

Abstract
In mammalian cells, the Ku heterodimer is involved in DNA double-strand-break recognition and repair. We have established in yeast a connection between Ku activity and DNA double-strand-break damage repair, and a connection between Ku activity and commitment to DNA replication. We generated double-stranded DNA breaks in yeast cells in vivo by expressing a restriction endonuclease and have shown that yeast mutants lacking Ku p70 activity died while isogenic wild-type cells did not. Moreover, we have discovered that DNA damage occurs spontaneously during normal yeast mitotic growth, and that Ku functions in repair of this damage. We also observed that mitotically growing Ku p70 mutants have an anomalously high DNA content, suggesting a role for Ku in regulation of DNA synthesis. Finally, we present evidence that Ku p70 function is conserved between yeast, Drosophila, and humans.
The Ku protein complex was first identified as an autoimmune antigen recognized by antisera from patients with polymyositis-scleroderma overlap syndrome (1). Further investigation showed Ku to be a heterodimer composed of 70-kDa and 80-kDa subunits that are localized in the nucleus (2). Originally, results of in vitro studies suggested that Ku has a role in DNA repair and transposition because the Ku complex displays sequence-independent double-strand DNA end-binding activity (3). Ku also binds to DNA nicks, gaps, and hairpins (4–7). The participation of Ku in double-strand DNA break repair, recombination, and transposition has since been documented (2, 8–12).
Mutant cultured hamster cell lines lacking Ku function have an increased sensitivity to ionizing radiation (IR) (ref. 13 and references therein). In addition to IR sensitivity, the cell lines are defective in double-strand-break repair and immunoglobulin V(D)J recombination (14–16). These DNA repair and recombination deficiencies as well as the IR sensitivity can be complemented by expressing the human gene encoding the Ku p70 or p80, depending on the cell line (refs. 8 and 9 and references therein). The Drosophila Ku p70 homologue is involved in the repair of DNA breaks formed during transposition of P transposable elements and is also involved in repair of DNA damage induced by the x-ray mimetic methyl methanesulfonate (MMS) (17). The mammalian Ku heterodimer has been shown to be a component of the double-stranded DNA-dependent protein kinase activity. The Ku p70 and p80 subunits confer the DNA dependence to the catalytic subunit (DNA PKcs) of the kinase (18–20).
The Ku proteins appear to be expressed in all eukaryotic cells, suggesting they play a fundamental role in general DNA metabolism. DNA binding activity attributed to homologues of the Ku p70 and p80 proteins has been found in organisms as diverse as Saccharomyces cerevisiae, mammals, and Drosophila melanogaster (10, 21, 22). The p80 homologues have been cloned from mammals and yeast and are 21% identical to each other (23). Mammalian, yeast, and Drosophila Ku p70 homologues have all been cloned. These Ku p70 homologues are also only 17–22% identical to each other (10). The low degree of sequence identity of the Ku p70 and Ku p80 homologues from these distantly related organisms raises questions about the conservation of Ku function in these organisms.
In this paper, we evaluated the conservation of function between the Ku proteins from humans, yeast, and Drosophila and tested whether yeast Ku function was required for DNA double-strand-break repair. In addition, we explored the impact that loss of Ku p70 function had on cell-cycle progression and the surveillance of the integrity of the genome.
MATERIALS AND METHODS
Yeast Methods.
Media for yeast growth and sporulation, and methods for mating, sporulation, and tetrad analysis, were as described (24, 25). Tetrads were dissected onto rich medium (yeast extract/peptone/dextrose) plates and grown at 20°C. The transformation of yeast with DNA was performed by the lithium acetate procedure as described (26).
Strains and Plasmids.
The following strains were used in this work: BRY1, MATa, his3Δ200, lys2-801, leu2-3,112, ura3-52, hdf1Δ1::LEU2; BRY2, MATa, ade2-101, ade3-130, ura3-52, leu2-3,112, hdf1Δ1::LEU2; BRY3, MATa, ade2-101, ade3-130, ura3-52, leu2-3,112; BRY4, MATα, his3D200, leu2-3,112, lys2-801, ura3-52; BRY5, MATα, rad9Δ::URA3, hdf1Δ1::LEU2, ade2-101, his3, lys2; and BRY6, MATa, rad9Δ::URA3, his3, leu2-3, 112, trp1-289, ura3-52. The following plasmids were used in this work: YCpGalRIb (27); pRS316ADE3YKu [pRS316 (28) containing the ADE3 gene and the HDF1 gene]; pRB1438 (pRS316 containing the GAL1/10 promoter element; from D. Botstein, Stanford University); pRB1438IRBP (pRB1438 containing the Drosophila IRBP gene); pCH37 [pRS313 (28) containing the GAL1/10 promoter element; from D. Drubin, University of California, Berkeley); pCH37HuKu [pCH37 containing the human Ku p70 cDNA (29)]; pCH37IRBP [pCH37 containing the Drosophila IRBP gene (10)]; YCp50GalHO (R. Jensen and I. Herskowitz, University of California, San Francisco). The following plasmids were a generous gift from Jasper Rine (University of California, Berkeley). All are based on a derivative of YEplac195, a 2-micron, URA3 plasmid-based vector into which different yeast promoters are fused to green fluorescent protein (GFP) coding sequence: pACA60 (RNR2 promoter), pACA74 (GPD1 promoter), pACA75 (RNR3 promoter), pACA84 (STE2 promoter), pACA102 (ADH1 promoter), pACA103 (ADH2 promoter), pACA119 (HIS4 promoter).
