

Abstract
In this review, we summarize the potential functional roles of transient receptor potential (TRP) channels in the vertebrate inner ear. The history of TRP channels in hearing and balance is characterized at great length by the hunt for the elusive transduction channel of sensory hair cells. Such pursuit has not resulted in unequivocal identification of the transduction channel, but nevertheless revealed a number of candidates, such as TRPV4, TRPN1, TRPA1, and TRPML3. Much of the circumstantial evidence indicates that these TRP channels potentially play significant roles in inner ear physiology. Based on mutations in the corresponding mouse genes, TRPV4 and TRPML3 are possible candidates for human hearing, and potentially also balance disorders. We further discuss the role of the invertebrate TRP channels Nanchung, Inactive, and TRPN1 and how the functional analysis of these channels provides a link to vertebrate hearing and balance. In summary, only a few TRP channels have been analyzed thus far for a prospective role in the inner ear, and this makes the search for additional TRPs associated with inner ear function quite a tantalizing endeavor.
Introduction
Inner ear disorders affect our senses of hearing and balance. Loss of hearing is one of the most prevailing disabilities, and it has been estimated that more than 275 million people worldwide have impairment of auditory function (http://www.who.int/mediacentre/factsheets/fs300/en/). Likewise, about 30% of people over the age of 65 are affected by dizziness or vertigo [1]. In the United States, about 30 million people are affected by hearing loss and more than 90 million Americans, age 17 and older, have experienced dizziness or balance problems (http://www.vestibular.org/vestibular-disorders/statistics.php) [2, 3].
The principal causes for hearing loss are genetic disposition (2-3 children in 1,000 are born with partial to profound compromise of auditory function), aging, noise exposure, certain infections, and ototoxic drugs (http://www.nidcd.nih.gov/health/hearing/). Balance disorders have similar causes, with infections or inflammations of the labyrinth or the vestibular nerve probably being the predominant reasons (http://www.nidcd.nih.gov/health/balance/) [4]. Degeneration and death of sensory hair cells is causal in greater than 80% of individuals with hearing loss and chronic balance disorders [5].
Sensory hair cells convert mechanical stimulation into electrical signals. This process, also known as mechanotransduction, happens in the mechanosensitive organelle, the hair bundle, which protrudes from the cell’s apical surface. The hair bundle is composed of a few tens to hundreds of actin-rich stereocilia that are arranged in a staircase fashion. Stereocilia are tapered at their base, which enables them to pivot near their basal insertion points. In addition, the stereocilia are organized in stepped rows of increasing length. Individual stereocilia are laterally linked along their longitudinal shafts, which allows the whole hair bundle to move as a unit. Mechanical deflection of the bundle toward the tallest stereocilia leads to shearing motions between adjacent stereocilia that are exerted by tip links, connectors between the tips of shorter stereocilia with its taller neighbor. The consequential increase of mechanical tension in the transduction apparatus increases the open probability of mechanically gated ion channels located at or near the tips of stereocilia, resulting in an influx of cations that depolarizes the hair cell, thereby generating a receptor potential (for reviews, see [6, 7]).
Invertebrates provide a link between TRP channels and hearing
The most prominent invertebrate mechanoreceptor organs are the mechanosensory tactile bristles and the chordotonal organs of Drosophila melanogaster, which are localized, for example, inside the antenna. The antennal chordotonal organ, also known as the Johnston’s organ, mediates the fly’s sense of hearing. Sound wave-evoked vibrations of the antenna are transmitted to the cilia of the chordotonal mechanosensory neurons. The chordotonal organs are quite similar in their architecture with the tactile bristle mechanosensors (for detailed diagrams, we refer the reader to references [8, 9]): movement of the hollow hair shaft of the tactile bristle acts as a lever arm and deflects the dendritic tip of the mechanosensory neuron that extends into the base of the shaft [10, 11]. This deflection of the bristle hair elicits a directionally sensitive and adapting receptor current that is very similar in its features to the mechanoreceptor current of vertebrate hair cells [12, 13]. These similarities are also manifested in the extracellular milieu surrounding the ciliated dendrites of the Drosophila mechanosensory neurons. This extracellular fluid is rich in K+ and has an unusual low concentration of Ca2+, which is very similar to the ionic composition of the endolymph surrounding the stereocilia of vertebrate hair cells [14]. Deflection of the bristle shaft toward the fly’s cuticula gates the mechanosensory transduction channels allowing influx of K+ and other ions into the neuron, which elicits a transduction current [12]. The very short latency of this response indicates a direct gating and argues against an involvement of second messenger systems. Overall, it has been proposed that the invertebrate mechanosensors display structural and molecular similarities with vertebrate hair cells [9].
