

Summary
p27Kip1 regulates G1 in normal and malignant cells. We present data that Src regulates p27 stability through phosphorylation of p27 at tyrosine 74 and tyrosine 88. Src-phosphorylated p27 inhibits cyclin E-Cdk2 poorly in vitro and Src transfection reduces cyclin E-Cdk2-bound p27. cSrc activation and Src-p27 co-precipitation precede the loss of p27-bound cyclin E-Cdk2 and total p27 during G1 progression. SiRNA to cSrc and Src inhibitors increase cellular p27, and Src induction increases pY74p27 and pT187p27 and decreases p27 half-life. Src-activated breast cancer lines show reduced p27. In 482 primary human breast cancers, Src activation correlates statistically with reduced nuclear p27. By impairing p27’s inhibition of Cdk2, p27 phosphorylation by Src would facilitate cyclin E-Cdk2-dependent p27 proteolysis. The Src inhibitor, AZD0530, together with tamoxifen increased p27 and restored G1 arrest in tamoxifen resistant breast cancer lines. These data provide a new rationale for Src inhibitors in cancer therapy.
Keywords: p27Kip1, Cdk2, Cyclin E, Src, breast cancer, antiestrogens
INTRODUCTION
Mammalian cell proliferation is tightly regulated by the sequential activation of cyclin dependent kinases (Cdks) (Sherr and Roberts, 1999). The Cdk inhibitor, p27, was initially identified in cells arrested by transforming growth factor-β, by contact inhibition, and by lovastatin (Polyak et al., 1994; Hengst et al., 1994; Slingerland et al., 1994; Koff et al., 1993). p27 is ubiquitously expressed and binds cyclin-Cdk2 to inhibit kinase activity.
p27 is maximal in quiescence and mitogen-stimulated p27 loss during G0 to S phase progression leads to cyclin E-Cdk2 activation. Several mechanisms regulate p27 levels to control cell proliferation. p27 translation is maximal in G0 and falls abruptly with G0 exit and during G1 progression(Gopfert et al., 2003; Hengst and Reed, 1996; Agrawal et al., 1996). At least four pathways regulate p27 degradation. In late G1 through S and into M phase, p27 is degraded by the SCFSkp2 ubiquitin ligase, whose interaction with p27 is activated by cyclin E-Cdk2 dependent p27 phosphorylation at T187 (Pagano et al., 1995; Sheaff et al., 1997; Vlach et al., 1997; Montagnoli et al., 1999). In early G1, T187-independent p27 proteolysis occurs by both Skp2-dependent (Malek et al., 2001) and Skp2-independent (Hara et al., 2001) pathways. In early G1, p27 phosphorylation at S10 promotes p27-CRM1-binding and nuclear export (Rodier et al., 2001; Ishida et al., 2002; Connor et al., 2003). Proteolysis of cytoplasmic p27 in G1 involves KPC1 ubiquitin ligase (Hara et al., 2005; Kamura et al., 2004). Rapid S10-independent p27 proteolysis may also occur in the nucleus in early G1 (Rodier et al., 2001). In quiescent cells, p27 proteolysis requires an intact p27-cyclin-Cdk binding motif (Besson et al., 2006).
p27 levels regulate cell proliferation and differentiation. p27 null animals exhibit multi-organ hyperplasia (Fero et al., 1996; Kiyokawa et al., 1996; Nakayama et al., 1996) and p27+/− heterozygous animals are tumor prone (Fero et al., 1998). While normal quiescent epithelial tissues express high p27 (Catzavelos et al., 1997), reduced p27 protein is observed in up to 60% of primary human breast and other cancers in association with poor patient outcome (Alkarain et al., 2004). Several oncogenic pathways activate p27 proteolysis in cancers (Liang and Slingerland, 2003). Human breast cancers frequently show EGFR overexpression or Her2/ErbB2 amplification (Tsutsui et al., 2002; Nicholson et al., 1990; Slamon et al., 1987). Overexpression of EGFR or Her2 increases p27 proteolysis in cell lines (Lane et al., 2000; Yang et al., 2000; Lenferink et al., 2000).
Activated EGFR family receptor tyrosine kinases (RTK) recruit and activate cSrc, and cSrc in turn further activates RTKs, stimulating cell proliferation (Ishizawar and Parsons, 2004). Drug mediated cSrc inhibition blocks the effects of EGFR and Her2 on cell proliferation (Belsches-Jablonski et al., 2001; Biscardi et al., 1999). cSrc is also activated by liganded estrogen receptor (ER) in human breast cancer cells. Estrogen:ER binding stimulates rapid transient recruitment of cSrc, Shc activation and MAPK signaling (Migliaccio et al., 1996). Estrogen:ER-stimulated Src further recruits receptor tyrosine kinases, Her2, EGFR (Chu et al., 2005) and IGF-1R (Song et al., 2004) to promote cell cycle progression.
We recently demonstrated a novel Lyn and Bcr-Abl-mediated tyrosine phosphorylation of p27 that contributes to p27 proteolysis (Grimmler et al., in print). Up to 60% of human breast cancers express the estrogen receptor and in these, estrogen is mitogenic. Estrogen-stimulated breast cancer proliferation requires a rapid loss of p27 through proteolysis (Cariou et al., 2000). Given the oncogenic role of Src in breast cancer and its rapid activation by RTKs and estrogen:ER, we investigated whether Src-mediated tyrosine phosphorylation of p27 may contribute to p27 proteolysis in breast cancer cell proliferation.
Here we present evidence that cSrc phosphorylates p27 on tyrosine 74 (Y74) and tyrosine 88 (Y88). p27 phosphorylation by Src reduced the cyclin E-Cdk2 inhibitory action of p27 in vitro. Src induction increased pT187p27 and reduced the t1/2 of p27. cSrc activation was correlated with low nuclear p27 in primary human breast cancers. Mitogen stimulation of quiescent cells rapidly activated cSrc, and p27 binding to cSrc preceded loss of cyclin E-Cdk2 from p27 in late G1. These data support a model in which cSrc-mediated tyrosine phosphorylation of p27 reduces its inhibitory action on cyclin E-Cdk2, facilitating subsequent p27 proteolysis.
RESULTS
Src phosphorylates p27 in vitro
Estrogen stimulates quiescent ER positive breast cancer cells to rapidly degrade p27 and enter cell cycle (Cariou et al., 2000). Estrogen bound ER stimulates rapid, transient cSrc activation (Migliaccio et al., 1996; Song et al., 2004). To investigate if cSrc contributes to estrogen-mediated p27 proteolysis, we assayed if Src could phosphorylate p27. Purified recombinant activated Src phosphorylated recombinant His-p27 and this was inhibited by the Src inhibitor, PP1 (Figure 1A). Recombinant WT Src could also phosphorylate p27 (not shown). To assay the relative contributions of three tyrosines, Y74, Y88 and Y89 to Src phosphorylation of p27, single, double and triple p27 mutations were generated converting tyrosines to phenylalanine. Mutant p27 and p27WT were compared as substrates in Src kinase assays. Src autophosphorylation indicates equal Src input (Figure 1B). Radioactivity in p27 was quantitated by phosphoimager (Figure 1C). Src-mediated p27 phosphorylation was reduced by 36% and 33% in p27Y74F and p27Y88F respectively, compared to p27WT, while that of p27Y89F was only reduced by 12% (Figure 1C). Loss of Y74 and Y88 (p27Y74FY88F) reduced p27 phosphorylation by Src by 78%. Thus, these two sites appear to be preferentially phosphorylated by Src in vitro (Figure 1C). Loss of potential to phosphorylate Y89 also reduced phosphorylation of Y74FY89F and Y88FY89F.
Figure 1. Src preferentially phosphorylates p27 at Y74 and Y88 in vitro.
