

Abstract
Virtually all measurements of biochemical kinetics have been derived from macroscopic measurements. Single-molecule methods can reveal the kinetic behavior of individual molecular complexes and thus have the potential to determine heterogeneous behaviors. Here we have used single-molecule fluorescence resonance energy transfer to determine the kinetics of binding of SNARE (soluble N-ethyl maleimide-sensitive fusion protein attachment protein receptor) complexes to complexin and to a peptide derived from the central SNARE binding region of complexin. A Markov model was developed to account for the presence of unlabeled competitor in such measurements. We find that complexin associates rapidly with SNARE complexes anchored in lipid bilayers with a rate constant of 7.0 × 106 M−1 s−1 and dissociates slowly with a rate constant of 0.3 s−1. The complexin peptide associates with SNARE complexes at a rate slower than that of full-length complexin (1.2 × 106 M−1 s−1), and dissociates much more rapidly (rate constant >67 s−1). Comparison of single-molecule fluorescence resonance energy transfer measurements made using several dye attachment sites illustrates that dye labeling of complexin can modify its rate of unbinding from SNAREs. These rate constants provide a quantitative framework for modeling of the cascade of reactions underlying exocytosis. In addition, our theoretical correction establishes a general approach for improving single-molecule measurements of intermolecular binding kinetics.
INTRODUCTION
Complexin (also known as synaphin) is a small cytoplasmic protein (∼15–21 kD mass) that binds to the SNARE (soluble N-ethyl maleimide-sensitive fusion attachment protein receptor) complex with high affinity (1,2) via a central, α-helical domain (3,4). High resolution structures derived from x-ray diffraction measurements show that this central domain of complexin selectively recognizes the interface between the coiled-coil domains of syntaxin and synaptobrevin within the SNARE complex (5,6). Recent in vitro experiments suggest that complexin clamps the trans-SNARE complex in a hemifusion state (7,8), which might be important to prime the SNARE complex for synaptotagmin binding (9).
The interaction between complexin and the SNARE complex is an essential step in Ca2+-dependent exocytosis. For example, neurotransmitter release is drastically reduced when complexin genes are knocked out (10) or point-mutated (11,12) or when binding-site peptides are used to inhibit the interaction of complexin with SNARE complexes (13). Complexin is also linked to some neurological disorders (14,15).
Although it is generally accepted that complexin is essential for neurotransmitter release, the timing and mechanism of complexin's role remain controversial (7–10,12,13,16). A key step toward understanding the physiological action of complexin within an in vivo signaling network is the characterization of the interaction between complexin and SNARE complexes in a simplified system.
Here we have determined the kinetics of complexin binding to membrane-anchored SNARE complexes by using single-molecule fluorescence resonance energy transfer (smFRET) measurements to directly monitor the time-dependent FRET signal generated by the interactions between individual SNARE complexes and complexin. We also have developed a theoretical approach based upon Markov modeling to extract kinetic information from such measurements despite the unavoidable presence of the unlabeled competitive molecules. With these approaches we also could determine the binding kinetics of a peptide, derived from the central, SNARE-binding domain of complexin (SBD; residues 46–74), which has been used to inhibit binding of full-length complexin to SNAREs (13,17). Our measurements of the kinetics of the SNARE complex binding to complexin and to the complexin peptide provide strong constraints for models of complexin-dependent exocytosis and provide biophysical insights into the dynamic regulation of membrane fusion. The combination of the smFRET approach and our Markov analysis can be generalized easily to study intermolecular interactions that occur in other biological settings.
MATERIALS AND METHODS
Sample preparation
Proteins: plasmids, mutations, expression, purification, and labeling
A cDNA construct for full-length complexin fused to a hexahistidine tag was provided by T. Abe (Niigata, Japan). Plasmids for glutathione S-transferase fusions of the cytoplasmic domain of synaptobrevin (1–94) and full-length SNAP-25 have been described previously (18). These fusion proteins were expressed and purified by glutathione-Sepharose with standard methods. Hexahistidine-tagged, full-length rat syntaxin-1A in pet28a and His-tagged, full length complexin were expressed, purified, and labeled as described earlier (19). Thrombin treatment followed by ion-exchange chromatography was used to remove glutathione S-transferase and hexahistidine tags.
In these plasmids, all cysteine residues in wild-type syntaxin-1A and complexin were mutated to serine to allow for site-specific dye labeling via engineered cysteine residues. The mutations E39C (complexin) and E41C (synaptobrevin) were created using the QuikChange Mutagenesis Kit (Stratagene, La Jolla, CA). Mutations were selected with guidance from the crystal structure of the truncated neuronal SNARE complex (20) and the structure of the complexin/SNARE complexes (5,6).
Proteins were labeled as described earlier (19) and labeling efficiency was determined by absorbance spectroscopy. The E41C synaptobrevin mutant was labeled with Alexa 647 maleimide (Invitrogen, Carlsbad, CA) at 84% efficiency. Full-length complexin with the E39C mutation was labeled with Alexa 555 maleimide at 30% ± 4% efficiency based upon the published extinction coefficients for the dyes (Invitrogen). We estimate the uncertainty of labeling from the observed spread of several independent absorption measurements of a single sample. Alexa-555-labeled complexin peptide was synthesized by a commercial source (Global Peptide, Fort Collins, CO) and dye-labeling efficiency of this peptide was >90%. The sequence of the peptide was: Alexa555-CERRKEKHRKMEEEREEMRQTIRDKYGLKK.
Formation of SNARE complexes
SNARE complexes were formed in solution as described previously (19). Briefly, syntaxin was mixed with full-length SNAP-25 at a 1:2 molar ratio followed by addition of a 1:5 molar ratio of the synaptobrevin cytosolic domain. After overnight incubation, SNARE complexes were purified away from free synaptobrevin by anion exchange on monoQ resin in Tris-buffered saline (TBS) (20 mM Tris at pH 8.2, 200 mM NaCl, 1 mM dithiothreitol) containing 100 mM β-d-octyl glucoside (Anatrace, Maumee, OH). Formation of the SNARE complex was confirmed by SDS-PAGE without boiling. Further boiling dissembled the SNARE complex (data not shown).