Mutations in the invertebrate TRP genes TRPN1 (also known as nompC), Inactive (iav), and Nanchung (nan) have been shown to affect transduction in bristle and chordotonal mechanoreceptors of Drosophila melanogaster [8, 12, 15].
Vertebrate orthologues of TRPN1 have been identified only in zebrafish and Xenopus. Interference with zebrafish TRPN1 affects hair cell function, suggesting a role in hair cell transduction [16, 17]. It is noteworthy that TRPN1 appears to be absent from all sequenced mammalian genomes. Iav and nan are essential for hearing in Drosophila melanogaster [8, 15] and are the closest homologues of TRPV4, a vertebrate TRP channel that has been cloned from a chicken cochlear duct cDNA library. TRPV4 is expressed in hair cells and marginal cells of the stria vascularis, a cochlear organ that is responsible for creating the unique ionic milieu of the endolymph, the inner ear fluid that is essential for proper hair cell function [18].
Naturally, vertebrate TRPN1 and TRPV4 genes have been considered candidates for the elusive mechanoelectrical transduction channel of vertebrate hair cells. A number of other TRP channels, such as TRPML3 and TRPA1 have also been put on the map, inter alia, based on their expression in sensory hair cells. In the following sections, we will review each of these channels, particularly with regard to their latent etiological potential to cause inner ear disorders.
TRPN1 – a shrouded mystery channel, or end of story?
TRPN1 was identified initially through positional cloning in fruitfly loss-of-function mutants [12]. It is expressed in sensory bristle cells, and contains 29 amino-terminal ankyrin repeats, followed by six predicted transmembrane-spanning domains. TRPN1 has a low, but significant homology to other TRP family members, and thus earned a place as a unique subfamily member of the TRPN subfamily [19].
TRPN1 fly mutants lacked receptor potentials and currents in their tactile bristles, but continued to display a small non-adapting mechanosensory current [12]. This non-adapting transduction current in chordotonal organs implies that either a second “primary” transduction channel exists, or that the TRPN1 channel acts as a mechanosensitive amplifier for another mechanically-gated transduction channel [9, 20]. It has been proposed that this other channel is composed of the TRPV4-related proteins Inactive and Nanchung [8, 15, 21].
Recently, vertebrate TRPN1 orthologues were discovered in zebrafish (Danio rerio) and frog (Xenopus laevis) [16, 17]. TRPN1 mRNA was detected in several zebrafish organs including the ear [16]. Moreover, Xenopus TRPN1 protein was immunologically detected in hair cells of the lateral line and the vestibular system. In these hair cells TRPN1 immunoreactivity was found in the kinocilium, which is the single axonemal cilium of hair cells [17]. To explore the functionality of TRPN1 in zebrafish, Sidi et al. [16] used antisense morpholino oligonucleotides, which disrupt the native expression of TRPN1 in fish larvae. As a consequence of this disruption, the TRPN1-deficient animals displayed abnormal acoustic startle response and atypical swimming behavior that is characteristic of auditory and vestibular dysfunctions, respectively. Even though this result is indicative that TRPN1 might be part of the mechanotransduction machinery in zebrafish, questions still remain. For example, TRPN1 immunoreactivity was only observed in kinocilia, but not in stereocilia [17]. Stereocilia are undisputedly the principal location of the mechanoelectrical transduction machinery, since not all hair cells have a kinocilium (for example mammalian cochlear hair cells) or when they do, they only possess them transiently during development. Moreover, small interfering RNAs or morpholino antisense oligonucleotides can have non-specific, off-target effects ([22, 23], but see also [24]).
TRPN1’s potential role for inner ear function in vertebrates remains enigmatic. In anticipation of an in-depth electrophysiological characterization of the Danio and Xenopus TRPN1 proteins, we await additional information buttressing a role of this potential mechanotransduction channel in hearing and balance. The spotty phylogenetic appearance of the TRPN1 gene in some vertebrate classes, in tunicates, in sea urchins, and in invertebrate species is puzzling (Fig. 1). We speculate that mammalian TRPN1 may play a highly successful hide-and-seek game with molecular biologists or the whole gene disappeared, possibly independently, during early evolution of mammals as well as birds and other animal classes. Unless a mammalian TRPN1 is identified, we conclude that a potential role of TRPN1 in human hearing and balance disorders is out of the question.