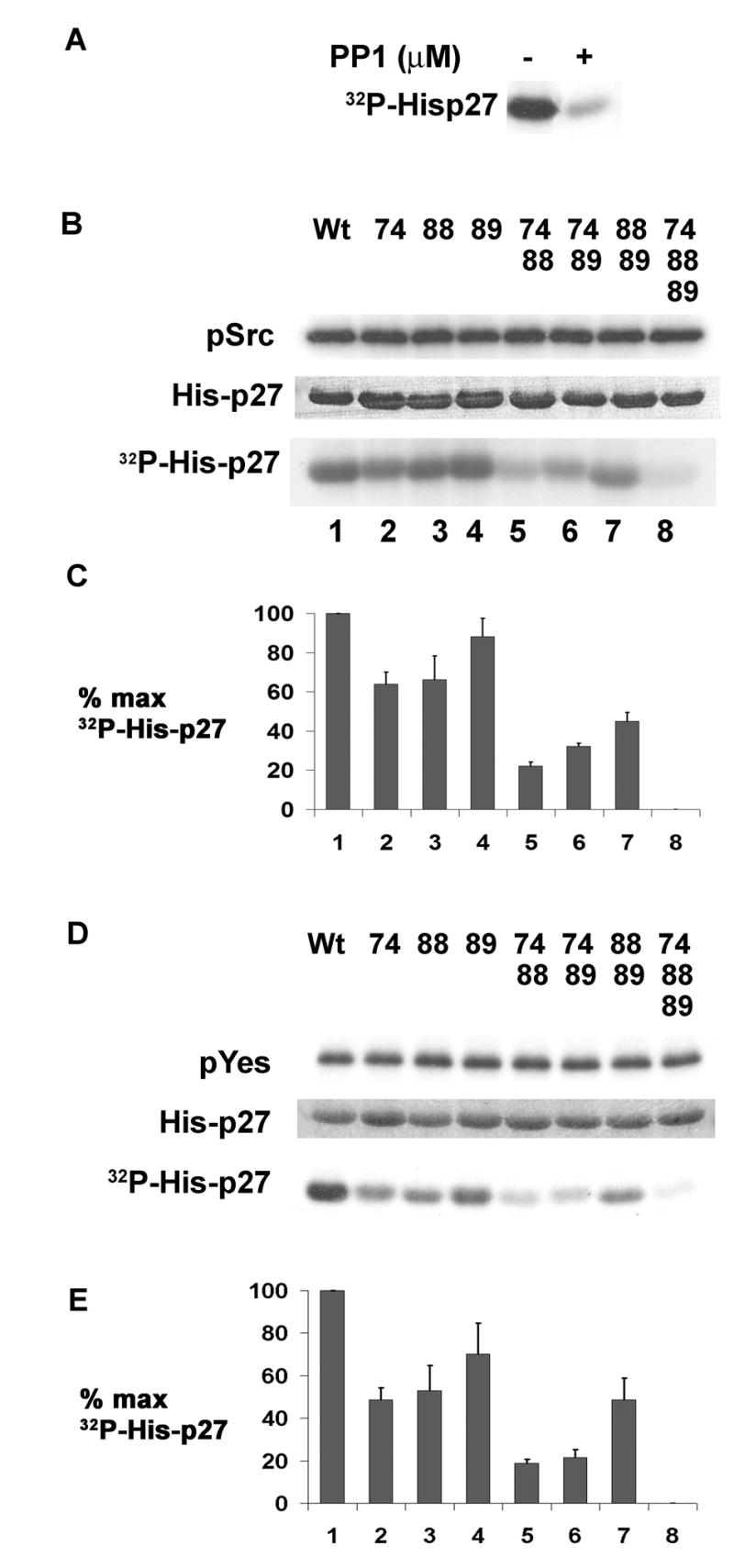
(A) Wt His-p27 reacted with Src kinase with or without PP1.
(B) His-p27WT and p27 mutants reacted with Src kinase per Exp. Procedures. Radioactivity in autophosphorylated Src (pSrc) or p27 (32P-His-p27) quantitated by phosphoimager, and His-p27 input are shown.
(C)Quantitation of triplicate Src kinase assays (means +/− SEM).
(D)His-p27WT and p27 mutants reacted with Yes kinase per Exp. Procedures. Radioactivity in Yes (pYes) or p27 (32P-His-p27) quantitated by phosphoimager, and His-p27 input are shown.
(E) Quantitation of triplicate Yes kinase assays (means +/− SEM).
Not all Src family kinases are expressed in mammary cells. Quantitative real time PCR of Src, Yes, Fyn and Lyn in three breast cancer lines, MCF-7, T47-D and MDA-MB-361 showed Yes expression was higher or comparable to that of cSrc. Fyn and Lyn mRNA were detectable but several logs lower in magnitude (Figure S1). As for Src, Yes kinase assays showed phosphorylation of p27 by Yes was reduced by mutational loss of Y74 and Y88 phosphorylation, while Y89F only modestly attenuated phosphorylation by Yes (Figure 1D & E).
Tyrosine phosphorylated p27 is a poor inhibitor of cyclin E-Cdk2
The crystal structure of p27-cyclin A-Cdk2 shows Y74, Y88, and Y89 of p27 interact with Cdk2 and not with N-terminal truncated cyclin A. Y88 is buried in the catalytic cleft of Cdk2, but Y74 and to a lesser extent Y89 also form contacts with Cdk2 (Russo et al., 1996). Since Y88 impedes ATP binding to Cdk2, structural data suggest that tyrosine phosphorylation of p27 would impair its inhibition of cyclin-bound Cdk2. To test this, increasing amounts of mock or Src-phosphorylated His-p27 were incubated with recombinant cyclin E-Cdk2, and Cdk2 activity was assayed. Src was inactivated by boiling the Src-p27 reactions. Mock treated His-p27 was also boiled. Tyrosine phosphorylated p27 (pY-p27) inhibited cyclin E-Cdk2 less efficiently than mock-phosphorylated p27 (Figure 2A).
Figure 2. Phosphorylation by Src reduces p27 inhibitory action on cyclin E-Cdk2.
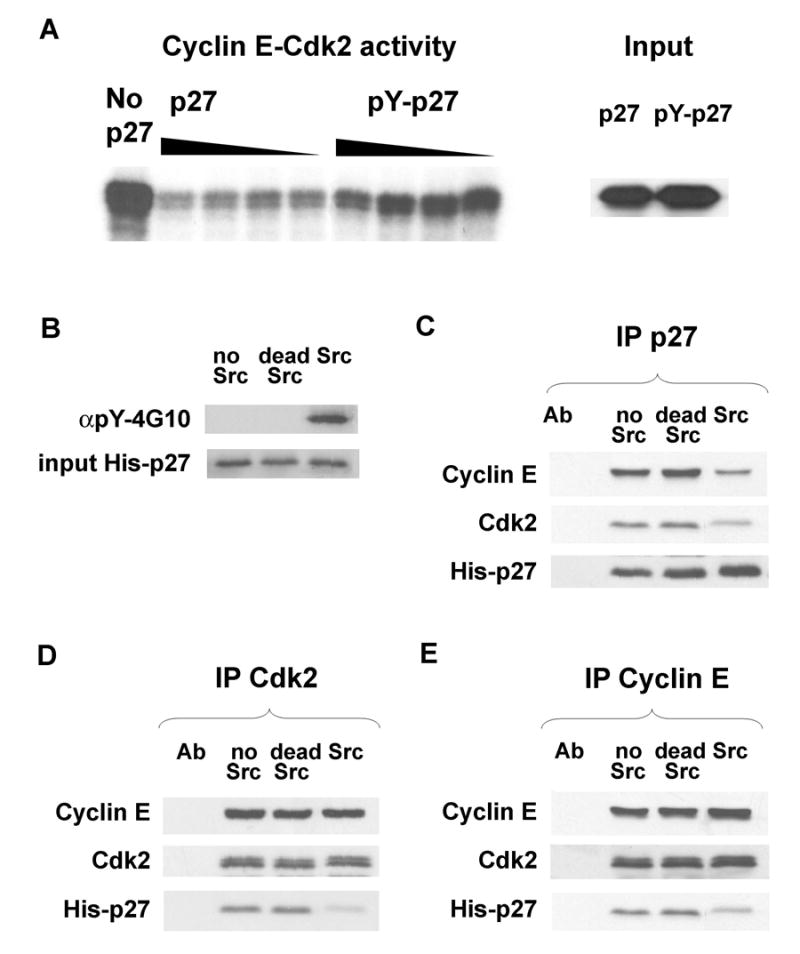
(A)His-p27WT was phosphorylated in vitro with activated Src. Mock treated (p27) or Src treated pY-p27 were incubated with cyclin E-Cdk2 and H1 kinase activity assayed. Equal input of mock vs. Src treated p27 shown.
(B)His-p27WT incubated without Src (no Src), with inactive Src (dead Src) and active Src are shown. pYp27 was precipitated with αpY-4G10.
Equal amounts of p27 from (B) were incubated with cyclin E-Cdk2. (C) p27, (D) Cdk2 and (E) Cyclin E were precipitated and associated proteins blotted.
To assay whether the impaired inhibitory function of pY-p27 correlated with decreased association with cyclin E-Cdk2, p27 was reacted in vitro with either active recombinant Src, Src that had been heat inactivated prior to reaction with p27 (dead Src), or subjected to a mock Src reaction. Only active Src treated p27 reacted with anti-phosphotyrosine antibody 4G10 (αpY-4G10, Figure 2B). Because the in vitro Src kinase reaction was not complete, pY-p27 was isolated by immunoprecipitation with αpY-4G10. Equal amounts of mock or Src treated p27 were incubated with recombinant cyclin E and Cdk2. Mock treated and dead-Src-treated p27 immunoprecipitates bound equal amounts of cyclin E and Cdk2, while pY-p27 precipitates bound less cyclin E and Cdk2 (Figure 2C). Less Src phosphorylated pYp27 was detected in Cdk2 and Cyclin E immunoprecipitates compared to mock or untreated p27 (Figure 2D&E). Thus, the reduced cyclin E-Cdk2 inhibitory function of Src phosphorylated p27 versus unphosphorylated p27 may result in part from its reduced steady state binding to this cyclin-Cdk2 target.
Intracellular tyrosine phosphorylation of p27
We and others recently identified tyrosine phosphorylated p27 in leukemic cells (Kardinal et al., 2006) (Grimmler et al., in print). Bcr-Abl and Lyn kinases phosphorylated p27 predominantly at Y88 in vitro, and in K562 cells, Y88 was the major tyrosine phosphorylated site in p27 (Grimmler et al., in print). Because in vitro kinase assays showed p27Y74F had reduced phosphorylation by Src, polyclonal antibody specific to Y74 phosphorylated p27 (p27pY74) was generated. αp27pY74 antibody recognized Src phosphorylated His-p27WT but not His-p27Y74F (Figure 3A, left), while αpY-4G10 reacted with both p27WT and p27Y74F after Src kinase reactions in vitro (Figure 3A, left). To detect intracellular Y74 phosphorylation of p27, MCF-7 cells were co-transfected with constitutively active Src or empty vector, and YFPp27. Tyrosine phosphorylated p27 was detectable with αpY-4G10 in p27 immunoprecipitates from Src-transfected but not from mock-transfected cells (Figure 3A, middle). Similarly, p27pY74 was detected with αp27pY74 antibody only after cSrc transfection (Figure 3A right). Endogenous cellular p27pY74 was also detected with this antibody in the absence of exogenous Src (see Figure 5E).
