

Abstract
We studied the secondary structure of the C-terminal domains of the histone H1 subtypes H1° (C-H1°) and H1t (C-H1t) in the presence of macromolecular crowding agents (Ficoll 70 and PEG 6000) by IR spectroscopy. The carboxyl-terminal domain has little structure in aqueous solution but became extensively folded in the presence of crowding agents. In 30% PEG, C-H1° contained 19% α-helix, 28% β-sheet, 16% turns, and 31% open loops. Similar proportions were observed in 30% Ficoll 70 and for C-H1t in both crowding agents. The proportions of secondary structure motifs were comparable to those of the DNA-bound domain. Kratky plots of the small-angle x-ray scattering showed that in crowding agents the C-terminus had the compaction of a globular state. Progressive dissipation of the secondary structure and a linear increase in partial heat capacity with temperature together with increased binding of ANS indicated that the C-terminus is not cooperatively folded in crowded conditions. Native-like secondary structure and compactness in absence of folding cooperativity indicate that the C-terminus in crowding agents is in a molten globule state. Folding of the C-terminus in crowded conditions may increase the rate of the transition toward the DNA-bound state and facilitate H1 diffusion inside cell nuclei.
INTRODUCTION
H1 linker histones are involved in chromatin structure and gene regulation. It is currently accepted that histone H1 could have a regulatory role in transcription through the modulation of chromatin condensation. H1 may mediate transcription on a more specific level, participating in complexes that either activate or repress specific genes (1–7). Binding to scaffold-associated regions (8) and participation in nucleosome positioning (9) are other mechanisms by which H1 could contribute to transcriptional regulation.
H1 contains three distinct domains: a short amino-terminal domain (20–35 amino acids), a central globular domain (∼80 amino acids) consisting of a three-helix bundle and a β-hairpin, and a long carboxy-terminal domain (∼100 amino acids) (10). The amino acid composition of the C-terminus is dominated by Lys (∼40%), Ala (∼17%), and Pro (∼12%). The C-terminal domain is the primary determinant of H1 binding to chromatin in vivo (11,12). Several studies indicate that the ability of linker histones to stabilize chromatin folding resides in the C-terminal domain of the molecule (13,14). Preferential binding of histone H1 to scaffold-associated regions and activation of apoptotic nuclease also appear to be determined by the C-terminal domain (15,16).
In aqueous solution, the C-terminal domain is dominated by the random coil and turn-like conformations in rapid equilibrium with the unfolded state, but on interaction with DNA, it folds cooperatively (17). The DNA-bound structure is extremely stable and includes α-helix, β-sheet, turns, and open loops. The H1 carboxyl-terminal domain thus appears to belong to the so-called intrinsically disordered proteins undergoing coupled binding and folding (17–22). Like most natively disordered proteins, the C-terminus is of low hydrophobicity, large net charge, and low sequence complexity (23–25).
Proteins are usually studied in dilute solution; however, in the cellular environment, macromolecules and small-molecule solutes are present at high concentrations so that a significant fraction of the intracellular space is not available to other macromolecules. The excluded volume effects are predicted to favor the adoption of compact as opposed to expanded macromolecular conformations, resulting in a reduction of the total excluded volume. However, experimental evidence has shown that although crowding agents in general favor refolding (26–29), in some cases they appear to be ineffective (30,31). A role of molecular crowding in accelerating α-synuclein fibrillation has been described (32,33).
In this study we have used IR spectroscopy to estimate the proportions of secondary structure motifs of the C-terminus of H1° (C-H1°) and H1t (C-H1t) in the presence of Ficoll 70 and polyethyleneglycol (PEG) 6000 as macromolecular crowding agents. Our results show that crowding is highly effective in promoting folding of the C-terminal domain. In the presence of 30% PEG or Ficoll, the proportions of secondary structure motifs appear to be similar to those of the DNA-bound domain. In these conditions, Kratky plots of the x-ray scattering indicate that the compactness of the C-terminus is that of a globular state. However, binding of 1-anilinonaphthalene-8-sulfonate (ANS), thermal melting, and differential scanning calorimetry (DSC) show that in a crowded environment the C-terminus is not cooperatively folded. The properties of the C-terminus in crowding agents are thus those of a molten globule state.
MATERIALS AND METHODS
Cloning, expression, and purification of the C-terminal domains of histone H1 subtypes
The C-terminal domains of histones H1° and H1t were obtained from recombinant Escherichia coli (M15) as described previously (17).
Circular dichroism spectroscopy
The C-terminal domains were at 0.3 mg/ml in 10 mM phosphate buffer, pH 7.0, plus 140 mM NaCl and in the presence of PEG 6000 or Ficoll 70. Spectra were obtained on a Jasco J-717 spectrometer in 1-mm cells at 20°C. The results were analyzed with Standard analysis software (JACSO) and expressed as mean residue molar ellipticity [θ]. The helical content was estimated from the ellipticity value at 222 nm (θ222), according to the empirical equation of Chen et al. (34): percentage helical content = 100[θ222/−39,500 × (1 − 2.57/n)], where n is the number of peptide bonds.