Figure 1. Occurrence of TRPN1 genomic DNA or expressed sequence tags in various animal classes.
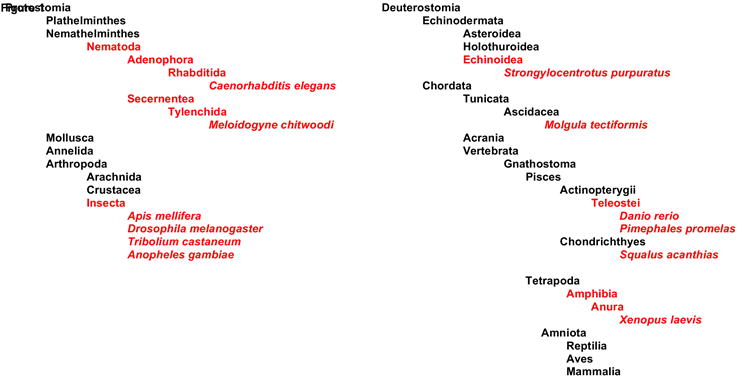
Classes, where existence of TRPN1 can be verified using GenBank database searches, are shown in red; individual species with a TRPN1 gene are italicized.
The sensory hair cell transduction channel candidate TRPA1 – functional relevance in hearing and balance to no avail?
Of the more than 30 known mammalian TRP channels, TRPA1 was put forward in 2004 as a compelling candidate for a component of the hair cell’s transduction machinery [25]. This candidacy was supported by an impressive accumulation of evidence starting with showing that expression of TRPA1 mRNA in vestibular hair cells is first detectable at embryonic day 15 (E15) and peaks at E17, which is the time point when vestibular hair cells display functional transduction. Corey and colleagues [25] also showed localization of TRPA1 protein to a place near the stereociliary tips of hair cells where the transduction apparatus is localized. When they disrupted the transduction apparatus by eliminating the tip links, they noticed that TRPA1 immunoreactivity at the stereociliary tips was reduced by up-to 85%. Knockdown of TRPA1 with morpholino oligonucleotides in the inner ears of zebrafish and with small interfering RNAs in mouse vestibular hair cells impeded hair cell transduction.
Despite these convincing, but circumstantial, findings and the demonstration that some of TRPA1’s electrophysiological features are similar with those of the transduction channel [26], a loss of gene function in the form of two independently generated knock out mouse lines revealed that TRPA1 is not essential for hair cell or inner ear function [27, 28]. In the face of this rather sobering evidence, it became obvious that TRPA1 might either be a redundant component of the transduction complex, or that the protein is outright irrelevant for hair cell transduction [24].
TRPV4 and a possible link to age-related hearing loss
TRPV4 has been cloned from a chicken cochlear duct cDNA library and is expressed in sensory hair cells and marginal cells of the stria vascularis, the cochlear organ that is generating the special ionic environment inside the scala media that is required for proper hair cell function (Fig. 2) [18, 29]. Similar to TRPA1, TRPV4 also has electrophysiological features reminiscent of those of the transduction channel. TRPV4 is gated by hypotonicity, a form of mechanosensation [18, 30], yet it has been suggested that the mechanical gating of TRPV4 is mediated by an indirect mechanism [31, 32], although direct mechanical gating of TRPV4 remains a possible activation modality. Reminiscent of a potential role for TRPV4 in hair cell mechanotransduction is: i) its homology with the invertebrate osmo- and mechanoreceptor OSM-9 [33-35], and ii) that TRPV4 is the closest vertebrate homologue of Inactive and Nanchung, two TRP genes that are essential for hearing in Drosophila melanogaster [8, 15].
Figure 2. Schematic picture of a cross section of the mammalian cochlea (adapted from [59, 60]).