Figure 3. pY74p27 antibody and in vivo p27 phosphorylation by Src.
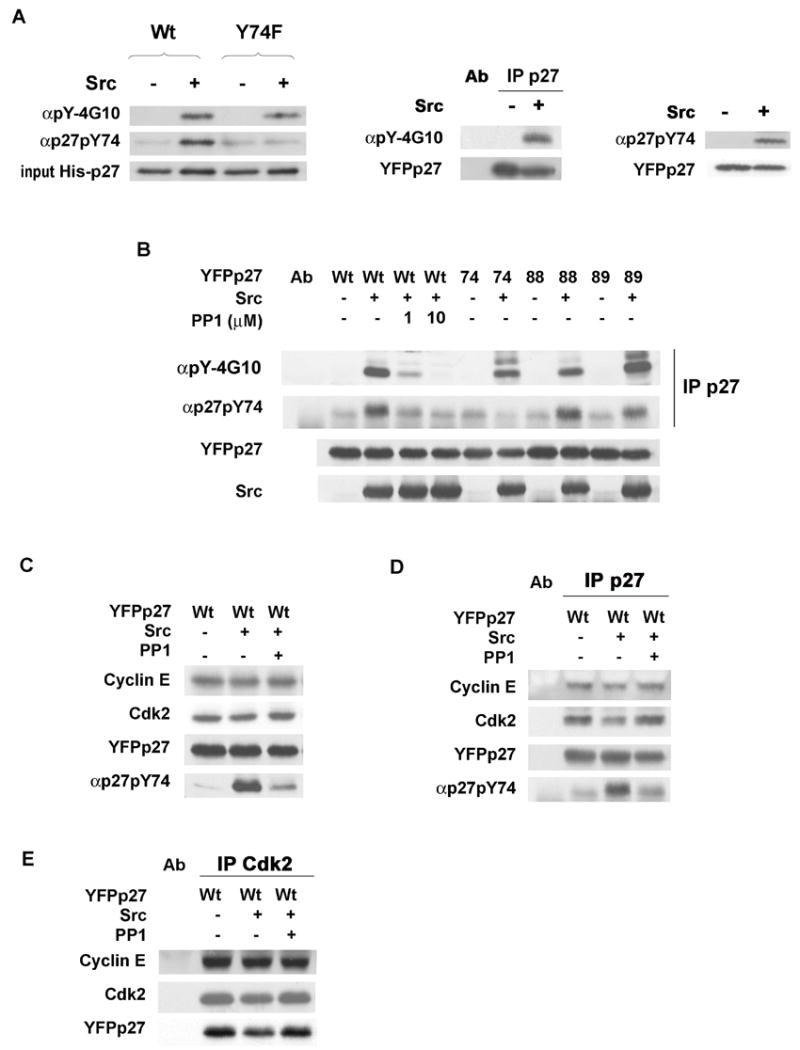
(A) His-p27WT or His-p27Y74F were reacted with Src in vitro. p27 precipitates were blotted with αpY-4G10 or αp27pY74 (left). MCF-7 was transfected with YFPp27WT and empty vector (Src −) or PCI-Src-Y530F (Src +). p27 precipitates were blotted with αpY-4G10, stripped and re-probed for p27 (middle panel). pY74p27 detected in Src transfected MCF-7 (right panel).
(B) MCF-7 was co-transfected with YFPp27WT or mutant p27 with or without Src. pYp27 was detected in p27 precipitates with αpY-4G10 or αp27pY74.
(C) Cyclin E, Cdk2 and YFPp27 levels in YFPp27WT transfected MCF-7 with or without Src cotransfection or PP1.
p27 (D) and Cdk2 (E) precipitates were resolved and proteins detected by blotting.
Figure 5. Src activation and p27-Src interaction increase in early G1.
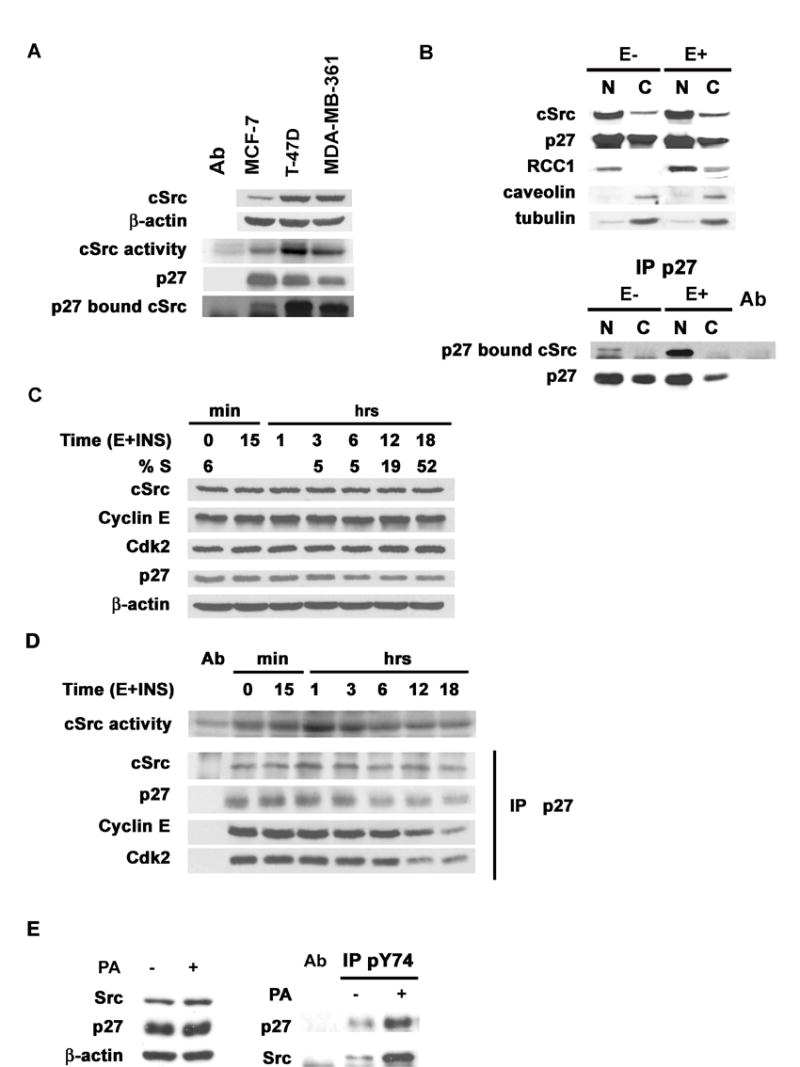
(A) p27, cSrc and β-actin, IP Src kinase activities and p27-bound cSrc in MCF-7, T-47D and MDA-MB-361.
(B) cSrc, p27, p27-bound cSrc and nuclear (RCC1), cytoplasmic (tubulin) and membrane (caveolin) markers in subcellular fractions from quiescent (E−) or asynchronous (E+) T-47D.
(C) Estrogen-deprived MCF-7 (t=0) were stimulated with estradiol and insulin and %S and protein levels assayed at indicated times.
(D) cSrc activity, p27-bound cSrc, cyclin E and Cdk2 in lysates from (C) above.
(E) Src, p27 and β-actin in estrogen starved cells treated with estrogen for 2 h with (+PA) or without (−PA) prior Src induction (left). p27 and Src in αp27pY74 precipitates (right).
To test the preference of Src for the different p27 tyrosines in cells, activated Src and YFPp27WT, p27Y74F, p27Y88F or p27Y89F were transfected into MCF-7. All p27 vectors were equally and stably overexpressed (Figure 3B). p27 precipitates were immunoblotted and reacted with αpY-4G10, αp27pY74, or antibody to total p27. Reactivity of p27WT with αpY-4G10 and with αp27pY74 following Src co-transfection was attenuated by the Src inhibitor, PP1 (Figure 3B).
Transfected p27Y74F and p27Y88F immunoprecipitates were less reactive than YFPp27WT with αpY-4G10, while αpY-4G10 reactivity of p27Y89F was not detectably reduced (Figure 3B). Y74 phosphorylation was detected in p27Y88F, p27Y89F and p27WT, but reactivity of αp27pY74 with p27Y74F showed no increase above non-specific baseline (Figure 3B). Thus, Y74 and Y88 appear to be the dominant cSrc phosphorylation sites in vivo. Loss of Y89 phosphorylation may attenuate Y74 phosphorylation in vivo and lead to a compensatory increase in Y88 phosphorylation.
The effect of Src overexpression on p27-cyclin E-Cdk2 complexes was tested. Src transfection with or without PP1 treatment did not affect levels of cellular cyclin E and Cdk2 or overexpressed YFPp27 (Figure 3C). YFPp27WT immunoprecipitates contained less bound cyclin E and Cdk2 following Src transfection and this was abrogated by PP1 (Figure 3D). Similarly, Src transfection modestly reduced p27 in Cdk2 precipitates, while Cdk2 bound cyclin E was unaffected (Figure 3E).