Reconstitution into liposomes
A chloroform solution of egg phosphatidylcholine (Avanti Polar Lipids, Alabaster, AL) was dried under flowing argon inside a glass culture tube and then placed in a vacuum for several hours. TBS was added to yield a final lipid concentration of 30 mg/ml and the solution was passed 21 times through 50-nm pore-size filters with the Mini-Extruder (Avanti Polar Lipids).
Preformed SNARE complexes were reconstituted into liposome solutions as described earlier (19). Briefly, protein solutions (80 nM) in TBS containing 100 mM β-d-octyl glucoside were mixed at a 1:4 ratio with 30 mg/ml liposome samples and incubated at 4°C for 30 min. These mixtures were then diluted 1:1 with detergent-free TBS and separated from detergent and unincorporated protein using size-exclusion chromatography on a Sepharose CL4B column (GE Healthcare Bio-Sciences, Piscataway, NJ) in detergent-free TBS.
Microscopy and data analysis
Fluorescence microscopy of supported lipid bilayers
Bilayers containing SNARE complexes were formed on the surface of a flow cell between a quartz microscope slide and a coverslip (see Fig. 1 b). Ultraviolet curing optical adhesive (Norland Products, Cranbury, NJ) sealed the edges of the chamber, and buffers were exchanged through holes drilled in the quartz slide. Bilayers formed by self-assembly during incubation of liposomes containing reconstituted SNARE proteins (3 mg/ml lipid for 10 min) in the flow channel. Liposomes reconstituted with SNAREs were diluted with protein-free liposomes before bilayer formation to make sure that the spacing between adjacent SNARE complexes in the bilayer was greater than the spatial resolution of the microscope. The incubation with SNARE-containing liposomes was followed by a second incubation with protein-free liposomes (15 mg/ml lipid for 10–30 min), which improved resistance to nonspecific binding of soluble proteins to the surface.
FIGURE 1.

Schematic illustrations of the labeling sites located in complexin and the SNARE complexes as well as the experimental system for smFRET assay. (a) Location of labeling sites on the SNARE complex, complexin, and complexin peptide. Synaptobrevin (VAMP) is blue, syntaxin is red, SNAP-25 (SN-25) is green, complexin is orange, and the binding-site peptide of complexin is pink. Residues that were mutated to cysteine for dye labeling are shown as yellow spheres along with their corresponding residue numbers (coordinates are taken from Chen et al. (5)). The unstructured C-terminal domain of complexin is indicated by the thin line. Position 45 corresponds to the labeling site at the N-terminus of the complexin peptide. Molecular graphics were generated using PyMOL (40). (b) Experimental setup for the detection of smFRET events.
These supported bilayers were illuminated by prism-type total internal reflection of coaxial 532-nm and 635-nm lasers. To allow sequential excitation of both donor and acceptor fluorophores, laser illumination was alternated in the sequence 635 nm for 1 s, 532 nm for 45 s, then 635 nm for 5 s. The illuminated region was observed by a 60× 1.2-NA water immersion objective (Olympus America, Center Valley, PA). A cy3/cy5 emission filter (Chroma Technology, Rockingham, VT) blocked laser excitation light in the emission path. A 645dcxr dichroic mirror (Chroma) split the emitted fluorescence light into two spectral bands that were relayed side by side onto a charged-coupled device detector (Cascade 512B, Roper Scientific, Tucson, AZ). Single fluorophores were identified based on their fluorescence intensity, quantized photobleaching, and spatial characteristics. All observations were conducted at room temperature in TBS buffer augmented with 2% glucose and an enzymatic oxygen scavenger system (100 units/ml glucose oxidase, 1000 units/ml catalase, and 200 μM cyclooctatetraene) to reduce photobleaching. FRET efficiency (E) was calculated from the background-subtracted intensities of the acceptor (Iacceptor) and donor (Idonor) as E = Iacceptor/(Iacceptor + Idonor) from fixed locations in the bilayer identified to contain an acceptor during the initial 635-nm illumination phase.
Measurement of dwell-time distributions
To examine the kinetics of complexin binding, fluorescence was measured at locations where preidentified SNARE complexes were present. The beginning of a binding event was defined by the rapid appearance (within a single image frame) of donor or acceptor emission with intensity signal levels typical of a single molecule, whereas the rapid disappearance of emission denoted unbinding. In a small subset of traces, fluorescence intensity recorded from a fixed location drifted or faded away without such rapid transitions. For example, the intensities for the recordings on the left-hand side of Fig. 2 b smoothly drift during the complexin-binding event. These cases presumably arise from the mobility of SNARE complexes reconstituted into the supported bilayers. In our measurements, ∼90% of the SNARE complexes were immobile, within our experimental resolution, whereas the remaining 10% exhibited detectable movement. This small mobile fraction is consistent with a previous fluorescence recovery after photobleaching study that observed that 3–7% of syntaxin molecules reconstituted into supported bilayers were mobile, with a diffusion coefficient of 0.07 μm2/s (21). All of the kinetic data reported in our article were derived from traces with stable intensities during binding events. However, including traces with drifting intensities did not produce significant changes in the measured kinetic parameters.
FIGURE 2.

smFRET assay for characterization of interactions between complexin and the SNARE complex. (a) Transient binding of complexin to the SNARE complex yields smFRET events characterized by increased fluorescence emission from the FRET acceptor on the SNARE complex during excitation of the FRET donor on complexin by green laser illumination. Measurements were made with complexin-39 (labeled with Alexa 555)/synaptobrevin-41 (labeled with Alexa 647). (b) Simultaneous measurements of fluorescence emission of both FRET acceptor and donor during smFRET. The FRET pair of Fig. 2 a was used. The sudden transitions upon binding and unbinding, as well as the overall intensity levels, are indicative of single-molecule level detection. (c) Simultaneous measurements of fluorescence emission of both FRET acceptor and donor during smFRET. Rare events are specially selected to illustrate the recovery of the donor emission after acceptor photobleaching or blinking. (d) Distribution of FRET efficiency for individual binding events between complexin and the SNARE complex. Labeled pair combinations are the same as in Fig. 2 a. The efficiency histogram was fit to a Gaussian distribution with a main peak of 0.65.
