.5.G1005. [DOI] [PubMed] [Google Scholar]
- 34.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 35.Iles KE, Forman HJ. Macrophage signaling and respiratory burst. Immunol Res. 2002;26:95–105. doi: 10.1385/IR:26:1-3:095. [DOI] [PubMed] [Google Scholar]
- 36.Nagy LE. Molecular mechanisms of alcohol metabolism. Annu Rev Nutr. 2004;24:55–78. doi: 10.1146/annurev.nutr.24.012003.132258. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Z, Bagby GJ, Stoltz D, Oliver P, Schwarzenberger PO, Kolls JK. Prolonged ethanol treatment enhances lipopolysaccharide/phorbol myristate acetate-induced tumor necrosis factor-α production in human monocytic cells. Alcohol Clin Exp Res. 2001;25:444–449. [PubMed] [Google Scholar]
- 38.White JE, Tsan MF. Differential induction of TNF-α and MnSOD by endotoxin: role of reactive oxygen species and NADPH oxidase. Am J Respir Cell Mol Biol. 2001;24:164–169. doi: 10.1165/ajrcmb.24.2.4169. [DOI] [PubMed] [Google Scholar]
- 39.Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferon-γ. J Neurosci Res. 2004;77:540–551. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- 40.Bonizzi G, Piette J, Schoonbroodt S, Merville MP, Bours V. Role of the protein kinase C λ/ι isoform in nuclear factor-κB activation by interleukin-1β or tumor necrosis factor-α: cell type specificities. Biochem Pharmacol. 1999;57:713–720. doi: 10.1016/s0006-2952(98)00353-0. [DOI] [PubMed] [Google Scholar]
- 41.Bey EA, Xu B, Bhattacharjee A, Oldfield CM, Zhao X, Li Q, Subbulakshmi V, Feldman GM, Wientjes FB, Cathcart MK. Protein kinase C δ is required for p47phox phosphorylation and translocation in activated human monocytes. J Immunol. 2004;173:5730–5738. doi: 10.4049/jimmunol.173.9.5730. [DOI] [PubMed] [Google Scholar]
- 42.Zhao X, Xu B, Bhattacharjee A, Oldfield CM, Wientjes FB, Feldman GM, Cathcart MK. Protein kinase Cδ regulates p67phox phosphorylation in human monocytes. J Leukoc Biol. 2005;77:414–420. doi: 10.1189/jlb.0504284. [DOI] [PubMed] [Google Scholar]
- 43.Stubbs CD, Slater SJ. Ethanol and protein kinase C. Alcohol Clin Exp Res. 1999;23:1552–1560. [PubMed] [Google Scholar]
- 44.Isayama F, Froh M, Yin M, Conzelmann LO, Milton RJ, McKim SE, Wheeler MD. TNF α-induced Ras activation due to ethanol promotes hepatocyte proliferation independently of liver injury in the mouse. Hepatology. 2004;39:721–731. doi: 10.1002/hep.20137. [DOI] [PubMed] [Google Scholar]
- 45.Guasch RM, Tomas M, Minambres R, Valles S, Renau-Piqueras J, Guerri C. RhoA and lysophosphatidic acid are involved in the actin cytoskeleton reorganization of astrocytes exposed to ethanol. J Neurosci Res. 2003;72:487–502. doi: 10.1002/jnr.10594. [DOI] [PubMed] [Google Scholar]
- 46.Qian Y, Luo J, Leonard SS, Harris GK, Millecchia L, Flynn DC, Shi X. Hydrogen peroxide formation and actin filament reorganization by Cdc42 are essential for ethanol-induced in vitro angiogenesis. J Biol Chem. 2003;278:16189–16197. doi: 10.1074/jbc.M207517200. [DOI] [PubMed] [Google Scholar]
- 47.Sebastian BM, Nagy LE. Decreased insulin-dependent glucose transport by chronic ethanol feeding is associated with dysregulation of the Cbl/TC10 pathway in rat adipocytes. Am J Physiol Endocrinol Metab. 2005;289:E1077–E1084. doi: 10.1152/ajpendo.00296.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Slater SJ, Cook AC, Seiz JL, Malinowski SA, Stagliano BA, Stubbs CD. Effects of ethanol on protein kinase C α activity induced by association with Rho GTPases. Biochemistry. 2003;42:12105–12114. doi: 10.1021/bi034860e. [DOI] [PubMed] [Google Scholar]
- 49.Marmillot P, Rao MN, Lakshman MR. Chronic ethanol exposure in rats affects rabs-dependent hepatic trafficking of apolipoprotein E and transferrin. Alcohol. 2001;25:195–200. doi: 10.1016/s0741-8329(01)00179-3. [DOI] [PubMed] [Google Scholar]
- 50.Bokoch GM. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 2005;15:163–171. doi: 10.1016/j.tcb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 51.Heo J, Campbell SL. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J Biol Chem. 2005;280:31003–31010. doi: 10.1074/jbc.M504768200. [DOI] [PubMed] [Google Scholar]
- 52.Heo J, Campbell SL. Superoxide anion radical modulates the activity of Ras and Ras-related GTPases by a radical-based mechanism similar to that of nitric oxide. J Biol Chem. 2005;280:12438–12445. doi: 10.1074/jbc.M414282200. [DOI] [PubMed] [Google Scholar]
- 53.Khoruts A, Stahnke L, McClain CJ, Logan G, Allen JI. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology. 1991;13:267–276. [PubMed] [Google Scholar]