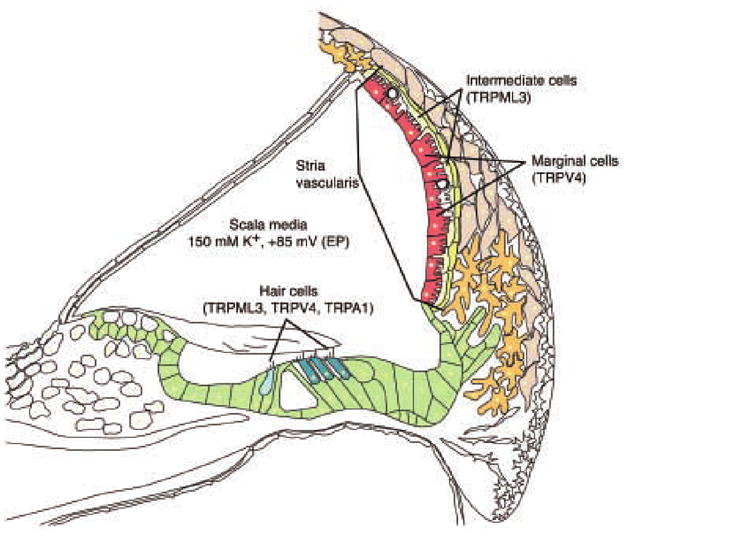
Specifically labeled are sensory hair cells and intermediate and marginal cells of the stria vascularis. The scala media, filled with potassium-rich endolymph and the endocochlear potential (EP) of +85 mV is indicated. Listed in parentheses after the individual cell type names are the TRP channels that were reported to be expressed in these cell types; expression of TRPML3 in the stria vascularis is supported by findings from our laboratory (Grimm and Heller, unpublished observations).
Consistent with an osmosensory function, TRPV4 is expressed in the epithelial cells of kidney tubules, and the circumventricular organ of the brain [36-38]. Beside activation by cell swelling and the cell swelling-induced arachidonic acid metabolite 5’,6’-epoxyeiconsatrienoic acid, TRPV4 is further activated by heat (>27 °C), by 4α-phorbol 12,13-didecanoate, and by bisandrographolide A, the active compound of an herbal plant extract used in traditional Eastern medicine [18, 31, 39-43].
Phenotypical analysis of TRPV4 homozygous knockout mice revealed deficiencies in fluid homeostasis regulation and cutaneous mechanosensation [37, 44, 45], but auditory brainstem responses (ABRs) and distortion product otoacoustic emissions (DPOAEs), two measures of auditory function, were indistinguishable between 3-month-old homozygous Trpv4 -/- animals and wild-type littermates (Liedtke and Heller, unpublished; [46]). Nevertheless, in a recent report it was shown that 6-month-old homozygous Trpv4 -/- animals have higher ABR thresholds and increased vulnerability to acoustic injury than their wild type littermates, suggesting that TRPV4 indeed plays an important role in the murine cochlea, but not likely in hair cell mechanotransduction [46]. Specifically, the 6-month-old Trpv4 -/- mice displayed significantly higher auditory thresholds than wild-type mice when hearing was assessed one week after exposure to noise at 128 dB sound pressure level.
It is interesting to note that the human TRPV4 gene is located within a 20-cM region of chromosome 12q21-24, a locus that was identified by genetic linkage analysis, for delayed-onset, progressive non-syndromic hearing loss codenamed DFNA25 [47]. Circumstantially, this human pathology coincides to a certain extent with the hearing problems reported in Trpv4 -/- mice. Mutations in human TRPV4 therefore may account for some rare forms of hereditary hearing loss.
In fruitflies, the TRPV4 orthologue, Nanchung, and its heteromeric partner, Inactive, are essential for hearing and are expressed in chordotonal neurons where they localize to their sensory cilia [8, 15]. Moreover, Nanchung and Inactive directly participate in mechanotransduction and have a modulatory role in auditory gain control of mechanical transduction [21]. In vertebrates, however, the high expression levels of TRPV4 in the stria vascularis [18] suggest that its function in the mammalian cochlea could be primarily to modulate ionic homeostasis. Nevertheless, we cannot completely exclude a role for TRPV4 in sensory hair cells, where the channel is expressed at low levels, particularly in older mammals.
TRPML3 – a clear link between a TRP and inner ear disorders
The discovery of Trpml3 mutations that confer the varitint-waddler (Va) mouse phenotype put this putative ion channel in the center stage of hearing transduction [48]. Classified as a member of the mucolipin (TRPML) subfamily of TRP ion channels, mucolipin 3 or TRPML3, is a protein with six predicted transmembrane domains, a putative pore loop sequence between transmembrane domains 5 and 6, and a distinctively long extracellular loop sequence between transmembrane domains 1 and 2 [48].