All data were acquired at 100 ms/frame with the exception of some complexin peptide results, which were acquired at 15 ms/frame where indicated. The koff rate was determined at 20–100 nM for complexin or complexin peptide. The duration of the bound state was taken from the dwell time in the high FRET efficiency state and koff was determined from the time constant of exponential fits to histograms of dwell times.
The kon rate constant was determined at 100–200 nM complexin or complexin peptide. Because the high background fluorescence associated with high concentrations of complexin or the complexin peptide obscured donor emission, we measured the time interval between consecutive high FRET efficiency events as the “dwell time” in the unbound state under these conditions. To test whether photobleaching affects determinations of binding kinetics, fluorescence dwell-time data obtained during the first 20 s of illumination were compared to data acquired from 20 to 40 s of illumination. No differences were observed (data not shown), which indicates that photobleaching of the surface immobilized acceptors did not distort the kinetic constants reported in this article.
RESULTS
FRET imaging of complexin-SNARE complex interactions
To interpret FRET signals in terms of specific intermolecular complexes, two different fluorophores must be incorporated into the interacting molecules at known locations. For this purpose, we introduced single cysteine mutations into synaptobrevin 2 (E41C) and complexin 1 (E39C) to allow the covalent attachment of maleimide derivative fluorophores (3). Synaptobrevin 2 (residues 1–94) labeled with Alexa 647 (the FRET acceptor) was incorporated into SNARE complexes containing full-length SNAP-25 and full-length syntaxin 1A (Fig. 1 a). To mimic the in vivo interaction between SNARE complexes and complexin, the SNARE complexes were anchored, through the single transmembrane domain of syntaxin, in lipid bilayers that were deposited on a quartz surface (Fig. 1 b). Complexin labeled with Alexa 555 (the FRET donor) was then introduced into the solution contacting the bilayer. The separation between these two dye attachment sites, as determined from crystallographic measurements of the static complexin-SNARE complex, is near the Förster radius (Ro) of our dye pair (5,6) and thus should yield FRET when the proteins bind.
A total internal reflection fluorescence microscopy system (TIRF) was used to excite and detect fluorescence emission from the lipid bilayers (Fig. 1 b). When monitoring the binding of complexin to SNAREs, the microscope field was first illuminated with 635 nm light for 1 s to excite acceptor dye emission on SNARE complexes. This signal allowed us to locate individual, anchored SNARE complexes. Next, the excitation light was switched to 532 nm to excite the donor fluorophore on complexin, whereas fluorescence emission was measured at the locations where SNARE complexes were found.
In such conditions, excitation of the donor fluorophore can produce several possible types of fluorescence signal that can be used to identify different binding conformations. If complexin binds to the SNARE complex in a way that causes the dyes to be positioned substantially outside the Ro of the dye pair, only emission from the donor on complexin will result. Conversely, if the binding of complexin to the SNARE complex causes the dyes to be separated by a distance of Ro or less, then the acceptor on the SNAREs will emit fluorescence due to FRET. In this case, dissociation of complexin from the SNARE complex will cause complexin to rapidly diffuse out of the evanescent field of the TIRF microscope, leading to loss of both donor and acceptor fluorescence. If the proteins bind but the FRET acceptor on the SNARE complex then photobleaches or enters a dark state (due to blinking), then disappearance of acceptor emission will be accompanied by a simultaneous increase in the emission from the donor on complexin.
Our measurements of spectrally resolved fluorescence emission from individual SNARE complexes revealed examples of most of these behaviors, as shown in Fig. 2. Fig. 2 a shows the time course of the emission signal from several FRET acceptors (SNAREs) during green laser excitation of the FRET donor (20 nM complexin). The transient increases in the emission of the acceptor indicate energy transfer from complexin to SNAREs. Such signals were not observed in control experiments where SNAREs were omitted from the bilayers or when complexin was absent (data not shown). Fig. 2 b shows typical examples of simultaneous fluorescence emission from both acceptor (SNAREs, red) and donor (complexin, green) fluorophores. Events such as those shown in Fig. 2 b, where the donor and acceptor fluorescence appeared and disappeared simultaneously, represented >95% of the total events observed and presumably reflect complexin binding to, and unbinding from the SNARE complex. Fig. 2 c shows rarer events that are presented to illustrate the anticorrelated recovery of donor emission after acceptor photobleaching or spontaneous blinking during the bound-state interval. These anticorrelated events confirmed that we were detecting single-molecule events. Note that the events in Fig. 2, b and c, had similar intensities of fluorescence emission, characteristic of signals from single molecules. In addition, independent photobleaching experiments demonstrated that emission signals of this magnitude were due to single molecules (data not shown). Our observations of single-molecule emission and anticorrelated behavior of fluorophores are in general agreement with a previous smFRET study of the SNARE-complexin interaction (22).
At low concentrations of complexin, where the emission of both acceptor and donor fluorophores could be resolved, FRET efficiency could be calculated by quantifying the percentage of energy transfer from the FRET donor to the FRET acceptor. The distribution of the FRET efficiency, calculated from many individual events such as those shown in Fig. 2, b and c, indicated that the mean FRET efficiency was ∼0.65 for the combination of synaptobrevin 2 (E41C) and complexin 1 (E39C (Fig. 2 d)). Although our lack of knowledge about the rotational freedom of the dyes and their local environment limits precision in converting the measured FRET efficiency into a distance, a mean FRET value of 0.65 corresponds to a separation between the dyes of just under 5 nm, given the published Förster radius of 5.1 nm for this dye pair (23). This distance compares favorably to crystallographic measurements of this complex, which suggest a distance of 5.7 nm between the Cα locations for these two labeling sites (5,6). The broad distribution of FRET efficiency is consistent with NMR and x-ray observations (5,12) that complexin 39 is in a region that remains somewhat flexible while complexin is bound to SNAREs. Higher-resolution data and studies with additional labeling sites will be useful to confirm this potential flexibility.