Construction of the hdf1 Deletion Allele.
The hdf1Δ1 deletion allele was constructed by the following method. Two DNA fragments were generated by conventional PCR using primers homologous to the HDF1 gene. Fragment 1 encompasses DNA starting 20 bp 5′ of the HDF1 ATG and extending 510 bp 5′. Fragment 2 encompasses DNA starting 4 bp 3′ of HDF1 stop codon and extending 563 bp 3′. Also by standard PCR techniques, XbaI and EcoRI restriction endonuclease sites were placed onto the 5′ end and 3′ ends, respectively, of fragment 1; BamHI and XhoI restriction endonuclease sites were placed onto the 5′ end and 3′ ends, respectively, of fragment 2. In a four-way ligation, the DNA sequences of fragments 1 and 2 were ligated onto the 5′ and 3′ ends, respectively, of a DNA fragment containing the LEU2 gene and the restriction sites at the 5′ and 3′ ends of the original fragments 1 and 2 (XbaI and XhoI) were used to ligate this new DNA fragment into the vector pHSX (30). The LEU2 gene flanked by EcoRI and BamHI restriction sites at the 5′ and 3′ ends was synthesized by conventional PCR using the pRS415 plasmid (28) as the source of the LEU2 coding information. This four-way ligation generated plasmid pHSX-YKuLEU2 from which a linear piece of DNA containing the LEU2 gene flanked 5′ by 510 bp and flanked 3′ by 563 bp of the chromosomal DNA that normally flanks the HDF1 gene was obtained by cleaving the plasmid with the restriction enzyme NotI. This linear piece of DNA was used to transform diploid cells and disrupt the wild-type HDF1 gene (31), yielding a strain heterozygous for the hdf1Δ1 null allele and the wild-type HDF1 allele. Leu+ colonies were selected and then verified for disruption at the HDF1 locus by DNA blot hybridization and PCR analysis (data not shown).
Fluorescence Microscopy.
Fluorescence microscopy was performed as described (32), using anti-α-tubulin antibody, YOL1/34 (33) and 4′,6-diamino-2-phenylindole (DAPI).
Measurement of DNA Content.
Cellular DNA was stained with propidium iodide as described (34), and DNA content was determined with a Becton Dickinson FACScan flow cytometer. At least 20,000 cells were counted per time point. When necessary, yeast cells were sorted by DNA content using a Becton Dickinson FACStarPlus machine. The cells collected from a given DNA content were then analyzed by light microscopy to determine their morphology.
GFP Quantitation.
The promoter/GFP fusion plasmids were transformed into BRY2 and BRY3 cells. The transformation reaction was plated onto supplemented minimal plates and incubated at 20°C. The GFP level of all transformants (50 to 100 colonies on average) present on the plate was quantitated using the FluorImagerSI (Molecular Dynamics).
α-Factor Arrest.
The α-factor arrest experiments were done by incubating exponential phase cells at 20°C in medium containing 3 μM α-factor for 6 h. The cells were then processed for fluorescence-activated cell sorter (FACS) analysis as described above.
RESULTS
The hdf1Δ1 Null Strain Exhibited Temperature-Sensitive Growth and a Phenotypic Lag.
In an effort to determine the exact biological function(s) of Ku, we took advantage of yeast genetics and cell biology and evaluated our hdf1Δ1 null mutant, which has a complete deletion of the HDF1 gene encoding the yeast Ku p70 homologue (21). A diploid strain heterozygous for the hdf1Δ1 deletion was sporulated at 20°C. All four spores from each of 30 tetrads were able to grow, demonstrating that a true hdf1 null was viable. Spores that inherited the complete hdf1Δ deletion were temperature-sensitive for growth. These cells grew as well as isogenic wild-type cells at 20°C, were impaired for growth at 30°C, and died at 37°C. The nongrowth at the nonpermissive growth temperature (37°C) was in agreement with the results of an earlier study using a partial hdf1 deletion mutant (21). However, in addition, we observed a phenotypic lag in the arrest phenotype. Cells with an hdf1Δ1 deletion mutation could form colonies on solid medium at 37°C, but the cells in these colonies were inviable and would not grow after being replica-plated to fresh medium at 37°C (see Fig. 1A). Assays of viability of cells grown in liquid culture at the permissive temperature, 20°C, and then shifted to 37°C revealed that the viability of the cells incubated at 37°C remained high for several generation times, but by 24 h viability had dropped to 20%. This finding is consistent with the phenotypic lag observed on plates.