Infrared spectroscopy measurements
The C-terminal domains were at 5 mg/ml in 10 mM HEPES, pH 7.0, plus 140 mM NaCl and in the presence of PEG 6000 or Ficoll 70 at 300 g/L. Data were collected on an FT600 Bio-Rad spectrometer equipped with an MCT detector. Typically, 1000 scans for each background and sample were collected, and the spectra were obtained with a nominal resolution of 2 cm−1 at 20°C. Secondary structure content was determined by curve fitting to the original spectrum using the component band positions identified by Fourier self-deconvolution as previously described (35,36). The baseline contributed by the solvent (10 mM HEPES, 140 mM NaCl, pH 7.0 in H2O or D2O and with or without PEG 6000 or Ficoll 70) was removed before fitting. Spectra were recorded both in H2O and D2O to distinguish between the overlapping contributions in D2O of random coil and open loops. The vibrations of loops and the α-helix are found at similar positions in H2O and D2O, the α-helix at ∼1652 cm−1 and loops at ∼1643 cm−1. In contrast, deuteration has a major effect on the position of the random coil so that in H2O it overlaps with the α-helix, whereas in D2O it overlaps with loops. The α-helix was, therefore, estimated directly in D2O, whereas loops were estimated directly in H2O. When both α-helix and loops are present in the protein structure, the percentage of random coil can be obtained either from the difference of the components around 1552 cm−1 in H2O and D2O or from the difference of the components around 1643 cm−1 in D2O and H2O, as previously described (17).
Small-angle x-ray scattering
Measurements were performed in an MBraun instrument equipped with a Siemens Kristalloflex (Graz, Austria) 760 (K-760) generator, producing radiation with a wavelength of 1.54 Å (CuKα), operating at 50 kV and 40 mA, and a Kratky Hecus camera. The collimator was a slit window, and the scattering was detected with a linear position sensitive detector OED-50M. The sample-to-detector distance was 268 mm. Samples were measured at 25°C. All samples were placed in glass capillaries of 1 mm diameter and 10 μm wall thickness. The background was subtracted by measuring blanks without protein. Both the samples and blanks were measured for 4 h unless otherwise stated. The concentration of protein was 4–10 mg/ml.
The scattering intensity I(Q) is represented as a function of the momentum transfer Q; Q = 4πsinθ/λ, where λ is the wavelength of x-rays and 2θ the scattering angle. The inner part of the scattering profile can be described by the Guinier approximation, 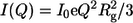 (37). The scattering at zero angle, I0, is proportional to the molecular mass of the particle and to the square of the contrast between the particle and the solvent. The radius of gyration, Rg, which is the root mean-square of the distances of all volume elements to the center of mass of the electronic volume of the particle, was obtained from the slope of a Guinier plot of lnI(Q) versus Q2.
(37). The scattering at zero angle, I0, is proportional to the molecular mass of the particle and to the square of the contrast between the particle and the solvent. The radius of gyration, Rg, which is the root mean-square of the distances of all volume elements to the center of mass of the electronic volume of the particle, was obtained from the slope of a Guinier plot of lnI(Q) versus Q2.
The Rg can also be measured with the Debye equation (38), 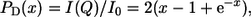 where
where 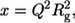 using the approximation given by Calmettes et al. (39): [PD(x)]−1 = 1 + 0.359 x1.103. The Rg is given by Rg = (a/0.359b)0.453, where a is the slope and b the intercept of the straight line.
using the approximation given by Calmettes et al. (39): [PD(x)]−1 = 1 + 0.359 x1.103. The Rg is given by Rg = (a/0.359b)0.453, where a is the slope and b the intercept of the straight line.
Thermal melting
For the analysis of the thermal stability of C-H1t in 30% Ficoll 70, the temperature of the sample was adjusted using a cover jack connected to a circulating thermostatic bath and monitored with a fitted external probe. Thermal analyses were performed by heating from 20 to 85°C at a rate of 1°C/min. For each degree of temperature interval, 400 interferograms were averaged, Fourier-transformed, and ratioed against background. Thermal analyses were performed in D2O. The Ficoll contribution at each temperature was subtracted using a Ficoll sample at the same temperature and concentration.
ANS fluorescence assays
1-Anilinonaphthalene-8-sulfonate (ANS) binding assays were performed in a Cary Eclipse spectrofluorometer. The spectra were measured in 140 mM NaCl, 10 mM phosphate buffer, pH 7.0, at 20°C. The concentration of ANS was 0.3 mM, and the concentration of protein was 10 mM. The ANS emission was scanned between 400 and 650 nm with an excitation wavelength of 380 nm.
Differential scanning calorimetry
DSC measurements were carried out on a Microcal MC-2 calorimeter supplied with an Origin software package for data analysis and curve fitting. Protein and background scans were performed at a scan rate of 1.5°C/min between 20 and 100°C. We used RNase A as a control at a concentration of 150 μM. C-H1t was analyzed at a concentration of 100 μM in buffer and in buffer plus 30% Ficoll 70. The buffer was 10 mM phosphate, pH 7.0, plus 140 mM NaCl.