Rate of complexin dissociation from SNARE complexes
Despite the diffuse background emission from FRET donor molecules (complexin) in solution, TIRF illumination combined with acceptor emission provided an adequate signal/noise ratio for clear detection of complexin/SNAREs binding events. Fig. 3 a illustrates a group of binding events at a 20 nM concentration of complexin. Those events are arranged in the style commonly associated with analysis of single ion channels, with the traces in this case representing random binding and unbinding of complexin.
FIGURE 3.

Kinetics of the complexin/SNARE complex interaction. (a) Time traces of acceptor emission for smFRET events between complexin-39 (labeled with Alexa 555) and synaptobrevin-41 (labeled with Alexa 647). (b) Dwell-time histogram for the bound state, defined as the duration of the high FRET level of acceptor fluorescence. A single exponential function fit yields a time constant (τ) of 3.1 s, corresponding to a dissociation rate of 0.33 s−1. Fit parameters were not significantly changed when data rebinned with time steps smaller by a factor of 4. (c) Dwell-time histogram for the unbound state, defined as the time interval between acceptor fluorescence events. Complexin concentration was 200 nM and labeling efficiency was 0.3. τ was 2.5 s, which yields a rate constant of 7.0 × 107 M−1 s−1 after applying the corrections described in the text.
The entire duration of fluorescence emission above the background level for either donor or acceptor channel were taken as the duration of a single binding event. The durations of bound states were measured for many such events and were compiled into the histogram shown in Fig. 3 b. The distribution of dwell times in the bound state could be described by an exponential function (Fig. 3 b). To test whether photobleaching might reduce the observed duration of the bound state, we compared the traces obtained in the first 20 s of acquisition with the traces from the last 20 s. No significant difference was found, indicating that the effect of acceptor photobleaching was minimal. The rate of transition between the bound and unbound states could be inferred from the time constant of this exponential distribution, and the resulting rate constant for dissociation of complexin from the SNARE complex was 0.33 s−1. This value compares favorably to the rate constant of 0.31 s−1 obtained in earlier measurements that used bulk stopped-flow fluorescence anisotropy (3), but differs from the rate of 2.5 s−1 obtained from previous smFRET measurements (22).
Bowen et al. (22) suggested that the location of the fluorophore might alter the kinetics of complexin binding to SNAREs. To address this issue, we measured off rates using several different labeling sites. If the location of the labeling sites does not alter the intermolecular interaction, then the measured binding kinetics should be similar for different labeling sites, even though the absolute FRET efficiency will differ. We analyzed bound-state kinetics for smFRET events between the sets of complexes with Cy3 and Cy5 labeling at complexin-105/syntaxin-193, complexin-105/syntaxin-228, and complexin-105/syntaxin-249 (see Fig. 1 a) (FRET intensity histograms for these data have been previously published (22)). Despite different FRET efficiencies with the different labeling combinations, the dwell-time histograms for the FRET pairs were all remarkably similar to each other and to the data for the complexin-39/synaptobrevin-41 pair shown in Fig. 3 b (Fig. 4). The inferred off-rate constant is in the range of 0.2–0.3 s−1, close to the rate of 0.33 s−1 obtained from Fig. 3 b and in agreement with previous fluorescence anisotropy measurements (3) using a fluorophore attached at complexin-39. Therefore, the presence of the fluorophore at these labeling sites does not alter the measured rate of dissociation.
FIGURE 4.

Kinetics for complexin/SNAREs interaction from experiments using several different labeling sites. (a) Dwell-time histogram of smFRET events for complexin bound to SNAREs, similar to Fig. 3 a, except that the labeling pair used was complexin-105/syntaxin-193. A single exponential function fit results in a time constant of 4.2 s, corresponding to a dissociation rate of 0.2 s−1. (b) Same as Fig. 4 a, except that the FRET pair complexin-105/syntaxin-228 was used. The exponential fitting obtained a similar time constant of 3.8 s, corresponding to a dissociation rate of 0.3 s−1. (c) Same as Fig. 4 a, except that the FRET pair complexin-105/syntaxin-249 was used. The exponential fitting obtained a similar time constant of 3.2 s, corresponding to a dissociation rate of 0.3 s−1.
Rate of complexin association with SNARE complexes
To measure the rate of complexin association with the SNARE complex, we increased the concentration of complexin to 200 nM. The higher concentration reduced the duration of the unbound state to a time sufficiently short to be measured with our experimental procedures. In this case, we used acceptor emission due to FRET to define binding events, because high fluorescence emission from complexin in solution made it difficult to discern the fluorescence emission associated with donor-labeled complexin binding to SNAREs at the membrane surface. Initial acceptor emission established the reference time point for determining the interval to the next binding event. As we were not measuring the duration of the bound state in such experiments, disappearance of the acceptor emission during dye blinking did not introduce significant errors. In addition, this measurement was not affected by acceptor bleaching: SNARE complexes with inactive acceptors (or no acceptor) will never produce high FRET and thus will not contribute to the distribution of dwell times.
Such measurements of the intervals between FRET events yielded the distribution of unbound states shown in Fig. 3 c. This distribution also could be described by an exponential function, with a time constant of 2.5 s. In principle, such measurements could be used to calculate the association rate constant for complexin to bind to SNAREs. However, the presence of unlabeled complexin in solution will cause the time constants of single exponential fits to the distribution of dark-state dwell times to differ from the actual on-rate constant. In our experiments, where the soluble ligand (complexin) carries the donor dye, there are two different kinetic states (see Fig. 5 a, inset) that are indistinguishable because they do not generate acceptor fluorescence emission via FRET: 1), when no complexin molecule is bound to the SNARE complex (state A); and 2), when an unlabeled complexin molecule is bound to the SNARE complex (state B′). An unlabeled complexin molecule bound to a SNARE complex will prevent a second labeled complexin molecule from binding. In this case, the measured values of dark intervals will underestimate the on rate for complexin binding to SNAREs because there will be undetected, or hidden, binding events (state B′) that occur between the observed binding events of labeled complexin (state B).
FIGURE 5.