Src effects on cellular p27 levels and stability
Since experiments above involved overexpressed p27, which could overwhelm cellular p27 proteolysis, effects of Src on cellular p27 were assayed. cSrc siRNA or Src inhibition for 48 hrs both increased cellular p27 (Figure 4A and B).
Figure 4. cSrc inactivation increases p27 levels and stability and Src induction decreases p27 t1/2.
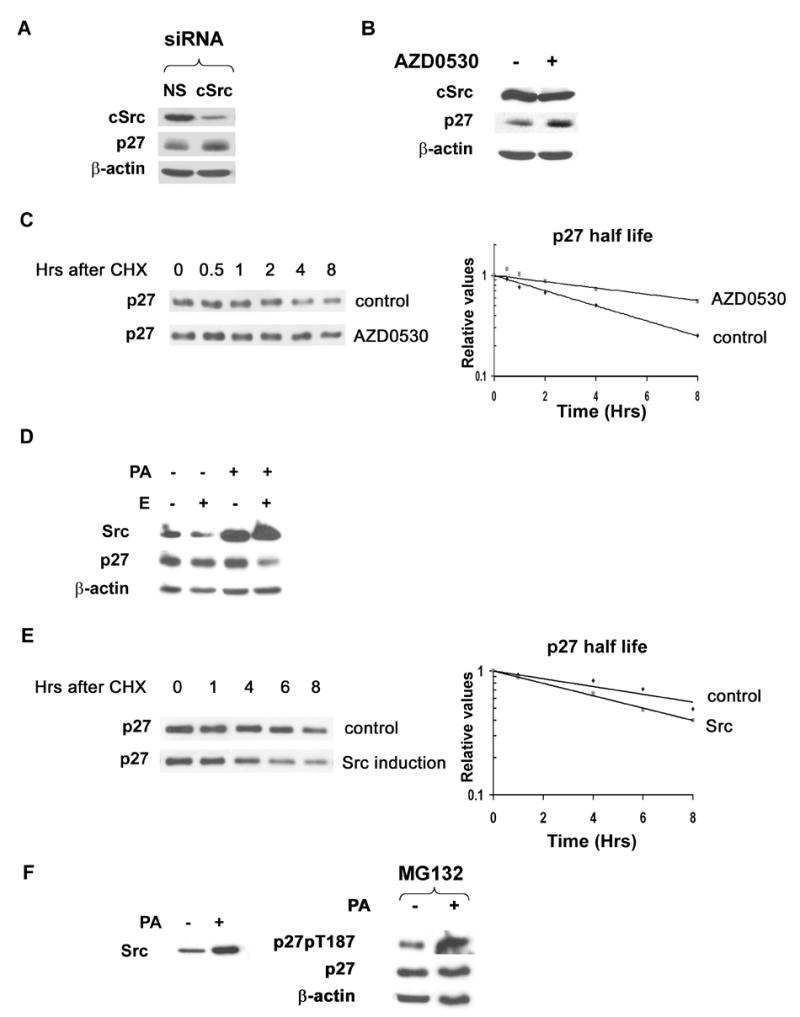
(A)MCF-7 cells transfected with cSrc siRNA or non-specific controls analyzed for cSrc, 27 and β-actin.
(B)cSrc, p27 and β-actin levels with (+) or without (−) AZD0530 in MDA-MB-361.
(C)Cycloheximide (CHX) chase of p27 in MDA-MB-361 before and 6h after AZD0530.
(D) Src was induced in estrogen starved MCF-pINDSrc2 (E −) by ponasterone A (PA+). 6h after estrogen addition (E+), Src, p27 and β-actin were assayed.
(E) CHX chase of p27 at 6 h after estrogen treatment, with or without prior Src induction.
(F) p27pT187, p27 and β-actin at 13 h after E stimulation, with or without prior Src induction. MG132 added 6 h prior to analysis.
Src is activated in MDA-MB-361 line due to Her2/ErbB2 activation (Belsches-Jablonski et al., 2001). p27 half-life was assayed by cycloheximide chase in asynchronous MDA-MB-361 and in cells treated with the Src family kinase inhibitor, AZD0530 for six hours. Src inhibition increased the p27 t1/2 from 4.0 to 9.7 hours (Figure 4C).
To test further the effect of Src on p27 stability, inducible Src expressing lines were generated. Cells were estrogen deprived and Src was induced or not with ponasterone A (PA) prior to re-addition of estrogen. Estrogen stimulation alone, in the absence of added growth factors had little effect on p27 levels over 6 hours, however, Src induction together with estrogen led to more rapid loss of p27 in two different Src inducible MCF-7 clones (shown for MCF-pINDSrc2, Figure 4D). Src induction reduced the p27 t1/2 at 6 hours after estrogen addition to 5.8 hours compared to the p27 t1/2 of 9.8 hours in controls treated with estrogen alone (Figure 4E). T187 phosphorylation of p27 was more readily detected when estrogen starved cells were treated with estrogen plus Src compared to estrogen alone (Figure 4F).
These findings support a model in which Src promotes p27 degradation by decreasing the inhibitory action of p27 on cyclin E-Cdk2, facilitating cyclin E-Cdk2 dependent p27 phosphorylation and SCFSKP2 mediated p27 proteolysis.
cSrc-p27 interaction is increased in cancer lines with activated Src
p27-bound cSrc was detected in three breast cancer lines MCF-7, T-47D and MDA-MB-361. Cells with higher cSrc activity had reduced p27 levels but higher p27-bound cSrc (Figure 5A).
In contrast to normal mammary epithelial cells where p27 shows nuclear localization (Catzavelos et al., 1997), T-47D and MDA-MB-361 have both nuclear and some cytoplasmic p27. Nuclear cSrc-p27 complexes were only minimally detectable in cells deprived of estrogen and growth factors in 0.1%cFBS for 48 hours (E−) but were readily detected in asynchronous cells in complete medium (E+) (data shown for T-47D Figure 5B). To confirm the nuclear localization of p27-Src complexes, p27S10A or p27WT were transfected into MCF-7 with or without transfection of Src. p27S10A is localized to the nucleus due to reduced nuclear export (Connor et al., 2003). p27 bound Src was equally detected in p27S10A and p27WT complexes (Figure S2).
cSrc-p27 interaction precedes p27 loss in G1 progression
Quiescent estradiol-depleted MCF-7 enter S phase within 12 hours of estrogen and insulin addition (Figure 5C) (Cariou et al., 2000). cSrc was rapidly activated on G0 exit, peaking at 1 hour, and fell to baseline by 3–6 hours. p27 levels fell during G1 progression while cSrc, cyclin E and Cdk2 levels did not vary (Figure 5C). Rapid Src activation correlated with increased Src-p27 co-precipitation, and preceded the reduction of total p27 and loss of p27-bound cyclin E and Cdk2 as cells entered S phase (Figure 5D).
To demonstrate Y74 phosphorylation of endogenous cellular p27, MCF-pINDSrc2 cells were estrogen deprived and the Src was induced or not by PA. Cells were then treated with estrogen for two hours to stimulate endogenous cSrc. αp27pY74 precipitates were resolved and immunoblotted for p27 and Src. Following estrogen stimulation of starved cells, Y74 phosphorylation of cellular p27 was detected and this was increased by Src induction (Figure 5E). Src-pYp27 complexes were increased in Src–induced cells (Figure 5E).
cSrc activation in human breast cancers is correlated with low nuclear p27
To test if cSrc activation is associated with reduced p27 in primary human breast cancers, p27 and phosphorylated, activated cSrc (detected with an anti-pY416-Src antibody) were assayed by immunohistochemistry in 482 tumors. 37% showed reduced nuclear p27 levels. Little or no cSrc activation (score 0) was detected in 23% of tumors. cSrc activation (scores of 2 or 3) was detected in 39% of tumors. Normal mammary duct tissue controls present on all tissue microarrays showed no cSrc activation. The correlation of low p27 with cSrc activation and other clinical and pathologic variables is shown in Table 1. As in prior studies, reduced p27 was inversely correlated with estrogen and progesterone receptor positive status and strongly associated with increased tumor size, high tumor grade (a measure of de-differentiation), and tumor invasiveness as indicated by more lymph nodes affected and intratumoral invasion of the lymphovascular spaces (LVI). Reduced p27 was statistically correlated with activated Src when all four Src scoring levels were used (p=0.05) and this was more significant when Src was categorized as high (scores 2–3) or low (scores 0–1), p=0.01 (Table 1). Cancers with low p27 and high pY416-Src immunoreactivity (left panels) and high p27 with low pY416-Src (right panel) are shown in Figure S3.
Table 1.