Figure 1.

Replica plating experiment showing the phenotypic lag of the temperature-sensitive growth of the hdf1 null strain, BRY1, at 37°C. BRY1 was transformed with (A) the vector (pCH37), (B) the HDF1 gene on a plasmid, or (C) the pCH37/IRBP (Drosophila Ku p70 cDNA) plasmid. Transformants were selected on medium containing glucose and then streaked to medium containing galactose and incubated at 37°C (plate on left). After 3 days, the plate was replica plated to a fresh galactose plate and the second plate was incubated at 37°C (plate on right).
The hdf1Δ1 null cells exhibited a previously unreported phenotype. These cells showed a complex terminal arrest phenotype at 37°C. At the restrictive temperature, the hdf1Δ1 null strain arrested as an almost equal mixture of large-budded cells and unbudded cells (Table 1). Antitubulin immunofluorescence microscopy and DAPI DNA-staining of hdf1Δ1 null cells revealed that the large-budded cells arrested with a short spindle and a single nucleus at the neck between mother cell and daughter cell indicative of a G2/M arrest (Fig. 2). The unbudded cells in the arrested population also had a single nucleus and had a microtubule astral array typical of cells in the G1 stage of the cell cycle. Therefore, by cell morphology and nuclear and microtubule organization, the population of cells had arrested at two positions in the cell cycle, half in G1 and the other half in G2/M (i.e., as unbudded and large-budded cells, respectively).
Table 1.
Phenotypes of Ku-deficient yeast cells
| Strain and temperature | Large bud,* % | No bud,* % | Small bud,* % | Viability,† % | Large budded cells
|
|
|---|---|---|---|---|---|---|
| 1 nuc. | 2 nuc. | |||||
| BRY3 (HDF1) | ||||||
| 20°C | 24 | 42 | 34 | 99 | 12 | 88 |
| 37°C | ||||||
| 1 h | 18 | 53 | 29 | ND | ND | ND |
| 4 h | 21 | 42 | 37 | 94 | ND | ND |
| 6 h | 29 | 40 | 31 | 92 | 14 | 86 |
| 24 h | 4 | 75 | 21 | 73 | ND | ND |
| BRY2 (hdf1Δ1) | ||||||
| 20°C | 33 | 25 | 42 | 97 | 22 | 77 |
| 37°C | ||||||
| 1 h | 37 | 30 | 33 | ND | ND | ND |
| 4 h | 43 | 23 | 34 | 90 | ND | ND |
| 6 h | 50 | 24 | 25 | 97 | 92 | 8 |
| 24 h | 50 | 43 | 7 | 21 | 97 | 3 |
| BRY5 (rad9Δ, hdf1Δ1) | ||||||
| 20°C | 35 | 43 | 22 | 97 | 9 | 91 |
| 37°C, 23 h | 24 | 56 | 20 | 14 | 19 | 81 |
| BRY6 (rad9Δ) | ||||||
| 20°C | 42 | 30 | 28 | 98 | ND | ND |
| 37°C 23 h | 23 | 53 | 24 | 82 | 16 | 84 |
| BRY1 + YCpGalRI (hdf1Δ1) | ||||||
| 20°C galactose, 0 h | 16 | 47 | 37 | 98 | ||
| 20°C galactose, 40 h | 46 | 44 | 10 | —‡ | ||
ND, not determined.
On average, 1000 cells were counted.
Viability was determined by counting cells using a Coulter Counter and then plating cells solid growth medium.
No growth.
Figure 2.

Immunofluorescence analysis of the hdf1Δ null (BRY2) cells incubated at 37°C for 8 h. (Upper) Antitubulin staining. (Lower) The same cells with DAPI staining to localize the nucleus. (Bar = 5 μm.)
The Morphologic Arrest Phenotype Was Rad9p-Dependent.
This arrest phenotype, characterized by microtubule morphology and by the presence of a single nucleus, was that expected of a cell sustaining DNA damage and triggering G1/S and G2/M cell-cycle check points, such as those controlled by RAD9 (35–38). To determine whether the terminal arrest phenotype of the hdf1Δ1 null cells was indeed Rad9p-dependent, a double-mutant strain (BRY5) containing the rad9 deletion mutation and the hdf1Δ1 deletion mutation was constructed. This double mutant died at 37°C with the same phenotypic lag kinetics as the RAD9+, hdf1Δ1 single mutant strain (BRY2). However, the double-mutant cells died as a morphologically heterogeneous population of cells distributed throughout the cell cycle (Table 1). In addition, at the nonpermissive temperature, the large-budded cells of the double-mutant strain (rad9Δ, hdf1Δ1), unlike those of RAD9+, hdf1Δ1 strain, were characterized by nuclei that were divided and segregated between the mother and daughter cells (Table 1). The rad9 deletion itself does not cause a temperature-sensitive lethality (Table 1, BRY6; and refs. 35 and 37). These results suggested that hdf1Δ1 cells sustained DNA damage at 37°C and triggered a Rad9p-dependent G1 or G2 checkpoint arrest.