RESULTS
CD and IR spectroscopy analysis of the C-terminal domain of histone H1 in the presence of crowding agents
We used CD to explore the effects of different concentrations of Ficoll 70 and PEG 6000 (10, 20, and 30%) on the secondary structure of the C-terminal domains of the histone H1 subtypes H1° and H1t in physiological salt (0.14 M NaCl) (Fig. 1). We found that in 30% of both crowding agents C-H1° and C-H1t became significantly folded. In 30% PEG, the estimated values of α-helix were 16% for C-H1° and 13% for C-H1t. In 30% Ficoll, the estimated helical content was 16% for both C-terminal domains.
FIGURE 1.

Conformational transition of the C-terminal domain of histone H1 measured by CD. C-H1° in Ficoll (A) and PEG (C). C-H1t in Ficoll (B) and PEG (D). The buffer was 10 mM phosphate, pH 7.0, plus 140 mM NaCl. (Solid line) C-terminal domains in buffer; (dotted line) buffer plus 10% PEG or Ficoll; (dashed-dotted line) buffer plus 20% PEG or Ficoll; and (dashed line) buffer plus 30% PEG or Ficoll.
IR spectroscopy was used to further characterize the secondary structure of the C-terminal domains in crowding agents. In the absence of crowding agents, the amide I (D2O) of both C-H1° and C-H1t was dominated by the unordered structure (at 1642 cm−1 in D2O) with ∼50% of the total intensity. The other main component (at 1663 cm−1), with 28–31% of the total area in both C-H1° and C-H1t, was assigned to turns in rapid equilibrium with the unfolded state. Well-defined structure was represented by 9–12% of β-sheet (at 1626–1630 cm−1) (Fig. 2 and Table 1).
FIGURE 2.

Amide I decomposition of the spectra of the C-terminal domain of histone H1. C-H1°, carboxy-terminal domain of histone H1°. C-H1t, carboxy-terminal domain of histone H1t. The spectra were recorded in D2O at 20°C. The buffer was 10 mM HEPES plus 140 mM NaCl, pH 7.0. The protein concentration was 5 mg/ml.
TABLE 1.
Percentages (%) of secondary structure of the carboxy-terminal domain of histones H1° (C-H1°) and H1t (C-H1t) in presence of crowding agents
| Buffer*
|
Ficoll†
|
PEG†
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| D2O
|
|
D2O
|
|
H2O
|
|
D2O
|
|
H2O
|
|
|
| Band (cm−1) | % | Band (cm−1) | % | Band (cm−1) | % | Band (cm−1) | % | Band (cm−1) | % | |
| Assignment C-H1° | ||||||||||
| Turns | 1676 | 3 | 1685 | 1 | 1685 | 3 | 1680 | 1 | 1680 | 7 |
| Turns | 1661 | 31 | 1667 | 26 | 1668 | 21 | 1668 | 15 | 1668 | 10 |
| α-Helix | 1655 | 15 | 1656 | 19 | ||||||
| α-Helix/random coil | 1655 | 15 | 1656 | 19 | ||||||
| Open loops | 1643 | 23 | 1642 | 31 | ||||||
| Random coil/open loops | 1642 | 52 | 1664 | 23 | 1645 | 31 | ||||
| Random coil‡ | 0 | 0 | ||||||||
| Random coil¶ | 0 | 0 | ||||||||
| β-Sheet | 1630 | 9 | 1635 | 14 | 1634 | 14 | 1635 | 18 | 1633 | 12 |
| β-Sheet | 1626 | 13 | 1625 | 13 | 1626 | 10 | 1623 | 11 | ||
| Low-frequency β-sheet | 1617 | 5 | 1616 | 8 | 1616 | 11 | 1616 | 6 | 1615 | 10 |
| C-H1t | ||||||||||
| Turns | 1685 | 2 | 1684 | 1 | 1681 | 8 | 1681 | 1 | 1681 | 8 |
| Turns | 1674 | 4 | 1668 | 29 | 1668 | 14 | 1666 | 23 | 1666 | 16 |
| Turns | 1663 | 28 | ||||||||
| α-Helix | 1656 | 15 | 1655 | 14 | ||||||
| α-Helix/random coil | 1656 | 15 | 1656 | 14 | ||||||
| Open loops | 1645 | 23 | 1643 | 27 | ||||||
| Random coil/open loops | 1643 | 53 | 1645 | 23 | 1645 | 27 | ||||
| Random coil‡ | 0 | 0 | ||||||||
| Random coil¶ | 0 | 0 | ||||||||
| β-Sheet | 1637 | 16 | 1635 | 15 | 1634 | 17 | 1634 | 13 | ||
| β-Sheet | 1626 | 12 | 1628 | 11 | 1625 | 16 | 1625 | 13 | 1624 | 11 |
| Low-frequency β-sheet | 1616 | 1 | 1616 | 5 | 1615 | 9 | 1615 | 5 | 1615 | 11 |
Buffer: 10 mM HEPES, 140 mM NaCl, pH 7.0.