Effect of nonfluorescent competitors on observed dark dwell times. (a) Theoretical distributions of non-FRET dwell time at different labeling efficiencies (colored curves). The data were computed using Eq. 1 with a total protein (complexin) concentration of 200 nM and with koff 0.33 s−1 and kon 7.0 × 106 M−1 s−1, and normalized so that the area under each curve is 1. (b) Underestimation of kon, resulting from not considering the effects of nonfluorescent competitors. The apparent kon was obtained by least-square fitting with a single exponential function (red circles, without correction) or with our solution (black circles, with correction). (c) The results in b are plotted as percent correction, demonstrating that even high protein-labeling efficiencies require significant corrections.
Calculation of on rate in the presence of unlabeled ligand
To take into account the contribution of unlabeled complexin, the kinetic scheme shown in the inset, Fig. 5 a, can be accurately modeled from the theory of Markov processes. The solution to this problem is known from the field of ion channel gating (24,25). Specifically, Eq. 2.32 in Colquhoun and Hawkes (24), which describes the distribution of shut-state lifetimes for ligand-gated ion channels, can be used to define the probability of observing intervals of length t between binding events of labeled complexin to the SNARE complex (A→B) in the presence of unlabeled complexin. This probability, P(t), is:
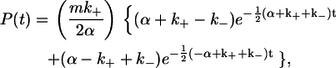 |
(1) |
where 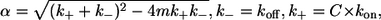 kon and koff are the usual association and disassociation rate constants, C is the molar concentration of total complexin (labeled + unlabeled), and m is the fraction of complexin with a dye label. An alternate derivation of Eq. 1 specific to the kinetic scheme shown in Fig. 5 a is presented in the Supplementary Material.
kon and koff are the usual association and disassociation rate constants, C is the molar concentration of total complexin (labeled + unlabeled), and m is the fraction of complexin with a dye label. An alternate derivation of Eq. 1 specific to the kinetic scheme shown in Fig. 5 a is presented in the Supplementary Material.
To estimate the magnitude of correction that results from ignoring the effect of an unlabeled protein population, we used Eq. 1 to compute the theoretical dwell-time histogram expected for different labeling efficiencies (Fig. 5 a). The error in the binding constants inferred from single exponential fits to these distributions is substantial in the presence of an unlabeled ligand population. As seen in Fig. 5, b and c, for 60% labeling efficiency, which is a common condition in many dye-labeled protein experiments, the correction is 20%. As the unlabeled fraction grows, the correction becomes even more significant.
We fit Eq. 1 to our smFRET data of the dark-state dwell times (Fig. 3 c) to extract the association rate. In this process, kon is the only free parameter, as m and C were independently determined from absorption spectroscopy and experimental conditions, whereas the value of koff was taken from the dissociation rate (0.33 s−1) measured above. We obtained an on-rate constant of 7.0 × 106 M−1 s−1. This value lies between the value determined from previous smFRET measurements (3.1 × 106 M−1 s−1 (22)) and previous bulk fluorescence anisotropy measurements (3 × 107 M−1 s−1 (3)).
Kinetics of the complexin peptide binding to SNARE complexes
We used the same smFRET assay to determine the kinetics of the interaction between membrane-incorporated SNARE complexes and SBD peptide derived from complexin. This peptide is a fragment of complexin (46–74) that includes the binding site for syntaxin (Fig. 1 a) and competes with full-length complexin for binding to the SNARE complex. Caged and noncaged versions of this peptide have been used to determine complexin's role in synaptic vesicle exocytosis (13,17). As before, Alexa 647 was attached to SNARE complexes via the E41C mutation of synaptobrevin 2, whereas the complexin peptide was labeled with Alexa 555 at its N-terminus.
Single-molecule FRET was observed from SBD peptide binding to the membrane-incorporated SNARE complex (Fig. 6 a). In marked contrast to the relatively long dwell time for binding of full-length complexin to the SNARE complex, the duration of FRET signal associated with binding of complexin peptide to SNARE complexes was 50–100 times shorter. In fact, the majority of binding events (Fig. 6 a, asterisks) did not extend beyond the time required to acquire a single image under our standard imaging conditions (100 ms). The histogram in Fig. 6 b shows the distribution of dwell times for complexin peptide bound to SNARE complexes. The measured time constant of 0.05 s, corresponding to a dissociation rate of 20 s−1, was limited by the speed of data acquisition. To better resolve the very brief time that complexin peptide was bound to SNARE complexes, we increased the data acquisition rate to 15 ms/image and also increased the illumination intensity by a factor of 5 to maintain the signal/noise ratio. This configuration allowed for the detection of single-molecule signals with higher time resolution, at the cost of a shorter time before photobleaching of the fluorophore. The fluorescence signals detected at this higher acquisition speed (Fig. 6 c) show that binding events were detected as brief bursts of FRET (asterisks). When this experiment was repeated with protein-free bilayers and the same analysis was applied to randomly selected locations, the rate of detection of such signals was at least 10 times lower than at locations containing a SNARE complex. We therefore conclude that these events were not due to the random collision of the fluorescent peptide or other contaminates, but instead were actual binding events. The exponential fit to the histogram of dissociation times yielded an off rate of 67 s−1 (Fig. 6 d). A similar rate (42 s−1) was obtained when the bin in the histogram representing shortest times was ignored during fitting. Most events still lasted for only one image, indicating that this result remains limited by the temporal resolution of our optical detection system. Thus, 67 s−1 is the lower limit for the very rapid rate for peptide to dissociate from the SNARE complex.
FIGURE 6.

Kinetics of the complexin peptide/SNARE complex interaction. (a) Time trajectories of smFRET events (asterisks), characterized by increased fluorescent emissions from the FRET acceptor (attached to the SNARE complex) during direct excitation of the FRET donor (attached to the complexin peptide). Image duration, 100 ms. (b) Dwell-time histogram for the bound state of complexin peptide/SNAREs. Similar plot as in Fig. 3 a, the dissociation rate from a single exponential fit is ∼20 s−1. (c) Time trajectories of smFRET events (asterisks) obtained at higher temporal resolution (15 ms/image). (d) Dwell-time histogram for the bound state of complexin peptide/SNAREs obtained at the higher frame rate (15 ms/frame). The dissociation rate obtained from a single exponential fit is 67 s−1. Note that this rate constant is limited by the time resolution of the instrument. (e) Dwell-time histogram for the unbound state of complexin peptide/SNAREs. The association rate from a single exponential fit is 1.2 × 106 M−1 s−1.