DNA Damage-Inducible Genes Are Activated in hdf1Δ1 Deletion Mutants.
If hdf1Δ1 null mutants have elevated levels of DNA damage, then the transcription from DNA damage-inducible genes, such as RNR2 and RNR3 (39, 40), should be elevated relative to their levels in wild-type cells. We analyzed the transcriptional activity from both the RNR2 and RNR3 promoters in hdf1Δ1 and isogenic wild-type strains. To monitor this activity, we used constructions in which the RNR2 and RNR3 promoters regulate the transcription of the GFP coding sequence in yeast (see Materials and Methods). As shown in Fig. 3, there was approximately a 4-fold and a 5-fold transcriptional induction of the RNR2 promoter and RNR3 promoter, respectively, in hdf1Δ1 cells relative to wild-type cells. Each of five control plasmids expressing GFP from non-DNA damage-inducible promoters showed approximately the same level of transcription in hdf1Δ1 null mutant and wild-type cells (Fig. 3). Interestingly, the RNR2 and RNR3 promoters had the same transcriptional induction in hdf1Δ1 cells relative to wild-type cells at the permissive growth temperature (20°C) and the nonpermissive growth temperature (37°C).
Figure 3.

Relative transcriptional activity in hdf1Δ null and wild-type strains as monitored by GFP activity. GFP activity expressed in yeast by seven different promoter fusion constructions. The promoter tested in each case is listed at the base of a pair of GFP levels. GFP levels have been normalized to the wild-type level for each promoter fusion construction. GFP levels in mutant hdf1Δ null cells (BRY2) are shown in striped boxes, and GFP levels in wild-type cells (BRY3) are shown in solid boxes. Identical results were obtained at 20°C and 37°C. Standard deviation for each data set was 10% or less.
Ku-Deficient Yeast Had Anomalous DNA Content.
FACS analysis of DNA content of haploid hdf1Δ1 null cells incubated at the permissive temperature of 20°C or at the nonpermissive temperature of 37°C revealed a new dimension of Ku-deficient cells not evident by morphological measures. At the permissive temperature (20°C), the majority of the hdf1Δ1 haploid cells in an exponentially growing population have a 2N DNA content (Fig. 4B), despite only ≈33% of the population being large-budded (Table 1). By cell sorting (see Materials and Methods), we determined that the composition of the 2N DNA content peak (Fig. 4B) was 60% large budded, 20% unbudded, and 13% small budded.
Figure 4.

FACS analysis of (A) wild-type cells (BRY3) growing exponentially at 20°C, (B) hdf1Δ null cells (BRY2) growing exponentially at 20°C, (C) hdf1Δ null cells shifted to 37°C and incubated for 3 h, and (D) hdf1Δ null cells shifted to 37°C and incubated for 22 h. The y-axis shows relative number of cells, and the x-axis shows relative DNA content.
Moreover, at the nonpermissive temperature (37°C), when the cells have terminally arrested for growth as ≈50% unbudded and 50% large-budded cells (Table 1), 50% of the population arrested with a DNA content of 2N and the rest with a DNA content of 4N (Fig. 4D), even though the cells were originally haploid. By cell sorting (see Materials and Methods), we were able to determine that the 2N DNA peak was composed of 70% unbudded cells and 30% large-budded cells, and that the 4N DNA peak was composed of 85% large-budded cells, 13% unbudded cells, and 2% small-budded cells. Therefore, at the terminal arrest, the majority of unbudded cells had a 2N DNA content and the majority of large-budded cells had a 4N DNA content.
The FACS results described above could not be explained by over replication of mitochondrial DNA because isogenic rho° hdf1Δ1 strains gave exactly the same profile as rho+ strains (data not shown).
Ku-Deficient Cells Arrest in α-Factor with 1N DNA.
To determine whether the originally haploid hdf1Δ1 null cells described above ever enter a normal haploid G1 cell-cycle stage with 1N DNA content, we analyzed these cells in two independent ways. First, we determined genetically whether the hdf1Δ1 null cells behaved as normal haploid cells. We found that they mated normally with haploid cells of the opposite mating type to generate diploid cells. We examined 10 independent diploid cells; upon sporulation, these diploids showed normal 2:2 segregation of all genetic markers present in the cross (data not shown). Triploid yeast (which would result from crossing a 2N strain to a 1N strain) would not show 2:2 segregation of markers and would have poor viability. Therefore, hdf1Δ1 cells, like normal cells, arrest during mating with a 1N DNA content. Second, we arrested hdf1Δ1 mutant cells and isogenic wild-type cells with α-factor and carried out FACS analysis. We found that the mutant hdf1Δ1 population which exhibited the 2N DNA content in exponential growth, arrested with 1N DNA content when treated with α-factor, and with exactly the same FACS profile as isogenic HDF1+ cells (Fig. 5). Thus hdf1Δ1 null cells, despite being mostly 2N during mitotic growth, enter G1 with a 1N DNA content.