The concentrations of Ficoll 70 and PEG 6000 were 30%.
The value corresponds to the difference between the components at ∼1643 in D2O and H2O.
The value corresponds to the difference between the components at ∼1655 in H2O and D2O.
Addition of either PEG or Ficoll forced the C-terminus to fold. In 30% PEG 6000, deconvolution of the amide I of C-H1° gave 19% α-helix (1656 cm−1), 28% β-sheet (1635, 1626 cm−1), 16% turns (1680, 1668 cm−1), and 31% open loops (1642 cm−1) (Fig. 3 and Table 1). Spectra were recorded both in H2O and D2O to distinguish between the overlapping contributions in D2O of random coil and open loops as described under Materials and Methods. The proportions of the different secondary structure motifs in C-H1t in the presence of PEG were similar to those found in C-H1°, although slight differences were present: a lower proportion of α-helix (14%) and open loops (27%) and a higher proportion of turns (24%); the proportion of β-sheet was very similar (30%) (Figs. 3 and 4). The amount of random coil was <1% in both C-H1° and C-H1t. The results in Ficoll 70 were similar to those found in PEG (Figs. 3 and 4, Table 1).
FIGURE 3.

Amide I decomposition of the spectra of C-H1° in the presence of macromolecular crowding agents. The left column shows the spectra measured in D2O, and the right column shows those measured in H2O. The buffer was 10 mM HEPES plus 140 mM NaCl, pH 7.0, at 20°C. The protein concentration was 5 mg/ml. The concentration of Ficoll 70 and PEG 6000 was 30%.
FIGURE 4.

Amide I decomposition of the spectra of C-H1t in the presence of macromolecular crowding agents. The left column shows the spectra measured in D2O, and the right column shows those measured in H2O. The buffer was 10 mM HEPES plus 140 mM NaCl, pH 7.0, at 20°C. The protein concentration was 5 mg/ml. The concentration of Ficoll 70 and PEG 6000 was 30%.
Small-angle x-ray scattering
The conformation of the C-terminus was examined by small-angle x-ray scattering (SAXS). The changes in compaction can be qualitatively analyzed by means of Kratky plots (Q2I(Q) versus Q). The scattering curve in Kratky plots has a characteristic bell shape when the protein is in the native state or in the molten globule state. Such a maximum is absent in completely unfolded proteins or in premolten globule states (20,40,41). In 30% Ficoll, the C-terminal domain of H1t showed the characteristic maximum of a well-developed globular structure (Fig. 5 A). The position of the maximum (0.1 Å−1) is that expected for the molecular mass of the C-terminus (Mr = 11,082) (41). The Guinier plot gave an Rg of 14.8 ± 0.7 Å (Fig. 5 B). In the absence of crowding agents, the Kratky plot lacked the maximum of globular conformations and exhibited the monotonic increase indicative of an expanded coil-like conformation (Fig. 5 A). The Rg in buffer solution was estimated with the Debye equation, which is more appropriate than the Guinier approximation in the case of unfolded proteins (39,42). In these conditions, the Rg was 25.0 ± 0.2 Å (Fig. 5 C). Although this value is considerably higher than the Rg in crowded conditions, it is smaller than the value of ∼31.5 Å that can be estimated for random coils with the expression: 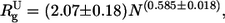 where N is the number of amino acid residues (24,43). This indicates that the C-terminus in crowded conditions is more compact than an unperturbed random coil polypeptide chain. This is consistent with the IR results, showing the presence of a residual proportion of turns and β-sheet in buffer solution. Turns may not contribute to a tightly folded core but may reduce the dimensions of the conformational ensemble.
where N is the number of amino acid residues (24,43). This indicates that the C-terminus in crowded conditions is more compact than an unperturbed random coil polypeptide chain. This is consistent with the IR results, showing the presence of a residual proportion of turns and β-sheet in buffer solution. Turns may not contribute to a tightly folded core but may reduce the dimensions of the conformational ensemble.
FIGURE 5.

Small-angle x-ray scattering of the C-terminal domain of histone H1t. (A) Kratky plots (Q2I(Q) as a function of Q) in 30% Ficoll (1) and in buffer solution (2). (B) Guinier plot (ln I(Q) as a function of Q2) of the protein in 30% Ficoll. (C) Representation of the x-ray scattering of the protein in buffer solution using the approximation to the Debye function (I(Q)−1 as a function of Q2.206); Q is in Å−1. The Q ranges used for the fits were 0.035 < Q < 0.084 for the protein in Ficoll and 0.0074 < Q < 0.0385 for the protein in buffer solution.
ANS binding
The C-terminal domains were tested for 1-anilinonaphthalene-8-sulfonate (ANS) binding in either the absence or presence of PEG and Ficoll (Fig. 6). This hydrophobic dye has been used as a probe for the formation of collapsed, partially folded states such as the premolten and molten globules (44,45). In the absence of crowding agents, the emission of ANS was not affected by the presence of the C-terminus. In contrast, in Ficoll, the λmax of fluorescence of protein-bound ANS shifted from 525 to 485 nm, and the fluorescence intensity increased ∼11-fold. In PEG, a shift from 525 to 501 nm and an ∼7-fold increase in fluorescence intensity were observed. (Fig. 6, A and B).