Finally, the on rate of peptide binding to SNARE complexes was determined from the distribution of dwell time in the non-FRET state under the standard illumination conditions with 100-ms time bins. This distribution had a time constant of 2.8 s (Fig. 6 e), indicating an on rate of 1.2 × 106 M−1 s−1. Because the labeled peptide used in this experiment was >90% labeled and was repurified by high-performance liquid chromatography, our modeling indicates that the error introduced by competitive binding of nonlabeled peptide should be <6%. This on rate is quite rapid, but it is ∼6 times slower than that of full-length complexin.
DISCUSSION
In this work, a smFRET assay was used to image SNARE complexes reconstituted into lipid bilayers and to quantify the kinetics of binding of these complexes to full-length complexin and to a complexin peptide. The measured rate constants indicate that both complexin and the complexin peptide bind rapidly to SNARE complexes (with on-rate constants 7.0 × 106 M−1 s−1 and 1.2 × 106 M−1 s−1, respectively). Our single-molecule measurements of the kinetic rates make clear that both the on and off rates contribute to this difference in the equilibrium binding of full-length complexin compared to the complexin SBD peptide. The steady-state affinity for binding of full-length complexin to SNARE complexes that can be calculated from our measurements of kinetic constants is 29–57 nM. This value is ∼3 orders of magnitude higher than the affinity constant of 56 μM calculated for the complexin peptide. These results are consistent with the high binding affinity (∼10 nM) of full-length complexin previously reported for the SNARE complex (3), as well as the low binding affinity of the complexin peptide for the SNARE complex (13,17). Full-length complexin binds more tightly to SNARE complexes, mainly because its dissociation rate constant (0.2 ∼ 0.4 s−1) is much slower than that of the complexin peptide (>67 s−1).
The off rate determined in our single-molecule studies compares favorably to results obtained with conventional bulk methods. Using fluorescence anisotropy and isothermal titration calorimetry to follow the interaction of full-length complexin with SNARE complexes by rapid stopped-flowed techniques, Pabst et al. (3) obtained an off-rate constant of 0.31 s−1 that agrees with the single-molecule results presented in this work (koff of 0.2 ∼ 0.4 s−1). The earlier bulk measurement used a truncated form of the SNARE complex that was soluble and therefore was not membrane-incorporated. The general agreement of the bulk and single-molecule kinetic studies thus indicates that the SNARE motif alone is the main determinant of binding to complexin, consistent with the conclusions of high-resolution structural studies (5,6). Detailed comparisons of the results indicate very good agreement in koff measured in the bulk experiments and this work, although the kon we measured is ∼4 times smaller than that reported by Pabst et al. It is possible that the anchoring of SNAREs in a bilayer in our experiment sterically restricts the interaction between complexin and the SNARE complex, although we cannot rule out contributions from other factors (such as the influence of the N-terminal three-helix regulatory bundle of syntaxin present only in the single-molecule work).
The use of single-molecule FRET to deduce kinetic binding parameters was first established by Bowen et al. (22) for binding of complexin to the SNARE complex. They obtained an off rate that was seven times faster and an on rate that was three times slower than those determined from our measurements. The dye-labeling sites used in this study (complexin-39/synaptobrevin-41) and in the macroscopic measurements of Pabst et al. (3) (complexin-39/ SNAP-25 residue 84) are not in the binding groove, whereas both the sites used in Bowen et al. (22) (complexin-64/synaptobrevin-61) are near the binding interface. It is likely that the difference in reported dissociation rate is due to the dye-labeling sites altering the interaction between SNAREs and complexin, as proposed by Bowen et al. (22).
An alternative explanation for this discrepancy is that proximity of the labeling sites at complexin-64 and synaptobrevin-61 might allow interactions between the two fluorophores that could alter the kinetic rates. To consider this explanation, we measured binding kinetics using proteins labeled at different sites and with different dyes. Complexin was labeled at a naturally occurring cysteine at residue 105 in the unstructured region with Cy5. Three different SNARE complex mutants were labeled with Cy3 at residues 193, 228, or 249 in syntaxin (see Fig. 1 a). In all three cases (Fig. 4), we obtained off-rates consistent with the results for the complexin-39/synaptobrevin-41 pair (Fig. 3 b), suggesting that the different results obtained with the complexin-64/synaptobrevin 61 labeling-site pair arises from label-induced alteration in binding kinetics. Further supporting this conclusion, biochemical characterization has demonstrated that a single amino substitution in the interface between complexin and SNARE complexes is sufficient to change binding affinity by >1 order of magnitude. Archer et al. (12) and Giraudo et al. (7) found that the complexin point mutation R59H disrupts complexin function in vivo and SNARE complex binding in vitro and Tokumaru et al. (17) have identified other point mutations that can have similar effects on binding affinity. Taken together, these diverse results highlight the importance of optimizing experimental conditions and applying caution when interpreting results utilizing mutagenesis and dye labeling.
The on rate previously obtained with smFRET is ∼2.4 times slower than the determination presented here (22). This difference might be due to the uncorrected presence of unlabeled complexin in the earlier work, which leads to an underestimation of association rate (Fig. 5 b). Indeed, application of our correction for the effect of 16% nonlabeled protein in the earlier studies modifies the on-rate constant from 3.1 × 106 M−1 s−1 to 4.8 × 106 M−1 s−1.
From our observations, it is evident that the complexin SBD peptide very rapidly binds to and dissociates from SNARE complexes. The rapid association and dissociation rates make this peptide a useful reagent for in vivo experiments (12,16) that block the reversible clamp of SNARE complexes by complexin (7), because equilibrium binding conditions are established rapidly. Although our current measurements do not fully resolve the peptide dissociation rate constant, we can still estimate the time required for the complexin peptide to reach equilibrium when binding to SNARE complexes. From the relationship τ = 1/(koff + kon × [Pep]), with 300 μM complexin peptide in solution, the timescale for association is on the order of 2 ms. This rapid binding could allow high concentrations of the complexin-binding-site peptide to effectively occupy the complexin binding site on the SNARE complex and thereby prevent the complexin-dependent release of neurotransmitter (13,17).