Figure 5.

Effect of α-factor on wild-type and Ku p70-deficient yeast. FACS profiles of wild-type and hdf1Δ null cells were incubated at 20°C before (A and C) and after (B and D) 6-h treatment with α-factor. Wild-type profiles are shown on the left (A and B); hdf1Δ null profiles on right (C and D). B and D show the 6-h α-factor arrest FACS profile in black superimposed on the FACS profiles from A and C, respectively, in gray.
hdf1 Null Cells Were Sensitive to Double-Stranded DNA Breaks.
The RNR2 and RNR3 promoter induction results above showed that DNA damage during mitotic growth at 20°C and at 37°C is elevated in the absence of Ku activity. We therefore tested whether Ku p70 function(s) was essential for repairing double-strand broken DNA molecules. To do this, we expressed the bacterial EcoRI restriction endonuclease in hdf1Δ1 null cells and isogenic HDF1 wild-type cells by transforming the cells with a plasmid that expresses the bacterial EcoRI endonuclease gene from the galactose-inducible promoter of the GAL1 gene (27). Previous studies using this plasmid have demonstrated that the EcoRI endonuclease is not expressed on medium containing glucose, but is expressed on medium containing galactose. Furthermore, the EcoRI endonuclease expressed from this plasmid generates double-strand breaks in chromosomal DNA at EcoRI recognition sites throughout the genome, but the cells remain viable if they retain the ability to repair these breaks (27). The hdf1Δ1 null cells containing the EcoRI endonuclease-expressing plasmid are able to grow normally on glucose-containing medium at 20°C, but were unable to grow at 20°C on medium containing galactose which induces EcoRI expression (Fig. 6A). Isogenic HDF1 wild-type cells containing the EcoRI-expressing plasmid were able to grow on medium containing galactose (Fig. 6C). Finally, hdf1Δ1 null cells containing a control plasmid were able to grow on medium containing galactose (Fig. 6B).
Figure 6.
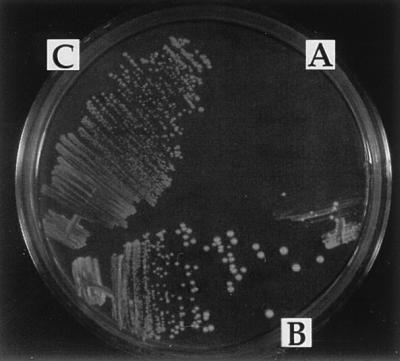
EcoRI endonuclease expression experiment. All strains were grown at 20°C on medium containing glucose and then streaked onto medium containing galactose and incubated at 20°C. (A) hdf1Δ null strain (BRY1) transformed with the EcoRI-producing plasmid YCpGalRIb. (B) hdf1Δ null strain (BRY1) transformed with the control vector. (C) Isogenic wild-type cells (BRY4) transformed with the EcoRI plasmid, YCpGalRIb.
The terminal arrest morphology of a population of hdf1Δ1 null cells expressing the EcoRI endonuclease in vivo, at the permissive temperature (20°C), was exactly the same as the temperature-sensitive arrest phenotype. That is, the culture of hdf1Δ1 null cells expressing the EcoRI endonuclease arrested as a mixture of ≈50% unbudded cells (G1 cells) and 50% large-budded cells (G2/M cells) (Table 1).
In a similar experiment, we transformed our hdf1Δ1 null haploid cells with a plasmid expressing the HO endonuclease under the control of the GAL1 promoter. The hdf1Δ1 null cells incubated at 20°C were not sensitive to the single double-strand break at the MAT locus caused by the HO endonuclease. We confirmed that these cells expressed HO endonuclease because they were able to complete the steps involved in mating type switching, forming zygotes and diploids, (data not shown).
Finally, in contrast to the significant sensitivity differences of a rad52 mutant and a RAD52 wild-type cell to standard DNA damaging agents such as MMS, UV, or ionizing radiation, we and others have determined that hdf1 mutant cells at their permissive growth temperature (20°C) are not dramatically more sensitive than are isogenic wild-type cells to these standard DNA damaging agents Although at the semirestrictive temperature of 30°C, there is a moderate MMS effect on hdf1 mutants (refs. 8 and 41–43; unpublished data).
Human and Drosophila Ku p70 Homologues Complement the Yeast hdf1 Null Mutation.
The Ku p70 homologues from D. melanogaster and human are ≈22% identical to each other and 18% and 17% identical, respectively, to the yeast Ku p70 encoded by the HDF1 gene (10). We investigated whether and to what extent the human and Drosophila Ku p70 Hdf1p homologues would function in yeast cells. Specifically, we tested whether the human and Drosophila Ku p70 homologues could complement the temperature-sensitive growth defect of the hdf1 null strain.