FIGURE 6.

Effects of macromolecular crowding on the C-terminal domain of histone H1. (A) Fluorescence emission spectra of 0.3 mM ANS in the presence of C-H1° without crowding agent (solid line) and with 30% PEG 6000 (dashed line) or 30% Ficoll 70 (dotted line). (B) Fluorescence emission spectra of 0.3 mM ANS in the presence of C-H1t without crowding agent (continuous line) and with 30% PEG 6000 (dashed line) or 30% Ficoll 70 (dotted line). (C) Thermal profiles of the secondary structure motifs obtained from amide I (D2O) decomposition in 140 mM NaCl, 30% Ficoll. (D) Temperature dependence of the partial heat capacity (Cp) in the absence (dashed line) and in the presence of 30% Ficoll 70 (continuous line). The DSC curve of ribonuclease A is shown for comparison (dotted line). Binding, thermal denaturation, and DSC experiments were performed in 10 mM phosphate buffer, pH 7.0, plus 140 mM NaCl.
Thermal melting and DSC
To better understand the features of the folded states of the C-terminus in crowding agents, we analyzed the thermal melting by IR spectroscopy and the change in partial heat capacity (Cp) with temperature by DSC of C-H1t in 30% Ficoll. The changes in the amide I (D2O) components were monitored in the 20-85°C interval. The α-helix and the β-sheet were lost fairly linearly as temperature was increased; all defined structure was lost by 85°C. Concomitantly with the decrease in α-helix and β-sheet, the random coil component increased from 23% to 56% (Fig. 6 C).
In parallel with the dissipation of secondary structure, the Cp increased almost linearly between 25°C and 85°C, where it leveled off coinciding with the complete dissipation of the α-helix and β-sheet (Fig. 6 D). Cooperatively folded proteins have a significant enthalpy difference between their native and unfolded states, which leads to large heat capacity peaks corresponding to heat absorption on denaturation (46). Thermal melting and Cp measurements indicate that, despite globular compaction and native-like secondary structure, the C-terminus is not cooperatively folded and, instead, has features characteristic of the molten globule state (47–50), which possesses an extensive secondary structure but does not have a fixed tertiary structure.
DISCUSSION
The C-terminal domain of histone H1 is mostly unstructured in dilute solution but folds extensively on interaction with DNA. The C-terminal domain of histone H1 can thus be considered an intrinsically disordered protein (18,19). Some intrinsically disordered proteins may adopt compact conformations in crowded environments (31). The C-terminal domain appears to belong to this category of intrinsically disordered proteins. In the presence of Ficoll 70 or PEG 6000, the C-terminal domain becomes extensively structured with 14–19% α-helix, 22–31% β-sheet, 16–26% turns, and 20–30% loops, depending on the subtype and crowding agent. Folding is associated with the differentiation of a limited hydrophobic core, as shown by the increased ANS binding by the C-terminus in crowded conditions. Interestingly, the high positive charge of the C-terminus (∼40% Lys) does not impair folding in crowded conditions.
The proportions of the secondary structure motifs of the C-terminus in crowded conditions are comparable to those of the DNA-bound domain (17), although the α-helix was proportionally more abundant in the DNA complexes, and the β-sheet more abundant in crowded conditions (Table 2).
TABLE 2.
Percentages of secondary structure of the C-terminal domain in crowded conditions and bound to DNA
| Complexes with DNA*
|
Ficoll
|
PEG
|
||||
|---|---|---|---|---|---|---|
| C-H1° | C-H1t | C-H1° | C-H1t | C-H1° | C-H1t | |
| Turns/310 helix | 33–34 | 40–42 | 24–27 | 22–30 | 16–17 | 24 |
| α-Helix | 24 | 21 | 15 | 15 | 19 | 14 |
| Open loops | 18 | 15 | 23 | 23 | 31 | 27 |
| β-Sheet | 16 | 13 | 27 | 27–31 | 22–28 | 24–30 |
| Random coil | <1 | <1 | <1 | <1 | <1 | <1 |
These data were taken from Roque et al. (17).
Despite the low sequence identity between C-H1° and C-H1t (∼30%), the proportions of their secondary structure motifs appear to be similar, as also occurs in the DNA-bound domains (17). This suggests that selective constraints may act to preserve the secondary structure of the C-terminus in the different subtypes.
The compactness of the C-terminus in the presence and in the absence of crowding agents was examined using SAXS. Global shape changes were analyzed by means of Kratky plots (Q2I(Q) versus Q). The Kratky representation has a characteristic maximum in globular states (native and molten globule). This maximum is absent in nonglobular states (random coil, premolten globule) (20). In the presence of crowding agents, the Kratky plot of the x-ray scattering by the C-terminus of H1t in 30% Ficoll exhibited the bell-shaped appearance typical of globular states. In the absence of crowding agents, the Kratky plot increased monotonously as observed for many unfolded proteins. The decrease of the Rg from 25.0 Å in buffer solution to 14.8 Å in crowding agents confirms the condensation of the C-terminus in crowded conditions.