In recent years, smFRET has matured into an important tool that has the ability to uncover functional states within diverse biochemical environments (reviewed in Weiss (26); Chu (27); and Myong et al. (28)). Some of the earliest success with smFRET was in revealing intramolecular conformational transitions among functional states within a single molecule (29–33). More recently, smFRET has been applied to systems of multiprotein interactions and has been used to directly visualize dynamic structural and functional rearrangements between interacting molecules, either in vitro (21,34) or inside living cells (35–37). Bowen et al. (21) established the usefulness of single-molecule FRET to examine the kinetics of interprotein binding. Our theoretical correction that corrects for nonfluorescent competitors extends the applicability of the single-molecule FRET technique to situations where incomplete dye labeling of protein is unavoidable (38,39). Our adaptation of the Markov analysis (24,25) can easily be generalized and will be useful in analysis of other single-molecule experiments where intermolecule interactions are measured.
In summary, our study resolves an inconsistency within the literature concerning the kinetics of binding of complexin to the SNARE complexes and defines the kinetics of binding of the SBD peptide to SNAREs. Our measurements provide kinetic parameters that strongly constrain models for complexin-dependent neurotransmitter release. Our work also provides a general approach to analyze intermolecular single-molecule FRET data and to extract accurate kinetic rate constants despite the presence of nonfluorescent competitors.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Axel Brunger and Mark Bowen for useful comments on this article. We thank T. Abe for providing a complexin construct and H. Tokumaru for assistance at the beginning of this project. We also thank T. Xiao for helpful discussions about smFRET analysis with Markov theories.
This work was supported by a National Institutes of Health grant to G.J.A., a Career Award at the Scientific Interface from the Burroughs Wellcome Fund to K.W., and an American Heart Association predoctoral fellowship to Y.L.
Yulong Li's present address is Dept. of Molecular and Cellular Physiology, Stanford University, Stanford, CA 94305.
Editor: David W. Piston.
References
- 1.McMahon, H. T., M. Missler, C. Li, and T. C. Sudhof. 1995. Complexins: cytosolic proteins that regulate SNAP receptor function. Cell. 83:111–119. [DOI] [PubMed] [Google Scholar]
- 2.Ishizuka, T., H. Saisu, S. Odani, and T. Abe. 1995. Synaphin: a protein associated with the docking/fusion complex in presynaptic terminals. Biochem. Biophys. Res. Commun. 213:1107–1114. [DOI] [PubMed] [Google Scholar]
- 3.Pabst, S., M. Margittai, D. Vainius, R. Langen, R. Jahn, and D. Fasshauer. 2002. Rapid and selective binding to the synaptic SNARE complex suggests a modulatory role of complexins in neuroexocytosis. J. Biol. Chem. 277:7838–7848. [DOI] [PubMed] [Google Scholar]
- 4.Pabst, S., J. W. Hazzard, W. Antonin, T. C. Sudhof, R. Jahn, J. Rizo, and D. Fasshauer. 2000. Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J. Biol. Chem. 275:19808–19818. [DOI] [PubMed] [Google Scholar]
- 5.Chen, X., D. R. Tomchick, E. Kovrigin, D. Arac, M. Machius, T. C. Sudhof, and J. Rizo. 2002. Three-dimensional structure of the complexin/SNARE complex. Neuron. 33:397–409. [DOI] [PubMed] [Google Scholar]
- 6.Bracher, A., J. Kadlec, H. Betz, and W. Weissenhorn. 2002. X-ray structure of a neuronal complexin-SNARE complex from squid. J. Biol. Chem. 277:26517–26523. [DOI] [PubMed] [Google Scholar]
- 7.Giraudo, C. G., W. S. Eng, T. J. Melia, and J. E. Rothman. 2006. A clamping mechanism involved in SNARE-dependent exocytosis. Science. 313:676–680. [DOI] [PubMed] [Google Scholar]
- 8.Schaub, J. R., X. Lu, B. Doneske, Y. K. Shin, and J. A. McNew. 2006. Hemifusion arrest by complexin is relieved by Ca2+-synaptotagmin I. Nat. Struct. Mol. Biol. 13:748–750. [DOI] [PubMed] [Google Scholar]
- 9.Tang, J., A. Maximov, O. H. Shin, H. Dai, J. Rizo, and T. C. Sudhof. 2006. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 126:1175–1187. [DOI] [PubMed] [Google Scholar]
- 10.Reim, K., M. Mansour, F. Varoqueaux, H. T. McMahon, T. C. Sudhof, N. Brose, and C. Rosenmund. 2001. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 104:71–81. [DOI] [PubMed] [Google Scholar]
- 11.Reim, K., H. Wegmeyer, J. H. Brandstatter, M. Xue, C. Rosenmund, T. Dresbach, K. Hofmann, and N. Brose. 2005. Structurally and functionally unique complexins at retinal ribbon synapses. J. Cell Biol. 169:669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Archer, D. A., M. E. Graham, and R. D. Burgoyne. 2002. Complexin regulates the closure of the fusion pore during regulated vesicle exocytosis. J. Biol. Chem. 277:18249–18252. [DOI] [PubMed] [Google Scholar]
- 13.Tokumaru, H., K. Umayahara, L. L. Pellegrini, T. Ishizuka, H. Saisu, H. Betz, G. J. Augustine, and T. Abe. 2001. SNARE complex oligomerization by synaphin/complexin is essential for synaptic vesicle exocytosis. Cell. 104:421–432. [DOI] [PubMed] [Google Scholar]
- 14.Morton, A. J., and J. M. Edwardson. 2001. Progressive depletion of complexin II in a transgenic mouse model of Huntington's disease. J. Neurochem. 76:166–172. [DOI] [PubMed] [Google Scholar]
- 15.Kinnunen, A. K., J. I. Koenig, and G. Bilbe. 2003. Repeated variable prenatal stress alters pre- and postsynaptic gene expression in the rat frontal pole. J. Neurochem. 86:736–748. [DOI] [PubMed] [Google Scholar]
- 16.Hu, K., J. Carroll, C. Rickman, and B. Davletov. 2002. Action of complexin on SNARE complex. J. Biol. Chem. 277:41652–41656. [DOI] [PubMed] [Google Scholar]
- 17.Tokumaru, H., T. Abe, K. R. Gee, L. F. Bonewald, and G. J. Augustine. 2002. Timing of synaphin/complexin action in neurotransmitter release probed by caged binding site peptide. Soc. Neurosci. 748.2. (Abstr.)