The cDNA for the Drosophila Ku p70 gene, IRBP (10), and one for the human gene (29) were each placed under the control of the galactose-inducible GAL1 promoter (see Materials and Methods). hdf1Δ null cells were transformed separately with these plasmids and also with the control plasmid vector, pCH37. The transformants, selected on medium containing glucose, were picked, restreaked, and allowed to grow into single colonies. The isolated single colonies were then tested for their ability to grow on medium containing galactose at 37°C. The cells transformed with either the Drosophila IRBP cDNA or the human Ku p70 cDNA were able to grow at 37°C (Figs. 1C and Fig. 7). These cells did not show a phenotypic lag and they remained viable. In contrast, cells containing the control vector plasmid were not able to grow (Figs. 1A and 7A). Thus the human and Drosophila proteins were able to function in yeast cells.
Figure 7.
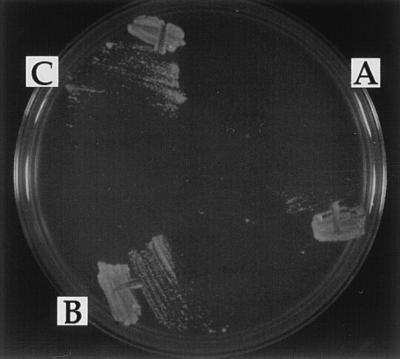
Complementation of the hdf1Δ null strain temperature-sensitive growth defect. (A) hdf1Δ null strain (BRY1) transformed with the control vector pCH37. (B) BRY1 transformed with pCH37/IRBP. (C) BRY1 transformed with pCH37/HuKu. Cells were grown at 20°C and then streaked onto medium containing galactose and incubated at 37°C.
DISCUSSION
The results from this study establish new dimensions to the function of the Ku p70 protein homologue encoded by the HDF1 gene. First, several lines of in vivo evidence showed that DNA damage occurred during normal mitotic growth and that Ku p70 was required for the in vivo repair of this damage. These conclusions are based on the following results. The hdf1Δ1 mutant had elevated levels of DNA damage (at both the permissive and nonpermissive growth temperatures) when compared with wild-type (HDF1+) cells, evident by the elevated transcriptional activity of the DNA damage-inducible genes, RNR2 and RNR3. In addition, the mutant had a temperature-sensitive terminal arrest phenotype (an equal mixture of unbudded cells and large-budded cells) at the nonpermissive temperature that was dependent on Rad9p function. The terminal arrest dependency on Rad9p suggested that the hdf1Δ1 null mutant died as a result of DNA damage. These results also suggest that the hdf1Δ1 mutant may be deficient for repair of damaged DNA; a suggestion we have confirmed. We found that Ku p70 was required for repair of double-strand DNA breaks because the hdf1 null mutants were sensitive to double-strand DNA breaks caused in vivo by the EcoRI endonuclease, whereas the HDF1+ wild-type cells are not sensitive. Finally, the population of EcoRI-expressing hdf1 null cells had the same terminal arrest morphology as the Rad9p-dependent terminal arrest morphology of hdf1 null cells incubated at 37°C. It should also be noted that these EcoRI lethality induction results are in agreement with the results of plasmid ligation assays in hdf1 mutants (23, 43).
The second novel dimension to Ku function not previously observed relates to DNA replication and was evident by the apparently high DNA content of the hdf1Δ1 mutants during mitotic growth. Because each hdf1Δ1 cell entered G1 with 1N DNA, yet the population is primarily 2N DNA, the fraction of the cell cycle spent as 1N was shorter in hdf1Δ1 mutants than in wild-type cells. The simplest explanation would be that the timing of replication was advanced relative to the beginning of G1 resulting in or resulting from a compression of G1 in the hdf1Δ1 mutants. This conclusion was supported by the morphological data which showed that a large fraction of cells in the 2N peak at the permissive growth temperature were unbudded. Thus it would appear that S phase was completed before bud emergence in a large fraction of the cells. If so, in the hdf1Δ1 mutant, the morphological cell division cycle and the DNA replication cycle have been uncoupled. Analysis of cells at the nonpermissive temperature (37°C) revealed an added complexity. The unbudded cells arrested with 2N DNA, but a majority of large-budded cells arrested with 4N DNA (or higher). At present, we are reluctant to offer an interpretation of these 4N cells as this phenotype was observed in a population of mostly dead cells. Additional study is needed to learn whether the 4N peak observed at the nonpermissive temperature results from endoreduplication, further replication, or some other phenomenon.
The 2N DNA content of the Ku-deficient mutant at the permissive temperature could explain why a phenotype expected for Ku-deficient yeast cells was not seen: a dramatic sensitivity to DNA damaging agents like MMS or ionizing radiation. This phenotype was expected of Ku-deficient yeast because it is seen with mammalian and Drosophila Ku-deficient cells (13, 17). The 2N DNA content of most haploid hdf1Δ1 null cells creates an opportunity for the Rad52-dependent homologous recombination repair pathway to repair the DNA damage and effectively mask most of the sensitivity of the hdf1 null mutant to damage caused by MMS or ionizing radiation. Indeed, the rad52, hdf1 double mutants have heightened MMS and ionizing radiation sensitivities compared with either single mutant alone (23, 41, 43). In contrast, in mammalian cells, direct break-fusion processes rather than homologous recombination are the predominant pathway for DNA repair (8, 9). Therefore, Ku-deficient mammalian cells would still die upon treatment with agents that cause random breaks in DNA independent of their DNA content or ploidy.