Analysis of the folded state of the C-terminus in crowding agents by thermal melting and DSC reveals the progressive dissipation of secondary structure in the temperature interval 20–85°C, which is paralleled by a linear increase in partial heat capacity, lacking the heat absorption peak typical of cooperatively folded proteins. These features indicate that the C-terminus in crowded conditions is not cooperatively folded despite native-like secondary structure and compaction and has, instead, the characteristics of the molten globule state (47,51). We have previously shown that on interaction with DNA, the secondary structure of the protein becomes extremely stable and melts cooperatively (17). The transition from the molten-globule state to the DNA-bound cooperatively folded state is probably driven by increased hydrophobicity after charge compensation of the positive charge of the abundant Lys residues of the C-terminus by the DNA phosphates.
The H1 globular domain is thought to be responsible for the location of H1 by interacting with a specific site in the nucleosome. The dependence of the tightening of the C-terminus molten globule structure on charge compensation may afford a mechanism to avoid the interference of the strong electrostatic binding of the C-terminus with binding site recognition by the globular domain because binding of the C-terminus to unspecific sites, such as nucleosomal DNA, already partially charge-compensated by core histones, may lead to partial folding and lower affinity.
The similarity of the proportions of secondary structure motifs of the domain in crowded conditions and bound to DNA suggests that in the presence of crowding agents the structure approximates that of the DNA-bound domain. An advantage related to chromatin dynamics of the condensation of the chain into a native-like structure could be an increased rate of the transition toward the native DNA-bound state (51). The folding of the C-terminus in crowded conditions might also facilitate diffusion of H1 inside cell nuclei. This effect would compensate for the slowing down of protein diffusion by crowding (52,53). Improved diffusion could have functional consequences because H1 molcules exchange in vivo between chromatin binding sites through a soluble intermediate (54).
In summary, the molten globule state of the C-terminus and the tightening of the structure on interaction with DNA could kinetically favor H1 binding and exchange.
Acknowledgments
We thank Dr. J. B. Cladera for providing facilities to perform the DSC measurements. We thank Dr. C. Solans, Dr. J. Esquena, and J. Caelles from IIQAB for their help with the SAXS experiments.
This work was supported by the Ministerio de Educación y Ciencia (Spain) grant BFU2005-02143.
Editor: Ruth Nussinov.
References
- 1.Khochbin, S., and A. P. Wolffe. 1994. Developmentally regulated expression of linker-histone variants in vertebrates. Eur. J. Biochem. 225:501–510. [DOI] [PubMed] [Google Scholar]
- 2.Bouvet, L., S. Dimitrov, and A. P. Wolffe. 1994. Specific regulation of Xenopus chromosomal 5S rRNA gene transcription in vivo by histone H1. Genes Dev. 8:1147–1159. [DOI] [PubMed] [Google Scholar]
- 3.Vermaak, D., O. C. Steinbach, S. Dimitrov, R. A. W. Rupp, and A. P. Wolffe. 1998. The globular domain of histone H1 is sufficient to direct specific gene repression in early Xenopus embryos. Curr. Biol. 8:533–536. [DOI] [PubMed] [Google Scholar]
- 4.Lee, H. L., and T. K. Archer. 1998. Prolonged glucocorticoid exposure dephosphorylates histone H1 and inactivates the MMTV promoter. EMBO J. 17:1454–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dou, Y., C. A. Mizzen, M. Abrams, C. D. Allis, and M. A. Gorovsky. 1999. Phosphorylation of linker histone H1 regulates gene expression in vivo by mimicking H1 removal. Mol. Cell. 4:641–647. [DOI] [PubMed] [Google Scholar]
- 6.Zlatanova, J., P. Caiafa, and K. van Holde. 2000. Linker histone binding and displacement: versatile mechanism for transcriptional regulation. FASEB J. 14:1697–1704. [DOI] [PubMed] [Google Scholar]
- 7.Koop, R., L. Di Croce, and M. Beato. 2003. Histone H1 enhances synergistic activation of the MMTV promoter in chromatin. EMBO J. 22:588–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Izaurralde, E., E. Käs, and U. K. Laemmli. 1989. Highly preferential nucleation of histone H1 assembly on scaffold-associated regions. J. Mol. Biol. 210:573–585. [DOI] [PubMed] [Google Scholar]
- 9.Pennings, S., G. Meersseman, and E. M. Bradbury. 1994. Linker histones H1 and H5 prevent the mobility of positioned nucleosomes. Proc. Natl. Acad. Sci. USA. 91:10275–10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartman, P. G., G. E. Chapman, T. Moss, and E. M. Bradbury. 1977. Studies on the role and mode of operation of the very-lysine-rich histone H1 in eukaryote chromatin. The three structural regions of the histone H1 molecule. Eur. J. Biochem. 77:45–51. [DOI] [PubMed] [Google Scholar]
- 11.Th'ng, J. P. H., R. Sung, M. Ye, and M. J. Hendzel. 2005. H1 family histones in the nucleus. Control of binding and localization by the C-terminal domain. J. Biol. Chem. 280:27809–27814. [DOI] [PubMed] [Google Scholar]