Correlation of p27 levels with Src activation and other clinical/pathologic variables in 482 primary human breast cancers
| Variables | Low p27 n=177 | High p27 n= 305 | p valuea |
|---|---|---|---|
| n (%) | n (%) | ||
| Tumor Grade
1 2 3 Missing |
9(6.8) 56(42.1) 68(51.1) 44 |
25(10.3) 126(52.1) 91(37.6) 63 |
0.04 |
| Nodal status
negative positive not tested |
55(35.5) 100 (64.5) 22 |
119(48.0) 129(52.0) 57 |
0.01 |
| ERb
negative positive missing |
78(44.3) 98(55.7) 1 |
60(19.9) 241(80.1) 4 |
<10−4 |
| PRc
negative positive missing |
84(53.9) 72(46.2) 21 |
77(29.0) 189(71.1) 39 |
<10−4 |
| LVId
not present present unknown/missing |
83(55.7) 66(44.3) 28 |
178(65.0) 96(35.0) 31 |
0.06 |
| Menopausal Status
post pre missing |
95(54.0) 81(46.0) 1 |
187(61.5) 117(38.5) 1 |
0.11 |
| Src
0 1 2 3 |
40(22.6) 55(31.1) 51(28.8) 31(17.5) |
73(23.9) 126(41.3) 72(23.6) 34(11.2) |
0.05 |
| Src
0 or 1 2 or 3 |
95(53.7) 82(46.3) |
199(65.3) 106(34.7) |
0.01 |
chi-square test to compare the frequency and t-test to compare the mean.
ER = estrogen receptor
PR = progesterone receptor
LVI= lymphovascular invasion
Src inhibition and tamoxifen cooperate to inhibit cell cycle progression
Tamoxifen is an ER blocking drug that arrests sensitive ER positive breast cancer cells in G1 by inhibiting estrogen stimulated p27 proteolysis. p27 expression is required for G1 arrest by tamoxifen (Cariou et al., 2000). Overactivation of receptor tyrosine kinases (RTKs) can confer antiestrogen resistance (Benz et al., 1992; Pietras et al., 1995; Donovan et al., 2001), and also recruit and activate Src (Biscardi et al., 1999; Tice et al., 1999). Since Src-dependent p27 phosphorylation impaired its cyclin-Cdk2 inhibitory action, we tested if Src inhibition could restore tamoxifen responsiveness to resistant cells. MDA-MB-361 has high cSrc activity (Figure 5A), fails to arrest upon estrogen deprivation and is resistant to tamoxifen-mediated G1 arrest. These cells showed only a partial G1 arrest after 48 hrs of treatment with an ER-saturating dose of tamoxifen. Increasing tamoxifen or treatment with the irreversible ER blocker, fulvestrant, failed to cause G1 arrest (not shown).
AZD0530 (AstraZeneca Ltd.) is a potent, specific drug inhibitor of Src family kinases (Ple et al., 2004) in clinical trials for cancer in humans. While AZD0530 inhibits both Src family and Abl kinases, Abl is not expressed in breast cancers. Asynchronous MDA-MB-361 cells were treated with AZD0530, tamoxifen alone, or both together. The 1 μM dose of each drug caused partial G1 arrest with the % S phase falling from 43% to 30% and 20%, respectively after 48 hours. AZD0530 and tamoxifen together reduced the % S phase cells to 4% (Figure 6A). Each drug alone increased p27 by less than 2-fold, whereas both drugs together increase p27 >4 fold (Figure 6B). Cyclin E-Cdk2 bound p27 increased 2.3-fold with AZD0530, 1.7-fold with tamoxifen alone, and 5 fold with both drugs together (Figure 6C) with loss of cyclin E-cdk2 activity (not shown). Thus, Src inhibition restored p27 action and tamoxifen mediated G1 arrest.
Figure 6. Src inhibitor cooperates with tamoxifen to arrest antiestrogen resistant breast cancer cells.
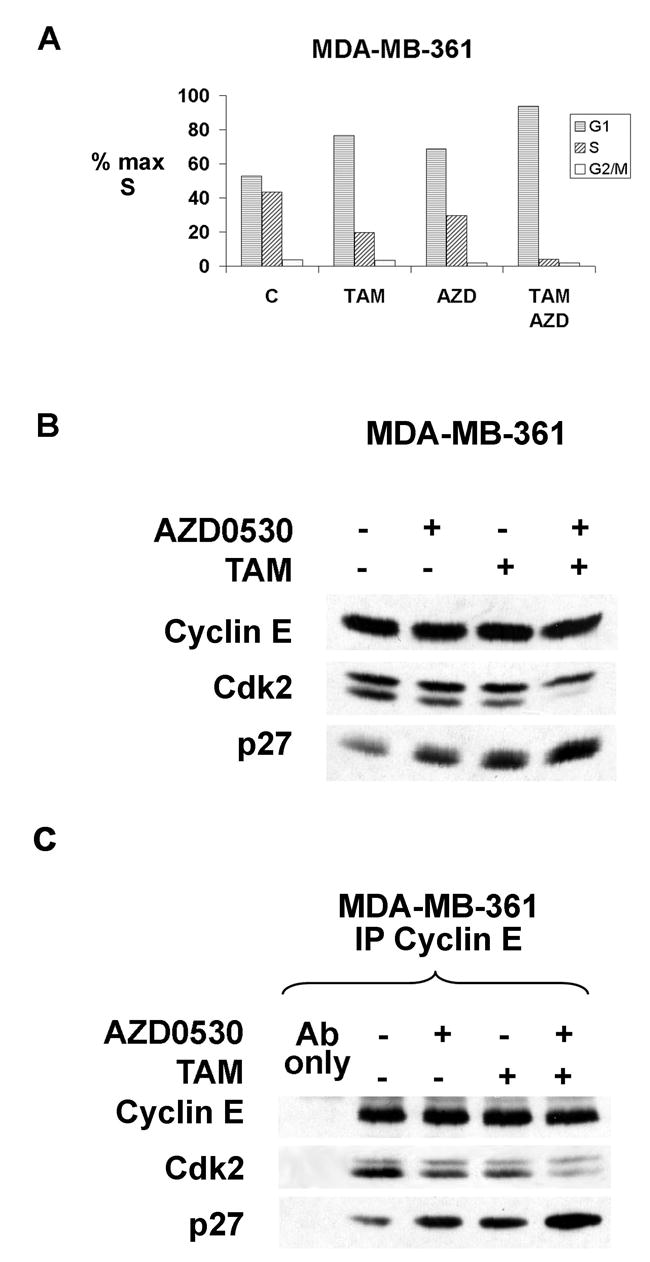
(A) Cell cycle profile in MDA-MB-361 treated with TAM, AZD0530 or both for 48 h.
(B) Cyclin E, Cdk2 and p27 in cells from (A).
(C) Cyclin E bound Cdk2 and p27.
Src inhibition with AZD0530 also restored response to tamoxifen in two other ER positive, partially resistant breast cancer lines, T-47D and BT474. In two lines with only modest Src activation (MCF-7 and ZR75-1), AZD0530 did not augment tamoxifen responses (See Table S1).
DISCUSSION
Regulation of p27 levels
p27 is frequently inactivated in human cancers through accelerated p27 proteolysis or sequestration in the cytoplasm. Reduced p27 is an independent marker of poor breast cancer prognosis (Alkarain et al., 2004). In normal cells, p27 levels fall rapidly as cell progress from G1-to-S phase. However, a major mechanism of p27 proteolysis involves its phosphorylation by the very kinase it inhibits. p27 acts as an inhibitor and as a substrate of cyclin E-Cdk2 (Sheaff et al., 1997). While cyclin E-Cdk2 phosphorylation of T187 targets p27 for SCFSkp2-mediated proteolysis in late G1, mechanisms permitting initial cyclin E-Cdk2 activation in early G1 despite the persistent abundance of its inhibitor have remained obscure. We and others have postulated that early G1 phosphorylation events may reduce the affinity of p27 for cyclin E-Cdk2 promoting p27 dissociation from Cdk2, binding to CRM1 and export driven proteolysis of p27 in the cytoplasm (Connor et al., 2003; Ishida et al., 2002; Hara et al., 2001). However it has been difficult to provide definitive evidence for this in vivo.
Our earlier work showed that TGF-β resistant cells (Florenes et al., 1996; Ciarallo et al., 2002) and cancer cell lines that overexpress oncogenes including ILK, Her2 or MEK exhibit altered p27 phospho-isoforms with reduced cyclin E-Cdk2 inhibitory function (Radeva et al., 1997; Donovan et al., 2001; Ciarallo et al., 2002). Such intermediate forms of p27 could facilitate the transition of p27 from inhibitor to substrate of cyclin E-Cdk2. Here we present evidence that Src-mediated tyrosine phosphorylation of p27 reduces its inhibitory function for cyclin E-Cdk2.
Src activation in cancer
cSrc activates mitogenic signaling in normal and malignant cells (Thomas and Brugge, 1997) to regulate cell proliferation, survival, metastasis, and angiogenesis (Ishizawar and Parsons, 2004). Increased cSrc levels or activity have been observed in many human cancers when compared to adjacent normal tissues, including breast, prostate, lung, colorectal, and ovarian carcinomas (Chen et al., 2006; Irby and Yeatman, 2000; Biscardi et al., 2000), and are often associated with increased disease stage (Frame, 2002). We observed Src activation in 39% of 482 primary human breast cancers.
Src activation in cancers results from oncogenic activation of receptor tyrosine kinases (RTK), including EGFR, Her2/ErbB2, IGF-1R, colony stimulating factor-1 and hepatocyte growth factor receptor. Reduced phosphorylation of its negative regulatory site, SrcY530, can also activate Src (Chen et al., 2006). Activating Src mutations are rare in human cancers and in colon cancer correlate with metastatic progression (Irby et al., 1999). EGFR and Her2/ErbB2 both bind Src to catalyze mutual kinase activation. cSrc and EGFR family members are frequently over-expressed in primary human breast cancers (Ishizawar and Parsons, 2004).