- 18.Poirier, M. A., J. C. Hao, P. N. Malkus, C. Chan, M. F. Moore, D. S. King, and M. K. Bennett. 1998. Protease resistance of syntaxin.SNAP-25.VAMP complexes. Implications for assembly and structure. J. Biol. Chem. 273:11370–11377. [DOI] [PubMed] [Google Scholar]
- 19.Weninger, K., M. E. Bowen, S. Chu, and A. T. Brunger. 2003. Single-molecule studies of SNARE complex assembly reveal parallel and antiparallel configurations. Proc. Natl. Acad. Sci. USA. 100:14800–14805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sutton, R. B., D. Fasshauer, R. Jahn, and A. T. Brunger. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 395:347–353. [DOI] [PubMed] [Google Scholar]
- 21.Bowen, M. E., K. Weninger, A. T. Brunger, and S. Chu. 2004. Single molecule observation of liposome-bilayer fusion thermally induced by soluble N-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs). Biophys. J. 87:3569–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bowen, M. E., K. Weninger, J. Ernst, S. Chu, and A. T. Brunger. 2005. Single-molecule studies of synaptotagmin and complexin binding to the SNARE complex. Biophys. J. 89:690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haugland, R. 2002. Handbook of Fluorescence Probes and Research Products Molecular Probes, 9th ed. Molecular Probes, Eugene, OR.
- 24.Colquhoun, D., and A. G. Hawkes. 1982. On the stochastic properties of bursts of single ion channel openings and of clusters of bursts. Philos. Trans. R. Soc. Lond. B. 300:1–59. [DOI] [PubMed] [Google Scholar]
- 25.Colquhoun, D., and A. G. Hawkes. 1981. On the stochastic properties of single ion channels. Proc. R. Soc. Lond. B Biol. Sci. 211:205–235. [DOI] [PubMed] [Google Scholar]
- 26.Weiss, S. 2000. Measuring conformational dynamics of biomolecules by single molecule fluorescence spectroscopy. Nat. Struct. Biol. 7:724–729. [DOI] [PubMed] [Google Scholar]
- 27.Chu, S. 2003. Biology and polymer physics at the single-molecule level. Philos. Trans. R. Soc. Lond. A. 361:689–698. [DOI] [PubMed] [Google Scholar]
- 28.Myong, S., B. C. Stevens, and T. Ha. 2006. Bridging conformational dynamics and function using single-molecule spectroscopy. Structure. 14:633–643. [DOI] [PubMed] [Google Scholar]
- 29.Michalet, X., A. N. Kapanidis, T. Laurence, F. Pinaud, S. Doose, M. Pflughoefft, and S. Weiss. 2003. The power and prospects of fluorescence microscopies and spectroscopies. Annu. Rev. Biophys. Biomol. Struct. 32:161–182. [DOI] [PubMed] [Google Scholar]
- 30.Zhuang, X., H. Kim, M. J. Pereira, H. P. Babcock, N. G. Walter, and S. Chu. 2002. Correlating structural dynamics and function in single ribozyme molecules. Science. 296:1473–1476. [DOI] [PubMed] [Google Scholar]
- 31.Zhuang, X., L. E. Bartley, H. P. Babcock, R. Russell, T. Ha, D. Herschlag, and S. Chu. 2000. A single-molecule study of RNA catalysis and folding. Science. 288:2048–2051. [DOI] [PubMed] [Google Scholar]
- 32.Lu, H. P., L. Xun, and X. S. Xie. 1998. Single-molecule enzymatic dynamics. Science. 282:1877–1882. [DOI] [PubMed] [Google Scholar]
- 33.Tinnefeld, P., and M. Sauer. 2005. Branching out of single-molecule fluorescence spectroscopy: challenges for chemistry and influence on biology. Angew. Chem. Int. Ed. Engl. 44:2642–2671. [DOI] [PubMed] [Google Scholar]
- 34.Dorywalska, M., S. C. Blanchard, R. L. Gonzalez, H. D. Kim, S. Chu, and J. D. Puglisi. 2005. Site-specific labeling of the ribosome for single-molecule spectroscopy. Nucleic Acids Res. 33:182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murakoshi, H., R. Iino, T. Kobayashi, T. Fujiwara, C. Ohshima, A. Yoshimura, and A. Kusumi. 2004. Single-molecule imaging analysis of Ras activation in living cells. Proc. Natl. Acad. Sci. USA. 101:7317–7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webb, S. E., S. R. Needham, S. K. Roberts, and M. L. Martin-Fernandez. 2006. Multidimensional single-molecule imaging in live cells using total-internal-reflection fluorescence microscopy. Opt. Lett. 31:2157–2159. [DOI] [PubMed] [Google Scholar]
- 37.Sako, Y., S. Minoguchi, and T. Yanagida. 2000. Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol. 2:168–172. [DOI] [PubMed] [Google Scholar]
- 38.Lieto, A. M., R. C. Cush, and N. L. Thompson. 2003. Ligand-receptor kinetics measured by total internal reflection with fluorescence correlation spectroscopy. Biophys. J. 85:3294–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lieto, A. M., and N. L. Thompson. 2004. Total internal reflection with fluorescence correlation spectroscopy: nonfluorescent competitors. Biophys. J. 87:1268–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeLano, W. L. 2002. The PyMOL Molecular Graphics System (http://www.pymol.org).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.