- 12.Hendzel, M. J., M. A. Lever, E. Crawford, and J. P. H. Th'ng. 2004. The C-terminal domain is the primary determinant of histone H1 binding to chromatin in vivo. J. Biol. Chem. 279:20028–20034. [DOI] [PubMed] [Google Scholar]
- 13.Allan, J., T. Mitchel, N. Harborne, L. Bohm, and C. Crane-Robinson. 1986. Roles of H1 domains in determining higher order chromatin structure and H1 location. J. Mol. Biol. 187:591–601. [DOI] [PubMed] [Google Scholar]
- 14.Lu, X., and J. C. Hansen. 2004. Identification of specific functional subdomains within the linker histone H1° C-terminal domain. J. Biol. Chem. 279:8701–8707. [DOI] [PubMed] [Google Scholar]
- 15.Roque, A., M. Orrego, I. Ponte, and P. Suau. 2004. The preferential binding of histone H1 to DNA scaffold-associated regions is determined by its C-terminal domain. Nucleic Acids Res. 32:6111–6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Widlak, P., M. Kalinowska, M. H. Parseghian, X. Lu, J. C. Hansen, and W. T. Garrard. 2005. The histone H1 C-terminal domain binds to the apoptotic nuclease, DNA fragmentation factor (DFF40/CAD) and stimulates DNA cleavage. Biochemistry. 44:7871–7878. [DOI] [PubMed] [Google Scholar]
- 17.Roque, A., I. Iloro, I. Ponte, J. L. R. Arrondo, and P. Suau. 2005. DNA-induced secondary structure of the carboxyl-terminal domain of histone H1. J. Biol. Chem. 280:32141–32147. [DOI] [PubMed] [Google Scholar]
- 18.Bracken, C., L. M. Iakoucheva, P. R. Romero, and A. K. Dunker. 2004. Combining prediction, computation and experiment for the characterization of protein disorder. Curr. Opin. Struct. Biol. 14:570–576. [DOI] [PubMed] [Google Scholar]
- 19.Dyson, H. J., and P. E. Wright. 2005. Intrinsically unstructured proteins and their functions. Nat. Rev. 6:197–208. [DOI] [PubMed] [Google Scholar]
- 20.Uversky, V. N. 2002. What does it mean to be natively unfolded? Eur. J. Biochem. 269:2–12. [DOI] [PubMed] [Google Scholar]
- 21.Uversky, V. N. 2002. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11:739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uversky, V. N. 2003. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: which way to go? Cell. Mol. Life Sci. 60:1852–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ponte, I., R. Vila, and P. Suau. 2003. Sequence complexity of histone H1 subtypes. Mol. Biol. Evol. 20:371–380. [DOI] [PubMed] [Google Scholar]
- 24.Munishkina, L. A., A. L. Fink, and V. N. Uversky. 2004. Conformational prerequisites for formation of amyloid fibrils from histones. J. Mol. Biol. 342:1305–1324. [DOI] [PubMed] [Google Scholar]
- 25.Hansen, J. C., X. Lu, E. D. Ross, and R. W. Woody. 2006. Intrinsic protein disorder, amino acid composition, and histone terminal domains. J. Biol. Chem. 281:1853–1856. [DOI] [PubMed] [Google Scholar]
- 26.Minton, A. P. 2000. Implications of macromolecular crowding for protein assembly. Curr. Opin. Struct. Biol. 10:34–39. [DOI] [PubMed] [Google Scholar]
- 27.van den Berg, B., R. Wain, C. M. Dobson, and R. J. Ellis. 2000. Macromolecular crowding perturbs protein refolding kinetics: implications for folding inside the cell. EMBO J. 19:3870–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qu, Y., and D. W. Bolen. 2002. Efficacy of macromolecular crowding in forcing proteins to fold. Biophys. Chem. 101:155–166. [DOI] [PubMed] [Google Scholar]
- 29.Sasahara, K., P. McPhie, and A. P. Minton. 2003. Effect of dextran on protein stability and conformation attributed to macromolecular crowding. J. Mol. Biol. 326:1227–1237. [DOI] [PubMed] [Google Scholar]
- 30.Flaugh, S. L., and K. J. Lumb. 2001. Effects of macromolecular crowding on the intrinsically disordered proteins c-Fos and p27 (Kip1). Biomacromolecules. 2:538–540. [DOI] [PubMed] [Google Scholar]
- 31.Dedmon, M. M., C. N. Patel, G. B. Young, and G. J. Pielak. 2002. FlgM gains structure in living cells. Proc. Natl. Acad. Sci. USA. 99:12681–12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uversky, V. N., M. E. Cooper, K. S. Bower, J. Li, and A. L. Fink. 2002. Accelerated alpha-synuclein fibrillation in crowded milieu. FEBS Lett. 515:99–103. [DOI] [PubMed] [Google Scholar]
- 33.Shtilerman, M. D., T. T. Ding, and P. T. Lansbury, Jr. 2002. Molecular crowding accelerates fibrillization of alpha-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson's disease? Biochemistry. 41:3855–3860. [DOI] [PubMed] [Google Scholar]
- 34.Chen, Y. J., J. T. Yang, and K. H. Chau. 1974. Determination of the helix and beta form of proteins in aqueous solution by circular dichroism. Biochemistry. 13:3350–3359. [DOI] [PubMed] [Google Scholar]
- 35.Arrondo, J. L. R., and F. M. Goñi. 1999. Structure and dynamics of membrane proteins as studied by infrared spectroscopy. Prog. Biophys. Mol. Biol. 72:367–405. [DOI] [PubMed] [Google Scholar]
- 36.Vila, R., I. Ponte, M. Collado, J. L. R. Arrondo, and P. Suau. 2001. Induction of secondary structure in a COOH-terminal peptide of histone H1 by interaction with the DNA: an infrared spectroscopy study. J. Biol. Chem. 276:30898–30903. [DOI] [PubMed] [Google Scholar]
- 37.Guinier, A., and G. Fournet. 1955. Small Angle Scattering of X-Rays. John Wiley & Sons, New York.