Src Regulates p27
Here we demonstrate cSrc mediated p27 phosphorylation in vitro and in vivo at both Y74 and Y88. Mutation converting p27 Y89 to F reduced in vitro Src phosphorylation of p27 by only 12% and p27Y89F co-transfected with Src showed no reduction in reactivity with αpY-4G10 antibody in vivo compared to p27WT. While loss of Src phosphorylation of Y89 showed little effect in vivo, this site may modulate phosphorylation of Y74 and Y88.
Both putative tyrosine phosphorylation sites, p27Y74 and p27Y88, form contacts with Cdk2. The partial crystal structure of cyclin A-Cdk2-bound p27 reveals that the region between Y74 and Y88 of p27 forms a clamp around the N-lobe of Cdk2 (Russo et al., 1996). Y74 is partially buried in the N-terminal β-sheet of Cdk2, forming hydrophobic interactions with L25, V30, L67 and V79 on the outer surface of the Cdk2 N-lobe. The aromatic ring of p27Y88 forms Van der Waals contacts to F80, F82 and L134 of Cdk2, and its hydroxyl group makes two hydrogen bonds to Cdk2 Glu 81 and L83 (Russo et al., 1996). The side chain of p27Y88 occupies the catalytic cleft of Cdk2 and mimics the contacts made by the purine base of ATP (Russo et al., 1996). NMR data indicate that Y88 phosphorylation of p27 ejects the 310 helix from the catalytic cleft of Cdk2, but this phosphorylation alone did not disrupt p27 binding to cyclin A-Cdk2. (Grimmler et al., in print). This would explain the reduced inhibitory action of p27 toward cyclin E-Cdk2 following Src kinase pre-treatment in vitro.
While Abl and Lyn appear to phosphorylate predominantly Y88, both Y74 and Y88 were phosphorylated by Src. Src phosphorylated p27 showed a decreased steady state association with cyclin E-Cdk2 both in vitro and following co-transfection of p27WT with activated Src in vivo. Phosphorylation of Y74 together with Y88 may not only mediate loss of p27 inhibitory activity against cyclin E-Cdk2, but also promote dissociation of p27 from the complex. Thus, different tyrosine kinases may have different consequences in vivo on p27 function and stability. Further studies will be required to elucidate the full structural implications of these tyrosine phosphorylation events on p27 function and to define the profile of tyrosine kinases that regulate these sites in vivo in response to mitogen stimulation. In certain cancers, constitutive Y88 phosphorylation, without concurrent Y74 phosphorylation may permit activation of p27 associated cyclin-Cdk2. NMR structural data support this model (Grimmler et al., in print).
Estrogen:
ER binding has been shown to recruit Src leading to MEK activation (Migliaccio et al., 1996; Song et al., 2004) and estrogen also reduces p27 (Cariou et al., 2000). The present data suggest that estrogen-dependent Src activation regulates p27 proteolysis. Src inhibition and siRNA increased cellular p27. Moreover, both cell lines and primary breast cancers with activated Src showed reduced p27. Src was rapidly activated by mitogen stimulation and cellular p27-Src co-precipitation preceded the reduction of p27 and loss of cyclin E-Cdk2 from p27 prior to S phase entrance. Src inhibition prolonged the p27 half-life and Src induction increased cellular pY74p27, pT187p27, and reduced p27 half-life. These data support a model in which tyrosine phosphorylation of p27 by Src would reduce p27-mediated inhibition of cyclin E-Cdk2, liberating this kinase to phosphorylate its approximated inhibitor at T187, promoting p27 ubiquitination by SCFSkp2, and its degradation (Pagano et al., 1995; Sheaff et al., 1997; Carrano et al., 1999). Tyrosine phosphorylation of p27 would thus regulate the transition of p27 from inhibitor to substrate of cyclin-Cdk2. p27 phosphorylation at T187 by cyclin A-Cdk2 in vitro was enhanced by prior Abl-phosphorylation of p27 and followed a pseudo-unimolecular mechanism, thus pY-p27 appears to be more readily phosphorylated by cyclin-Cdk2 compared to non-phosphorylated p27 (Grimmler et al., in print). Src transfection in NIH3T3 and Rat-1 fibroblasts also increased Cdk2 activity and reduced p27 (Riley et al., 2001; Johnson et al., 1998). Moreover, induced overexpression of dominant negative Src increased p27 t1/2 in MCF-7 (Gonzalez et al., 2006). The role of cYes in response to RTKs and estrogen is less well characterized than that of cSrc. Given our observation that cYes can also phosphorylate p27 in vitro, the potential for cooperative effects of cYes and cSrc on p27 function warrants further investigation.
A role for Src inhibitors in treatment of antiestrogen resistant breast cancer
ER blockade by tamoxifen or estrogen deprivation by aromatase inhibitors is of therapeutic utility in a majority of breast cancers that express the ER. p27 is required for G1 arrest by tamoxifen or estrogen deprivation (Cariou et al., 2000). Despite initial responses, the development of antiestrogen resistance limits treatment efficacy. EGFR and Her2/ErbB2 over-expression can confer tamoxifen resistance in cultured lines (Benz et al., 1992; Pietras et al., 1995; Donovan et al., 2001). Since these RTKs activate Src and given our observation that Src impairs the Cdk2 inhibitory action of p27, we tested if Src inhibition could restore antiestrogen responsiveness to resistant cells.
AZD0530 is a potent, orally available dual inhibitor of Abl and Src family kinases (Ple et al., 2004). It showed potent (<uM IC50) antiproliferative activity in 17 human cancer cell lines and inhibited growth of 6/13 xenograft tumor models tested. AZD0530 inhibits cell motility and invasion in vitro (Hiscox et al., 2005) and inhibits metastasis in animal models in vivo (Green et al, AACR abstract, 2005)
In three different Src-activated ER positive breast cancer lines, AZD0530 or ER-saturating doses of tamoxifen each, when given alone, caused partial cell cycle inhibition. AZD0530 together with tamoxifen increased p27, inhibited cyclin E-Cdk2, and caused G1 arrest. In contrast, in lines lacking Src activation, addition of AZD0530 did not enhance the antiproliferative effect of tamoxifen. Our analysis showed a statistical association between low nuclear p27 and Src activation in 482 primary human breast cancers. Of 392 ER positive cancers assayed, 127/339 (37%) showed Src activity scores of 2–3. This proportion is similar to the fraction of ER positive breast cancers that manifest de novo antiestrogen resistance. These data raise the provocative possibility that Src inhibitors could restore response to tamoxifen in breast cancers with Src activation. AZD0530 has shown good bioavailability (AstraZeneca, unpublished) and is in phase II trials for human cancers. The present data not only identify a novel mechanism whereby RTKs via Src promote p27 phosphorylation and proteolysis, they also provide a rationale for testing Src inhibitors together with antiestrogens in xenografts and to further explore their therapeutic potential in human breast cancer.
EXPERIMENTAL PROCEDURES
Cell Culture
MCF-7, MDA-MB-361, BT474, T-47D and ZR75-1 were cultured as in (Cariou et al., 2000). MCF-7 were depleted of estradiol using 5% charcoal-stripped FBS for 48 h. Cell cycle entry was stimulated by estradiol (1x10−8 M) and insulin (10 μg/ml).
Plasmids, site-directed mutagenesis, recombinant protein and transfection
Conversion of tyrosine 74, 88, 89, 74/88, 74/89 and 74/88/89 to phenylalanine in pET28a p27WT used QuickChange mutagenesis kit (Stratagene). Recombinant p27 proteins were purified by nickel-agarose FPLC.
The WT and mutant p27 cDNAs were subcloned into pEYFP-C1 and transfected into MCF-7 as in(Connor et al., 2003). 50 nM cSrc siRNA and non-specific control oligos (Dharmacon) were transfected using lipofectamineTM 2000 (Invitrogen). Activated human cSrc PCI-Src Y530F, was provided by D. Fujita (U of Calgary). Human Cdk2 and cyclin E were produced from baculovirus-infected Sf9 cells as in (Ciarallo et al., 2002).
Antibodies
Antibody sources were: p27 (Transduction Labs and Santa Cruz); anti-PSTAIRE (Cariou et al., 2000); Cdk2 (Santa Cruz); cyclin E1 mAbs (E12 and E172) from E. Harlow (Boston, MA), anti-phosphotyrosine Ab4G10 (Upstate); Src GD11 (Upstate) and β-actin (Sigma). The polyclonal αpY74p27 antibody was developed by Invitrogen/Zymed using a cys.-KLH-coupled peptide HKPLEGKpYEWQE for rabbit immunization and purified by column chromatography.
Assays of immunoprecipitable and recombinant Src kinase activity
Cells were lysed in 40C NP40 lysis buffer (Cariou et al., 2000) and Src precipitated with GD11 (Upstate). IP cSrc kinase assays were as described (Egan et al., 1999).
For in vitro Src kinase reactions, 400 ng recombinant cSrc (Upstate) was reacted with (10 μg) his-p27WT or pYp27 mutant with 2 mM ATP for 2 h in Src kinase buffer as in (Egan et al., 1999). Radioactivity in p27 was quantitated by Molecular Dynamics PhosphoImager and ImageQuant software.