Homozygous Va mice are profoundly deaf, display vestibular defects (circling behavior, imbalance, head-bobbing, waddling), pigmentation deficiencies, sterility, and perinatal lethality [48, 49] (see also a more detailed review by Atiba-Davies and Noben-Trauth in this issue). Di Palma and colleagues [48] discovered that Va is an allele of Trpml3 where an alanine residue at amino acid position 419 is substituted with proline (A419P). This mutation is located within putative transmembrane domain 5, just proximal to the pore region. A second allele (VaJ) that arose in the Va background was found to carry an additional missense mutation resulting in an isoleucine to threonine amino acid substitution at position 362 (I362T) located between predicted transmembrane domains 3 and 4. VaJ animals display a phenotype with reduced severity, particularly in heterozygous animals [50].
Ultrastructural studies showed that deafness in Va and VaJ mice is due to progressive cellular aberration of sensory hair cells and intermediate cells in the stria vascularis (see also Fig. 2). Intermediate cells are melanocytes that play an essential role in maintaining the ionic composition of the endolymph, which generates the endocochlear potential (EP) in the inner ear fluid that is indispensable for proper sensory hair cell function. The cellular defects and the potential the loss of intermediate cells have been linked to a small or absent endocochlear potential (EP) in VaJ mice that live long enough to allow measurement of the EP [49]. Furthermore, sensory hair cell defects in varitint-waddler mice already become visible during embryonic development in the form of distinctive disorganization of stereociliary bundles [48, 51].
The TRPML subfamily consists of two more members, namely, TRPML1 and TRPML2. Recent observations indicate that TRPML1 is a functional ion channel with preference to monovalent cations [52]. The functional properties of TRPML2 and TRPML3, however, remain to be investigated. Interestingly, TRPML1 and TRPML2 proteins have been reported to localize to the lysosomal compartment of cells, and heterologous coexpression of either TRPML1 or TRPML2 with TRPML3 changes the subcellular distribution of TRPML3 from the endoplasmic reticulum to the lysosomes [53]. This observation suggests that TRPML3 may have distinct roles in cells, depending on the influence of TRPML1 or TRPML2. Note, however, that in hair cells, TRPML3 has been detected in vesicle-like intracellular structures as well as in the plasma membrane, particularly in stereocilia [48]. Heterologous expression of TRPML3 in HEK293, NIH 3T3, and HeLa cells also shows protein localization both in the plasma membrane and intracellularly, consistent with its observed distribution in sensory hair cells (Grimm and Heller, unpublished observations). These observations suggest that TRPML3 may play multiple roles at different subcellular locations.
Since the discovery of the A419P substitution in TRPML3, no further evidence has been reported as to the mechanism behind the vestibular abnormalities or hearing loss in the Va mice. Nonetheless, we speculate that the pathological correlates of the A419P mutation may be due to proline-induced structural anomaly in the TM5 domain that also could alter the pore helix, pore loop, and TM6 region. Prolines are known to break alpha-helical protein conformations, creating “kinks, hinges, or swivels” of the otherwise rigid helical structure [54]. While molecular modeling has obvious limitations, theoretical models are useful tools to further analyze mutational effects of amino acid substitutions within the core structure of protein domains, and it would be useful to further dissect the proline-induced structural change in TRPML3. Molecular modeling has been successfully used to investigate effects of amino acid substitions on distinct channel domains, e.g. pore helix, selectivity filter, etc.; see [55-58]). Nonetheless, a potential modification of the complete TM5-pore loop-TM6 core structure of TRPML3 manifested by a single amino acid deviation quite distant from the predicted central pore region warrants further investigation.
In summary, the relatively distinct varitint-waddler phenotype associated with mutations in Trpml3 indicates that human hereditary hearing and balance disorders may as well be linked to the human TRPML3 gene. The human TRPML3 locus, located at 1p22.3, therefore is a bona fide candidate for syndromic hearing loss, but perhaps also for non-syndromic types of hearing impairment.
Concluding Remarks
The history of TRP channels in hearing and balance disorders is characterized mainly by the hunt for the elusive sensory hair cell’s transduction channel. Although the vertebrate transduction channel still remains a mystery, a number of TRPs are connected to a potential physiological function in the inner ear. To date, TRPML3 and TRPV4 are the two most prevailing candidates for human hearing and balance disorders. Nevertheless, PCR-based expression analyses show that many TRP channels are expressed in the mammalian organ of Corti (Fig. 3). We consequently conclude that in-depth analysis of additional TRPs may reveal more connections of TRP channels with inner ear disorders. Likewise, we remain optimistic that a member of the TRP superfamily may turn out to be the mammalian sensory hair cell transduction channel, but as the saying goes, “it ain’t over until the fat lady sings.”