The 2N content of the hdf1 mutant could save the yeast cell from MMS-induced DNA damage but would not be expected to save it from EcoRI-induced damage. This is because MMS damage is randomly distributed in the genome and therefore, a lesion in one chromatid would not be expected to occur at exactly the same location on the sister chromatid. Therefore, the Rad52-dependent repair pathway would be able to repair the random MMS damage using the wild-type information of the duplicate chromosome. In contrast, the site-specific nature of restriction enzymes ensures that breaks would occur at exactly the same position on both sister chromatids, precluding recombination-based repair mechanisms. In the case of the HO endonuclease experiment, wild-type information is still available from the HML or HMR loci to repair the damage at the MAT locus.
When considering the temperature-sensitive phenotype of the hdf1Δ1 mutant, it was not immediately obvious why a null mutant would be temperature sensitive for growth rather than dead at all temperatures. There are at least two different ways to explain this phenotype. It is likely that DNA damage occurs equally frequently in the hdf1Δ1 null mutant at low and high temperatures because the RNR2 and RNR3 promoter-driven GFP transcription was induced to the same levels in the hdf1Δ1 null cells at the permissive temperature (20°C) or at the nonpermissive temperature (37°C). One possibility is a thermolabile repair function that is redundant with Ku p70 in yeast cells. A second possibility is that the shorter cell-cycle time at 37°C (90 min) relative to 20°C (4–5 h) allows less time for the redundant function to repair the damage. In either case, it appears that replication of damaged DNA ultimately causes the death of the cell at 37°C. The phenotypic lag of the temperature sensitivity implied that within a single cell cycle, DNA damage occurred sufficiently infrequently or is repaired efficiently enough, that multiple cell divisions must occur before a substantial fraction of the population accumulates lethal damage.
Finally, our data established that Ku function has been conserved during evolution because the human and Drosophila Ku p70 proteins can complement the temperature-sensitive phenotype of the hdf1 null mutant. This suggests that lessons learned about Ku p70 in yeast apply to all eukaryotes.
Acknowledgments
We are indebted to David Drubin and Jasper Rine for their encouragement and support of this work. We also gratefully acknowledge C. Chan, D. Drubin, J. Rine, M. Botchan, K. Collins, P. Kaufman, N. Machin, J. Lee, A. Dillon, and E. Beall for numerous discussions and critical readings of the manuscript. We are also indebted to William Huyn (Director of the University of California, San Francisco, Laboratory for Cell Analysis) for assistance with flow cytometry experiments. We also thank the reviewers for helpful comments. This work obtained Pilot Project support from a National Institute on Environmental Health Sciences Mutagenesis Center Grant (B. Ames) and was also supported by grants from the National Institutes of Health GM-47842 (G.B.) and GM-48862 (D.R.).
Footnotes
Abbreviations: MMS, methyl methanesulfonate; GFP, green fluorescent protein; DAPI, 4′,6-diamino-2-phenylindole; FACS, fluorescence-activated cell sorter.
References
- 1.Mimori T, Akizuki M, Yamagata H, Inada S, Yoshida S, Homma M. J Clin Invest. 1981;68:611–620. doi: 10.1172/JCI110295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mimori T, Hardin J A, Steitz J A. J Biol Chem. 1986;261:2274–2278. [PubMed] [Google Scholar]
- 3.Mimori T, Hardin J A. J Biol Chem. 1986;261:10375–10379. [PubMed] [Google Scholar]
- 4.Griffith A J, Blier P R, Mimori T, Hardin J A. J Biol Chem. 1992;267:331–338. [PubMed] [Google Scholar]
- 5.Paillard S, Strauss F. Nucleic Acids Res. 1991;19:5619–5624. doi: 10.1093/nar/19.20.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang W W, Yaneva M. Biochem Biophys Res Commun. 1992;186:574–579. doi: 10.1016/s0006-291x(05)80847-2. [DOI] [PubMed] [Google Scholar]
- 7.Blier P R, Griffith A J, Craft J, Hardin J A. J Biol Chem. 1993;268:7594–7601. [PubMed] [Google Scholar]
- 8.Weaver D T. Trends Genet. 1995;11:388–392. doi: 10.1016/s0168-9525(00)89121-0. [DOI] [PubMed] [Google Scholar]
- 9.Jackson S P, Jeggo P A. Trends Biochem Sci. 1995;20:412–415. doi: 10.1016/s0968-0004(00)89090-8. [DOI] [PubMed] [Google Scholar]