Src Effects on p27-cyclin E-Cdk2 binding and Cyclin E-Cdk2 activity
pYp27 was isolated from Src kinase reactions by precipitation with mAb4G10. Recombinant cyclin E-Cdk2 was mixed with equal amounts of pYp27 or with p27 that was either mock treated or treated with Src inactivated by PP1 and boiled for 5 minutes (dead Src). Cyclin E, Cdk2 and p27 immune complexes were analyzed by Western. Cyclin E-Cdk2 activity was assayed per (Cariou et al., 2000).
Immunoblotting and immunoprecipitation
Cells were lysed in 40C 0.1% NP40, 1% NP40 or RIPA buffer. Westerns of cyclin E, p27, Cdk2, and IP-Western assays of cyclin E, Cdk2 and p27 complexes were as in (Donovan et al., 2001). Cellular Src was detected in p27 precipitated from 1mg lysate. Nuclear and cytoplasmic subcellular fractionation was as described (Connor et al., 2003).
Inducible Src lines and cycloheximide chase
Src Y530F cDNA was cloned into pIND (Invitrogen) and transfected into MCF-7 with an integrated pVgRXR vector (Invitrogen). Src was induced with 2 μM ponasterone A (PA). Two clones (MCF-pINDSrc2 and MCF-pINDSrc12) had no Src expression above baseline without PA, but high Src levels 8–24 h after PA. These were arrested by estrogen deprivation as above for 72 hours and 2 μM PA added or not for the last 10 h of estrogen deprivation. Cells were then transferred to 0.1% cFBS and 10nM estradiol was added for 6h and p27 t1/2 assayed by cycloheximide. Cycloheximide 100 μg/ml was added and the decrease in p27 quantitated over time. PA alone did not alter p27 levels in parental MCF-7.
For detection of T187 phosphorylated p27, at 6 h after estrogen addition with or without prior induction of Src, cells were treated with MG132 for a further 6 h and then pT187p27 blotted.
For Src effects on p27 stability, MDA-MB361 were treated for 6 h with 1 μM AZD0530 prior to CHX chase. p27 was quantitated by densitometry in multiple ECL exposures of different biologic experiments and p27 t1/2 quantitated by linear regression.
Breast cancer patient population and tumor immunohistochemistry
The Henrietta Banting Breast Tumor Bank contains paraffin embedded breast cancers resected between 1987 and 1997 at Women’s College Hospital, Toronto, Canada. The database includes clinical and pathologic features at diagnosis and patient outcome. A tissue microarray containing over 500 primary cancers was constructed (Kononen et al., 1998), using three different, 0.6 mm diameter cylinders from each original tumor block.
p27 immunohistochemistry was as described (Catzavelos et al., 1997) using monoclonal p27Kip1 antibody and biotin-labeled anti-mouse IgG, preformed avidin-biotin-peroxidase complex and diaminobenzidine(AB) substrate. p27 was scored as % tumor nuclei positive (0–24% = low p27 or 25% = high p27) as in (Catzavelos et al., 2006).
Activated Src was detected by blocking with 5% H2O2, and 10% normal goat serum followed by reaction with 1:33 dilution of anti-phospho-cSrc (pY416-Src) (Stressgen) overnight at 40 C, reaction with biotinylated goat anti-rabbit antibody (Zymed) and then streptavidin peroxidase conjugate and diaminibenzidnine. Of 523 cases retrieved, 16 lacked Src staining, and in 25, the p27 stain was inadequate due to poor tissue fixation or no tumor on the TMA. The predominant staining intensity was scored as: 0, no staining; 1, weak staining; 2, moderate staining; and 3, intense staining. The mean score for three cores for each tumor was used in statistical analysis. Institutional Ethics Board approval was obtained prior to study initiation. Correlations between p27 and pY416-Src scores and other clinical-pathologic data in 482 tumors was assayed using Fischer’s exact test to compare means and chi-square analyses to compare frequencies.
Flow cytometric analysis
BrdU pulse labeling and flow cytometry were as in (Cariou et al., 2000).
Cell cycle effects of tamoxifen and AZD0530
AZD0530 (AstraZeneca) was used as described (Ple et al., 2004). The lowest dose of 4-OH-TAM or AZD0530 to cause maximal cell cycle inhibition when used alone was used for assays of both drugs together. Cells were incubated with vehicle alone, 1 μM 4-OH-TAM, 1 μM AZD0530 or both and recovered 48 hours later for flow cytometry and protein analysis.
Supplementary Material
Acknowledgments
This work was supported by DOD W81XWH-04-1-0392 to IC, by NIH RO1-CA105118 to JMS and by the Braman Family Breast Cancer Institute of UM Sylvester Comprehensive Cancer Center. LH is supported by FWF grant P18873.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Agrawal D, Hauser P, McPherson F, Dong F, Garcia A, Pledger WJ. Repression of p27kip1 synthesis by platelet-derived growth factor in BALB/c 3T3 cells. Mol Cell Biol. 1996;16:4327–4336. doi: 10.1128/mcb.16.8.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkarain A, Jordan R, Slingerland J. p27 deregulation in breast cancer: prognostic significance and implications for therapy. J Mammary Gland Biol Neoplasia. 2004;9:67–80. doi: 10.1023/B:JOMG.0000023589.00994.5e. [DOI] [PubMed] [Google Scholar]
- Belsches-Jablonski AP, Biscardi JS, Peavy DR, Tice DA, Romney DA, Parsons SJ. Src family kinases and HER2 interactions in human breast cancer cell growth and survival. Oncogene. 2001;20:1465–1475. doi: 10.1038/sj.onc.1204205. [DOI] [PubMed] [Google Scholar]
- Benz CC, Scott GK, Sarup JC, Johnson RM, Tripathy D, Coronado E, Shepard HM, Osborne CK. Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res Treat. 1992;24:85–95. doi: 10.1007/BF01961241. [DOI] [PubMed] [Google Scholar]
- Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 2006;20:47–64. doi: 10.1101/gad.1384406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscardi JS, Ishizawar RC, Silva CM, Parsons SJ. Tyrosine kinase signalling in breast cancer: Epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000;2:203–210. doi: 10.1186/bcr55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274:8335–8343. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- Cariou S, Donovan JC, Flanagan WM, Milic A, Bhattacharya N, Slingerland JM. Down-regulation of p21WAF1/CIP1 or p27Kip1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. Proc Natl Acad Sci U S A. 2000;97:9042–9046. doi: 10.1073/pnas.160016897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nature Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- Catzavelos C, Arnaout A, Bull SB, Pinnaduwage D, Hanna WM, Prithcard KI, Andrulis I, Slingerland JM. p27 is prognostic for node negative breast cancer, predicts tamoxifen resistance and may enhance prognostic utility of Her2/ErdB2 utitlity. In review by the Journal of the National Cancer Institute 2006 [Google Scholar]
- Catzavelos C, Bhattacharya N, Ung YC, Wilson JA, Roncari L, Sandhu C, Shaw P, Yeger H, Morava-Protzner I, Kapusta L, Franssen E, Pritchard KI, Slingerland JM. Decreased levels of the cell-cycle inhibitor p27Kip1 protein: prognostic implications in primary breast cancer. Nat Med. 1997;3:227–230. doi: 10.1038/nm0297-227. [DOI] [PubMed] [Google Scholar]
- Chen T, George JA, Taylor CC. Src tyrosine kinase as a chemotherapeutic target: is there a clinical case? Anticancer Drugs. 2006;17:123–131. doi: 10.1097/00001813-200602000-00002. [DOI] [PubMed] [Google Scholar]
- Chu I, Blackwell K, Chen S, Slingerland J. The dual ErbB1/ErbB2 inhibitor, lapatinib ( GW572016), cooperates with tamoxifen to inhibit both cell proliferation- and estrogen-dependent gene expression in antiestrogen-resistant breast cancer. Cancer Res. 2005;65:18–25. [PubMed] [Google Scholar]
- Ciarallo S, Subramaniam V, Hung W, Lee JH, Kotchetkov R, Sandhu C, Milic A, Slingerland JM. Altered p27(Kip1) phosphorylation, localization, and function in human epithelial cells resistant to transforming growth factor beta-mediated G(1) arrest. Mol Cell Biol. 2002;22:2993–3002. doi: 10.1128/MCB.22.9.2993-3002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor MK, Kotchetkov R, Cariou S, Resch A, Lupetti R, Beniston RG, Melchior F, Hengst L, Slingerland JM. CRM1/Ran-Mediated Nuclear Export of p27(Kip1) Involves a Nuclear Export Signal and Links p27 Export and Proteolysis. Mol Biol Cell. 2003;14:201–213. doi: 10.1091/mbc.E02-06-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan JC, Milic A, Slingerland JM. Constitutive MEK/MAPK activation leads to p27(Kip1) deregulation and antiestrogen resistance in human breast cancer cells. J Biol Chem. 2001;276:40888–40895. doi: 10.1074/jbc.M106448200. [DOI] [PubMed] [Google Scholar]
- Egan C, Pang A, Durda D, Cheng HC, Wang JH, Fujita DJ. Activation of Src in human breast tumor cell lines: elevated levels of phosphotyrosine phosphatase activity that preferentially recognizes the Src carboxy terminal negative regulatory tyrosine 530. Oncogene. 1999;18:1227–1237. doi: 10.1038/sj.onc.1202233. [DOI] [PubMed] [Google Scholar]
- Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Polyak K, Firpo E, Tsai L, Broudy V, Perlmutter RM, Kaushansky K, Roberts JM. A syndrome of multi-organ hyperplasia with features of gigantism, tumorigenesis and female sterility in p27Kip1-deficient mice. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- Florenes VA, Bhattacharya N, Bani MR, Ben-David Y, Kerbel RS, Slingerland JM. TGF-beta mediated G1 arrest in a human melanoma cell line lacking p15INK4B: evidence for cooperation between p21Cip1/WAF1 and p27Kip1. Oncogene. 1996;13:2447–2457. [PubMed] [Google Scholar]
- Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta. 2002;1602:114–130. doi: 10.1016/s0304-419x(02)00040-9. [DOI] [PubMed] [Google Scholar]
- Gonzalez L, Agullo-Ortuno MT, Garcia-Martinez JM, Calcabrini A, Gamallo C, Palacios J, Aranda A, Martin-Perez J. Role of c-Src in human MCF7 breast cancer cell tumorigenesis. J Biol Chem. 2006;281:20851–20864. doi: 10.1074/jbc.M601570200. [DOI] [PubMed] [Google Scholar]
- Gopfert U, Kullmann M, Hengst L. Cell cycle-dependent translation of p27 involves a responsive element in its 5'-UTR that overlaps with a uORF. Hum Mol Genet. 2003;12:1767–1779. doi: 10.1093/hmg/ddg177. [DOI] [PubMed] [Google Scholar]
- Hara T, Kamura T, Nakayama K, Oshikawa K, Hatakeyama S, Nakayama KI. Degradation of p27Kip1 at the G0-G1 transition mediated by a Skp2-independent ubiquitination pathway. J Biol Chem. 2001;276:48937–48943. doi: 10.1074/jbc.M107274200. [DOI] [PubMed] [Google Scholar]
- Hara T, Kamura T, Kotoshiba S, Takahashi H, Fujiwara K, Onoyama I, Shirakawa M, Mizushima N, Nakayama KI. Role of the UBL-UBA protein KPC2 in degradation of p27 at G1 phase of the cell cycle. Mol Cell Biol. 2005;25:9292–9303. doi: 10.1128/MCB.25.21.9292-9303.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengst L, Dulic V, Slingerland JM, Lees E, Reed SI. A cell cycle-regulated inhibitor of cyclin-dependent kinases. Proc Natl Acad Sci U S A. 1994;91:5291–5295. doi: 10.1073/pnas.91.12.5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengst L, Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996;271:1861–1864. doi: 10.1126/science.271.5257.1861. [DOI] [PubMed] [Google Scholar]
- Hiscox S, Morgan L, Green TP, Barrow D, Gee J, Nicholson RI. Elevated Src activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cells. Breast Cancer Res Treat. 2005:1–12. doi: 10.1007/s10549-005-9120-9. [DOI] [PubMed] [Google Scholar]
- Irby RB, Mao WG, Coppola D, Kang J, Loubeau JM, Trudeau W, Karl R, Fujita DJ, Jove R, Yeatman TJ. Activating SRC mutation in a subset of advanced human colon cancers. Nature Genetics. 1999;21:187–190. doi: 10.1038/5971. [DOI] [PubMed] [Google Scholar]
- Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636–5642. doi: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip 1on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem. 2002;277:14355–14358. doi: 10.1074/jbc.C100762200. [DOI] [PubMed] [Google Scholar]
- Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell. 2004;6:209–214. doi: 10.1016/j.ccr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Johnson D, Frame MC, Wyke JA. Expression of the v-Src oncoprotein in fibroblasts disrupts normal regulation of the CDK inhibitor p27 and inhibits quiescence. Oncogene. 1998;16:2017–2028. doi: 10.1038/sj.onc.1201727. [DOI] [PubMed] [Google Scholar]
- Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, Yoshida M, Nakayama K, Nakayama KI. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004;6:1229–1235. doi: 10.1038/ncb1194. [DOI] [PubMed] [Google Scholar]
- Kardinal C, Dangers M, Kardinal A, Koch A, Brandt DT, Tamura T, Welte K. Tyrosine phosphorylation modulates binding preference to cyclin-dependent kinases and subcellular localization of p27Kip1 in the acute promyelocytic leukemia cell line NB4. Blood. 2006;107:1133–1140. doi: 10.1182/blood-2005-05-1771. [DOI] [PubMed] [Google Scholar]
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27Kip1. Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- Koff A, Ohtsuki M, Polyak K, Roberts JM, Massague J. Negative regulation of G1 in mammalian cells: inhibition of cyclin E-dependent kinase by TGF-beta. Science. 1993;260:536–539. doi: 10.1126/science.8475385. [DOI] [PubMed] [Google Scholar]
- Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP. Tissue microarrays for high-throughput molecular profiling of tumor specimens 1. Nat Med. 1998;4:844–847. doi: 10.1038/nm0798-844. [DOI] [PubMed] [Google Scholar]
- Lane HA, Beuvink I, Motoyama AB, Daly JM, Neve RM, Hynes NE. ErbB2 potentiates breast tumor proliferation through modulation of p27(Kip1)-Cdk2 complex formation: receptor overexpression does not determine growth dependency. Mol Cell Biol. 2000;20:3210–3223. doi: 10.1128/mcb.20.9.3210-3223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenferink AEG, Simpson Jf, Shawver LK, Coffey RJ, Forbes JT, Arteaga CL. Blockade of the epidermal growth factor receptor tyrosine kinase suppresses tumorigenesis in MMTV/Neu plus MMTV/TGF-alpha bigenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:9609–9614. doi: 10.1073/pnas.160564197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- Malek NP, Sundberg H, McGrew S, Nakayama K, Kyriakidis TR, Roberts JM. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature. 2001;413:323–327. doi: 10.1038/35095083. [DOI] [PubMed] [Google Scholar]
- Migliaccio A, DiDomenico M, Castona C, DeFalco A, Bontempo P, Nola E, Auricchio F. Tyrosine kinas/p21ras/MAP-kinase pathway activation by estradiol receptor complex in MCF-7 cells. EMBO J. 1996;15:1292–300. [PMC free article] [PubMed] [Google Scholar]
- Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, Pagano M. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes & Dev. 1999;13:1181–1189. doi: 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- Nicholson S, Wright C, Sainsbury JR, Halcrow P, Kelly P, Angus B, Farndon JR, Harris AL. Epidermal growth factor receptor (EGFR) as a marker for poor prognosis in node-negative breast cancer patients: neu and tamoxifen failure. Journal of Steroid Biochemistry and Molecular Biology. 1990;37:811–814. doi: 10.1016/0960-0760(90)90424-j. [DOI] [PubMed] [Google Scholar]
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- Pietras RJ, Arboleda J, Reese DM, Wongvipat N, Pegram MD, Ramos L, Gorman CM, Parker MG, Sliwkowski MX, Slamon DJ. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene. 1995;10:2435–2446. [PubMed] [Google Scholar]
- Ple PA, Green TP, Hennequin LF, Curwen J, Fennell M, Allen J, Lambert-Van Der Brempt C, Costello G. Discovery of a new class of anilinoquinazoline inhibitors with high affinity and specificity for the tyrosine kinase domain of c-Src 1. J Med Chem. 2004;47:871–887. doi: 10.1021/jm030317k. [DOI] [PubMed] [Google Scholar]
- Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- Radeva G, Petrocelli T, Behrend E, Leung-Hagesteijn C, Filmus J, Slingerland J, Dedhar S. Overexpression of the integrin-linked kinase promotes anchorage-independent cell cycle progression. J Biol Chem. 1997;272:13937–13944. doi: 10.1074/jbc.272.21.13937. [DOI] [PubMed] [Google Scholar]
- Riley D, Carragher NO, Frame MC, Wyke JA. The mechanism of cell cycle regulation by v-Src. Oncogene. 2001;20:5941–5950. doi: 10.1038/sj.onc.1204826. [DOI] [PubMed] [Google Scholar]
- Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, Meloche S. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J. 2001;20:6672–6682. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes & Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Slingerland JM, Hengst L, Pan CH, Alexander D, Stampfer MR, Reed SI. A novel inhibitor of cyclin-Cdk activity detected in transforming growth factor beta-arrested epithelial cells. Mol Cell Biol. 1994;14:3683–3694. doi: 10.1128/mcb.14.6.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song RX, Barnes CJ, Zhang ZG, Bao YD, Kumar R, Santen RJ. The role of Shc and insulin-like qrowth factor 1 receptor in mediating the translocation of estrogen receptor a to the plasma membrane. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2076–2081. doi: 10.1073/pnas.0308334100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci U S A. 1999;96:1415–1420. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui S, Ohno S, Murakami S, Hachitanda Y, Oda S. Prognostic value of epidermal growth factor receptor (EGFR) and its relationship to the estrogen receptor status in 1029 patients with breast cancer. Breast Cancer Research and Treatment. 2002;71:67–75. doi: 10.1023/a:1013397232011. [DOI] [PubMed] [Google Scholar]
- Vlach J, Hennecke S, Amati B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J. 1997;16:5334–5344. doi: 10.1093/emboj/16.17.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HY, Zhou BP, Hung MC, Lee MH. Oncogenic signals of HER-2/neu in regulating the stability of the cyclin-dependent kinase inhibitor p27. J Biol Chem. 2000;275:24735–24739. doi: 10.1074/jbc.C000147200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.