Figure 3. Expression analysis of TRP channels in the murine organ of Corti.
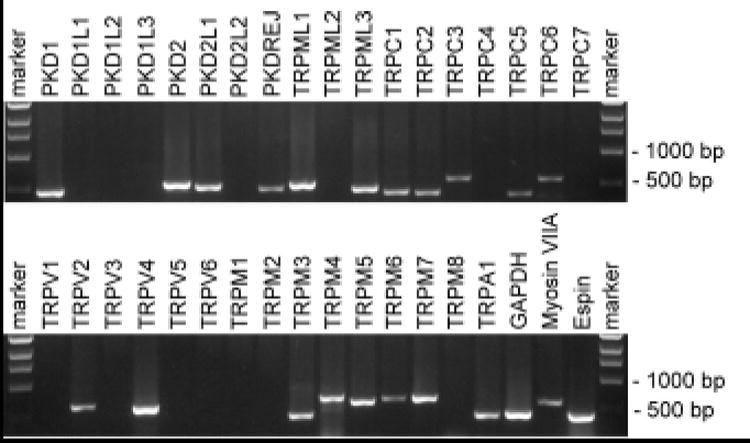
As a template, cDNA from an organ of Corti library (a gift from Dr. Bechara Kachar, NIH, Bethesda, USA) was used to PCR amplify TRP channel fragments with specific sense and antisense primers. Primers that did not give signals in this analysis were functionally verified with cDNA from other mouse tissues (not shown).
Acknowledgments
We thank Dr. Anthony Ricci and the members of our laboratory for critically reading the manuscript. SH is funded by NIH grant DC6167 and by Research Grant RPG0032/2004 from the Human Frontier Science Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Colledge N, Lewis S, Mead G, Sellar R, Wardlaw J, Wilson J. J Neurol Neurosurg Psychiatry. 2002;72:587–9. doi: 10.1136/jnnp.72.5.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li H, Corrales CE, Edge A, Heller S. Trends Mol Med. 2004;10:309–15. doi: 10.1016/j.molmed.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Caban AJ, Lee DJ, Gomez-Marin O, Lam BL, Zheng DD. Am J Public Health. 2005;95:1940–2. doi: 10.2105/AJPH.2004.056671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kerber KA. Semin Neurol. 2006;26:484–91. doi: 10.1055/s-2006-951620. [DOI] [PubMed] [Google Scholar]
- 5.Davis A. In: Hearing Science and Hearing Disorders. Lutman M, Haggard M, editors. Academic Press; New York: 1993. [Google Scholar]
- 6.Howard J, Roberts WM, Hudspeth AJ. Annu Rev Biophys Biophys Chem. 1988;17:99–124. doi: 10.1146/annurev.bb.17.060188.000531. [DOI] [PubMed] [Google Scholar]
- 7.Hudspeth AJ. Science. 1985;230:745–52. doi: 10.1126/science.2414845. [DOI] [PubMed] [Google Scholar]
- 8.Gong Z, Son W, Chung YD, Kim J, Shin DW, McClung CA, Lee Y, Lee HW, Chang DJ, Kaang BK, Cho H, Oh U, Hirsh J, Kernan MJ, Kim C. J Neurosci. 2004;24:9059–66. doi: 10.1523/JNEUROSCI.1645-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gillespie PG, Walker RG. Nature. 2001;413:194–202. doi: 10.1038/35093011. [DOI] [PubMed] [Google Scholar]
- 10.Thurm U. Cold Spring Harb Symp Quant Biol. 1965;30:75–82. doi: 10.1101/sqb.1965.030.01.011. [DOI] [PubMed] [Google Scholar]
- 11.Thurm U. Cold Spring Harb Symp Quant Biol. 1965;30:83–94. doi: 10.1101/sqb.1965.030.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Walker RG, Willingham AT, Zuker CS. Science. 2000;287:2229–34. doi: 10.1126/science.287.5461.2229. [DOI] [PubMed] [Google Scholar]
- 13.Hudspeth AJ. Nature. 1989;341:397–404. doi: 10.1038/341397a0. [DOI] [PubMed] [Google Scholar]
- 14.Grunert U, Gnatzy W. J Comp Physiol [A] 1987;161:329–33. doi: 10.1007/BF00615253. [DOI] [PubMed] [Google Scholar]
- 15.Kim J, Chung YD, Park DY, Choi S, Shin DW, Soh H, Lee HW, Son W, Yim J, Park CS, Kernan MJ, Kim C. Nature. 2003;424:81–4. doi: 10.1038/nature01733. [DOI] [PubMed] [Google Scholar]
- 16.Sidi S, Friedrich RW, Nicolson T. Science. 2003;301:96–9. doi: 10.1126/science.1084370. [DOI] [PubMed] [Google Scholar]