- 10.Beall E L, Admon A, Rio D C. Proc Natl Acad Sci USA. 1994;91:12681–12685. doi: 10.1073/pnas.91.26.12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knuth M W, Gunderson S I, Thompson N E, Strasheim L A, Burgess R R. J Biol Chem. 1990;265:17911–17920. [PubMed] [Google Scholar]
- 12.Stuiver M H, Coenjaerts F E, van der Vliet P C. J Exp Med. 1990;172:1049–1054. doi: 10.1084/jem.172.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins A R. Mutat Res. 1993;293:99–118. doi: 10.1016/0921-8777(93)90062-l. [DOI] [PubMed] [Google Scholar]
- 14.Lee S E, Pulaski C R, He D M, Benjamin D M, Voss M, Um J, Hendrickson E A. Mutat Res. 1995;336:279–291. doi: 10.1016/0921-8777(95)00002-2. [DOI] [PubMed] [Google Scholar]
- 15.Taccioli G E, Rathbun G, Oltz E, Stamato T, Jeggo P A, Alt F W. Science. 1993;260:207–210. doi: 10.1126/science.8469973. [DOI] [PubMed] [Google Scholar]
- 16.Pergola F, Zdzienicka M Z, Lieber M R. Mol Cell Biol. 1993;13:3464–3471. doi: 10.1128/mcb.13.6.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beall E L, Rio D C. Genes Dev. 1996;10:921–933. doi: 10.1101/gad.10.8.921. [DOI] [PubMed] [Google Scholar]
- 18.Dvir A, Peterson S R, Knuth M W, Lu H, Dynan W S. Proc Natl Acad Sci USA. 1992;89:11920–11924. doi: 10.1073/pnas.89.24.11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dvir A, Stein L Y, Calore B L, Dynan W S. J Biol Chem. 1993;268:10440–10447. [PubMed] [Google Scholar]
- 20.Gottlieb T M, Jackson S P. Cell. 1993;72:131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 21.Feldmann H, Winnacker E L. J Biol Chem. 1993;268:12895–12900. [PubMed] [Google Scholar]
- 22.Jacoby D B, Wensink P C. J Biol Chem. 1994;269:11484–11491. [PubMed] [Google Scholar]
- 23.Milne G T, Jin S, Shannon K B, Weaver D T. Mol Cell Biol. 1996;16:4189–4198. doi: 10.1128/mcb.16.8.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherman F, Fink G R, Hicks J B. Methods in Yeast Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1986. [Google Scholar]
- 25.Rose M D, Winston F, Hieter P. Methods in Yeast Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1990. [Google Scholar]
- 26.Ito H, Fukuda Y, Murata K, Kimura A. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barnes G, Rine J. Proc Natl Acad Sci USA. 1985;82:1354–1358. doi: 10.1073/pnas.82.5.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reeves W H, Sthoeger Z M. J Biol Chem. 1989;264:5047–5052. [PubMed] [Google Scholar]
- 30.Jones K R, Rubin G M. Neuron. 1990;4:711–723. doi: 10.1016/0896-6273(90)90197-n. [DOI] [PubMed] [Google Scholar]
- 31.Rothstein R J. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- 32.Pringle J R, Preston R A, Adams A E, Stearns T, Drubin D G, Haarer B K, Jones E W. Methods Cell Biol. 1989;31:357–435. doi: 10.1016/s0091-679x(08)61620-9. [DOI] [PubMed] [Google Scholar]
- 33.Kilmartin J, Wright B, Milstein C. J Cell Biol. 1982;93:576–582. doi: 10.1083/jcb.93.3.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loo S, Fox C A, Rine J, Kobayashi R, Stillman B, Bell S. Mol Biol Cell. 1995;6:741–756. doi: 10.1091/mbc.6.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weinert T A, Hartwell L H. Genetics. 1993;134:63–80. doi: 10.1093/genetics/134.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siede W, Friedberg A S, Friedberg E C. Proc Natl Acad Sci USA. 1993;90:7985–7989. doi: 10.1073/pnas.90.17.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinert T A, Kiser G L, Hartwell L H. Genes Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- 38.Garvik B, Carson M, Hartwell L. Mol Cell Biol. 1995;15:6128–6138. doi: 10.1128/mcb.15.11.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elledge S J, Davis R W. Genetics. 1989;9:4932–4940. doi: 10.1128/mcb.9.11.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elledge S J, Davis R W. Genes Dev. 1990;4:740–751. doi: 10.1101/gad.4.5.740. [DOI] [PubMed] [Google Scholar]
- 41.Siede W, Friedl A A, Dianova I, Eckardt-Schupp F, Friedberg E C. Genetics. 1996;142:91–102. doi: 10.1093/genetics/142.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porter S E, Greenwell P W, Ritchie K B, Petes T D. Nucleic Acids Res. 1996;24:582–585. doi: 10.1093/nar/24.4.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boulton S J, Jackson S P. EMBO J. 1996;15:5093–5103. [PMC free article] [PubMed] [Google Scholar]