- 38.Debye, P. 1947. Molecular-weight determination by light scattering. J. Phys. Colloid Chem. 51:18–32. [DOI] [PubMed] [Google Scholar]
- 39.Calmettes, P., D. Durand, M. Desmadril, P. Minard, V. Receveur, and J. C. Smith. 1994. How random is a highly denatured protein? Biophys. Chem. 53:105–113. [DOI] [PubMed] [Google Scholar]
- 40.Glatter, O., and O. Kratky. 1982. Small Angle X-Ray Scattering. Academic Press, London.
- 41.Semisotnov, G. V., H. Kihara, N. V. Kotova, K. Kimura, Y. Amemiya, K. Wakabayashi, I. N. Serdyuk, A. A. Timchenko, K. Chiba, K. Nikaido, T. Ikura, and K. Kuwajima. 1996. Protein globularization during folding. A study by synchrotron small-angle x-ray scattering. J. Mol. Biol. 262:559–574. [DOI] [PubMed] [Google Scholar]
- 42.Pérez, J., P. Vachette, D. Russo, M. Desmadril, and D. Durand. 2001. Heat-induced unfolding of neocarzinostatin, a small all-beta protein investigated by small-angle x-ray scattering. J. Mol. Biol. 308:721–743. [DOI] [PubMed] [Google Scholar]
- 43.Millett, I. S., S. Doniach, and K. W. Plaxco. 2002. Toward a taxonomy of the denatured state: small angle scattering studies of unfolded proteins. Adv. Protein Chem. 62:241–262. [DOI] [PubMed] [Google Scholar]
- 44.Goto, Y., and A. L. Fink. 1989. Conformational states of beta-lactamase: molten-globule states at acidic and alkaline pH with high salt. Biochemistry. 28:945–952. [DOI] [PubMed] [Google Scholar]
- 45.Semisotnov, G. V., N. A. Rodionova, O. I. Razgnlyaev, V. N. Uversky, A. F. Gripas, and R. I. Gilmanshin. 1991. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 31:119–128. [DOI] [PubMed] [Google Scholar]
- 46.Privalov, L. P., and S. J. Gill. 1988. Stability of protein structure and hydrophobic interaction. Adv. Protein Chem. 39:191–234. [DOI] [PubMed] [Google Scholar]
- 47.Griko, Y. V., and P. L. Privalov. 1994. Thermodynamic puzzle of apomyoglobin unfolding. J. Mol. Biol. 235:1318–1325. [DOI] [PubMed] [Google Scholar]
- 48.Peng, Z. Y., and P. S. Kim. 1994. A protein dissection study of a molten globule. Biochemistry. 33:2136–2141. [DOI] [PubMed] [Google Scholar]
- 49.Redfield, C., R. A. G. Smith, and C. M. Dobson. 1994. Structural characterization of a highly-ordered ‘molten globule’ at low pH. Nat. Struct. Biol. 1:23–29. [DOI] [PubMed] [Google Scholar]
- 50.Ptitsyn, O. B. 1995. Molten globule and protein folding. Adv. Protein Chem. 47:83–229. [DOI] [PubMed] [Google Scholar]
- 51.Privalov, L. P. 1996. Intermediate states in protein folding. J. Mol. Biol. 258:707–725. [DOI] [PubMed] [Google Scholar]
- 52.Muramatsu, N., and A. P. Minton. 1988. Tracer diffusion of globular proteins in concentrated protein solutions. Proc. Natl. Acad. Sci. USA. 85:2984–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou, H. X. 2004. Protein folding and binding in confined spaces and in crowded solutions. J. Mol. Recognit. 17:368–375. [DOI] [PubMed] [Google Scholar]
- 54.Lever, M. A., J. P. H. Th'ng, X. Sun, and M. J. Hendzel. 2000. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature. 408:873–876. [DOI] [PubMed] [Google Scholar]