- 17.Shin JB, Adams D, Paukert M, Siba M, Sidi S, Levin M, Gillespie PG, Grunder S. Proc Natl Acad Sci U S A. 2005;102:12572–7. doi: 10.1073/pnas.0502403102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S. Cell. 2000;103:525–35. doi: 10.1016/s0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montell C. Sci STKE 2005. 2005:re3. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- 20.Eberl DF, Hardy RW, Kernan MJ. J Neurosci. 2000;20:5981–8. doi: 10.1523/JNEUROSCI.20-16-05981.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gopfert MC, Albert JT, Nadrowski B, Kamikouchi A. Nat Neurosci. 2006;9:999–1000. doi: 10.1038/nn1735. [DOI] [PubMed] [Google Scholar]
- 22.Achenbach TV, Brunner B, Heermeier K. Chembiochem. 2003;4:928–35. doi: 10.1002/cbic.200300708. [DOI] [PubMed] [Google Scholar]
- 23.Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, Linsley PS. Rna. 2006;12:1179–87. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corey DP. J Physiol. 2006 [Google Scholar]
- 25.Corey DP, Garcia-Anoveros J, Holt JR, Kwan KY, Lin SY, Vollrath MA, Amalfitano A, Cheung EL, Derfler BH, Duggan A, Geleoc GS, Gray PA, Hoffman MP, Rehm HL, Tamasauskas D, Zhang DS. Nature. 2004;432:723–30. doi: 10.1038/nature03066. [DOI] [PubMed] [Google Scholar]
- 26.Nagata K, Duggan A, Kumar G, Garcia-Anoveros J. J Neurosci. 2005;25:4052–61. doi: 10.1523/JNEUROSCI.0013-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ, Corey DP. Neuron. 2006;50:277–89. doi: 10.1016/j.neuron.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 28.Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D. Cell. 2006;124:1269–82. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 29.Takumida M, Kubo N, Ohtani M, Suzuka Y, Anniko M. Acta Otolaryngol. 2005;125:929–34. doi: 10.1080/00016480510038572. [DOI] [PubMed] [Google Scholar]
- 30.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. Nat Cell Biol. 2000;2:695–702. doi: 10.1038/35036318. [DOI] [PubMed] [Google Scholar]
- 31.Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Proc Natl Acad Sci U S A. 2004;101:396–401. doi: 10.1073/pnas.0303329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. Am J Physiol Cell Physiol. 2004;286:C195–205. doi: 10.1152/ajpcell.00365.2003. [DOI] [PubMed] [Google Scholar]
- 33.Colbert HA, Smith TL, Bargmann CI. J Neurosci. 1997;17:8259–69. doi: 10.1523/JNEUROSCI.17-21-08259.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liedtke W, Tobin DM, Bargmann CI, Friedman JM. Proc Natl Acad Sci U S A. 2003;100(Suppl 2):14531–6. doi: 10.1073/pnas.2235619100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tobin D, Madsen D, Kahn-Kirby A, Peckol E, Moulder G, Barstead R, Maricq A, Bargmann C. Neuron. 2002;35:307–18. doi: 10.1016/s0896-6273(02)00757-2. [DOI] [PubMed] [Google Scholar]
- 36.Liedtke W. J Physiol. 2005;567:53–8. doi: 10.1113/jphysiol.2005.088963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liedtke W, Friedman JM. Proc Natl Acad Sci U S A. 2003;100:13698–703. doi: 10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen J, Harada N, Kubo N, Liu B, Mizuno A, Suzuki M, Yamashita T. Neuroreport. 2006;17:135–9. doi: 10.1097/01.wnr.0000199459.16789.75. [DOI] [PubMed] [Google Scholar]
- 39.Alessandri-Haber N, Yeh JJ, Boyd AE, Parada CA, Chen X, Reichling DB, Levine JD. Neuron. 2003;39:497–511. doi: 10.1016/s0896-6273(03)00462-8. [DOI] [PubMed] [Google Scholar]
- 40.Chung MK, Lee H, Mizuno A, Suzuki M, Caterina MJ. J Biol Chem. 2004;279:21569–75. doi: 10.1074/jbc.M401872200. [DOI] [PubMed] [Google Scholar]
- 41.Smith PL, Maloney KN, Pothen RG, Clardy J, Clapham DE. J Biol Chem. 2006;281:29897–904. doi: 10.1074/jbc.M605394200. [DOI] [PubMed] [Google Scholar]
- 42.Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, Vriens J, Cairns W, Wissenbach U, Prenen J, Flockerzi V, Droogmans G, Benham CD, Nilius B. J Biol Chem. 2002;277:13569–77. doi: 10.1074/jbc.M200062200. [DOI] [PubMed] [Google Scholar]
- 43.Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Nature. 2003;424:434–8. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- 44.Mizuno A, Matsumoto N, Imai M, Suzuki M. Am J Physiol Cell Physiol. 2003;285:C96–101. doi: 10.1152/ajpcell.00559.2002. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki M, Mizuno A, Kodaira K, Imai M. J Biol Chem. 2003;278:22664–8. doi: 10.1074/jbc.M302561200. [DOI] [PubMed] [Google Scholar]
- 46.Tabuchi K, Suzuki M, Mizuno A, Hara A. Neurosci Lett. 2005;382:304–8. doi: 10.1016/j.neulet.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 47.Greene CC, McMillan PM, Barker SE, Kurnool P, Lomax MI, Burmeister M, Lesperance MM. Am J Hum Genet. 2001;68:254–60. doi: 10.1086/316925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Palma F, Belyantseva IA, Kim HJ, Vogt TF, Kachar B, Noben-Trauth K. Proc Natl Acad Sci U S A. 2002;99:14994–9. doi: 10.1073/pnas.222425399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cable J, Steel KP. Hear Res. 1998;123:125–36. doi: 10.1016/s0378-5955(98)00107-5. [DOI] [PubMed] [Google Scholar]
- 50.Lane PW. J Hered. 1972;63:135–40. doi: 10.1093/oxfordjournals.jhered.a108252. [DOI] [PubMed] [Google Scholar]
- 51.Steel KP. Proc Natl Acad Sci U S A. 2002;99:14613–5. doi: 10.1073/pnas.232585699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kiselyov K, Chen J, Rbaibi Y, Oberdick D, Tjon-Kon-Sang S, Shcheynikov N, Muallem S, Soyombo A. J Biol Chem. 2005;280:43218–23. doi: 10.1074/jbc.M508210200. [DOI] [PubMed] [Google Scholar]
- 53.Venkatachalam K, Hofmann T, Montell C. J Biol Chem. 2006;281:17517–27. doi: 10.1074/jbc.M600807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cordes FS, Bright JN, Sansom MS. J Mol Biol. 2002;323:951–60. doi: 10.1016/s0022-2836(02)01006-9. [DOI] [PubMed] [Google Scholar]
- 55.Dodier Y, Banderali U, Klein H, Topalak O, Dafi O, Simoes M, Bernatchez G, Sauve R, Parent L. J Biol Chem. 2004;279:6853–62. doi: 10.1074/jbc.M310534200. [DOI] [PubMed] [Google Scholar]
- 56.Nilius B, Prenen J, Janssens A, Owsianik G, Wang C, Zhu MX, Voets T. J Biol Chem. 2005;280:22899–906. doi: 10.1074/jbc.M501686200. [DOI] [PubMed] [Google Scholar]
- 57.Owsianik G, Talavera K, Voets T, Nilius B. Annu Rev Physiol. 2006;68:685–717. doi: 10.1146/annurev.physiol.68.040204.101406. [DOI] [PubMed] [Google Scholar]
- 58.Yeh BI, Kim YK, Jabbar W, Huang CL. Embo J. 2005;24:3224–34. doi: 10.1038/sj.emboj.7600795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heller S. J Physiol. 2002;543:3–12. doi: 10.1113/jphysiol.2002.018911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kikuchi T, Kimura RS, Paul DL, Adams JC. Anat Embryol (Berl) 1995;191:101–18. doi: 10.1007/BF00186783. [DOI] [PubMed] [Google Scholar]