

Abstract
Objective
The antipsychotic drug haloperidol (HAL) has been linked to apoptosis and to inhibition of prosurvival Akt signalling in pheochromocytoma (PC12) and neuronal cell cultures. However, the mechanism involved is unclear.
Methods
We used HAL to induce cytotoxicity in preneuronal PC12 cells. The expression and the subcellular localization of selected components of the PI3K–Akt survival cascade were monitored with standard biochemical approaches, such as subcellular fractionation, western blot analysis, gene transfer and fluorescence microscopy.
Results
PC12 cell stimulation with the epidermal growth factor (used as a control) results in normal processing of phosphatidylinositol 3'-kinase (PI3K)–Akt signalling (e.g., localization of PI3K to the plasma membrane and phosphorylation of Akt (Ser473). Surprisingly, HAL induces PI3K-generated phosphoinositol [phosphatidylinositol-3,4,5-triphosphate (PIP3), which conflicts with its ability to inhibit Akt. In fact, the production of PIP3s is nuclear, as assessed by the localized concentration of a fluorophore-tagged PIP3-targeting pleckstrin homology protein and a fluorophore-tagged substrate-trapping mutant of the phosphoinositide phosphatase, phosphatase and tensin homologue deleted on chromosome 10 (PTEN). However, phosphoinositide-dependent protein kinase 1 (PDK1, the activating kinase of Akt) does not colocalize to the nucleus with the PI3K complex. This effectively inactivates both cytoplasmic and nuclear pools of Akt.
Conclusion
The differential compartmentalization of effectors of the PI3K–PDK1–Akt pathway is a unique means by which HAL disrupts Akt functioning in PC12 cells.
Medical subject headings: haloperidol, EGF, phospholipids
Abstract
Objectif
Il existe un lien entre l'halopéridol, un antipsychotique, et l'apoptose et l'inhibition du signal Akt prosurvie dans les phéochromocytomes (PC12) et les cultures de cellules neuronales. Le mécanisme en cause n'est toutefois pas clair.
Méthodes
Nous avons utilisé l'halopéridol pour provoquer la cytotoxicité dans des cellules PC12 à l'état préneuronal. On a surveillé l'expression et la localisation sous-cellulaire d'éléments de la voie de survie PI3K/Akt au moyen de méthodes biochimiques standards comme le fractionnement sous-cellulaire, le transfert de Western, le transfert de gènes et la microscopie par fluorescence.
Résultats
La stimulation des cellules de PC12 au moyen du facteur de croissance épidermique (utilisé comme témoin) provoque un effet normal sur le signal phosphatidylinositol 3'-kinase (PI3K)/Akt (p. ex., localisation de la PI3K sur la membrane plasmatique et phosphorylation de l'Akt [Ser473]). L'halopéridol induit le phosphoïnositol [phosphatidylinositol-3,4,5-triphosphate (PIP3)] généré par la PI3K, ce qui entre en conflit avec sa capacité d'inhiber l'Akt. En fait, la production de PIP3 se fait dans le noyau de la cellule telle qu'évaluée par la concentration localisée, d'une part, d'une protéine homologue à la pleckstrin ciblant PIP3 et marquée par un fluorophore et, d'autre part, d'un mutant de la phosphoïnositide phosphatase, de la phosphatase et du PTEN (phosphatase and tensin homologue deleted on chromosome ten), capable de piéger le substrat et marqué par un fluorophore. La protéine kinase 1 dépendante du phosphoïnositide (PDK1, la kinase activant l'Akt) ne se colocalise toutefois pas dans le noyau avec le complexe PI3K, ce qui inactive en fait l'Akt cytoplasmique et nucléaire.
Conclusion
La compartimentalisation différentielle des effecteurs de la voie de signalisation PI3K-PDK1-Akt est un moyen unique par lequel l'halopéridol perturbe le fonctionnement de l'Akt dans les cellules de PC12.
Introduction
Many survival mechanisms rely on activation of phosphatidylinositol 3′-kinase (PI3K), which exists as a heterodimer consisting of a 110 kDa catalytic subunit (p110) and a constitutively associated 85 kDa (p85) regulatory subunit. The p110 subunit generates the D3-phosphoinositide PI(3,4,5)P3 (PIP3) at the inner surface of the plasma membrane, which helps to recruit the phosphoinositide-dependent protein kinase-1 (PDK1) via its pleckstrin homology (PH) domain.1,2 PDK1, in turn, phosphorylates and activates the serine/threonine kinase Akt3, another protein anchored to PIP3s at the plasma membrane by virtue of a PH domain.4 The balance between PI3K and the phosphoinositide phosphatase PTEN (phosphatase and tensin homologue deleted on chromosome 10) frequently determines PIP3 levels5 and Akt function. Positive regulation of PI3K–Akt function can also rely on other phosphatases, including the Src homology-2 domain-containing protein-tyrosine phosphatase (SHP-2)6 that associates with the PI3K complex via the docking protein, Gab1.7 The PI3K– PDK1–Akt pathway is sufficient and, in some cases, obligatory for the survival of several neuronal cell types.1 Downstream targets of cytoplasmic PI3K generally appear to influence cell metabolism, cytoskeletal rearrangements, vesicle transport and apoptosis.
PI3K-mediated antiapoptotic events require Akt and its ability to phosphorylate and, thereby, inactivate diverse pro-apoptotic substrates, including caspase-9, forkhead transcription factor and several Bcl-2 homologues.4,8 PI3K and Akt localize predominantly to the cytoplasm, but they can also exist in the nucleus or translocate there upon stimulation.9 Cytoplasmic PI3K–Akt signalling has been well characterized, yet relatively little is known about its role in the nucleus. Nuclear PIP3s may mediate a broad range of processes, including cell cycle progression and nuclear response to DNA damage.10,11 Although PIP3s will bind to and inhibit caspase-3,12 most nuclear PIP3-mediated events are not exerted by the lipids themselves but by downstream effectors such as the obligate Akt.13
The typical antipsychotic drug haloperidol (HAL) is cytotoxic,14–16 which mitigates its widespread use in the clinical setting. The cytotoxicity of HAL in PC12 cells comprises an oxidative stress component as well as an apoptotic component.14,16 Our work links the apoptotic component, which consistently accounts for approximately 25%–30% of HAL-induced toxicity, to the sigma2 (σ2) receptor system; we have also precluded any involvement of the dopamine D2 receptor system,16 which has been classically associated with the function of typical antipsychotic drugs such as HAL. The activation of the σ2 receptor system by HAL induces an Akt-sensitive mitochondrial translocation of proapoptotic Bcl-xS (Wei, Dai and Mousseau, unpublished data). We demonstrate that HAL treatment of PC12 cells results in the activation and nuclear translocation of the PI3K complex; its immediate downstream effector PDK1, however, remains in the cytosol. As a result, PDK1 is effectively isolated and unable to benefit from the newly generated nuclear PIP3s. The consequence of this differential compartmentalization of pivotal contributors to the PI3K/PDK1/Akt pathway is a disruption of prosurvival processes.
Materials and methods
Chemicals and antibodies
We purchased the antibodies directed to Gab1, poly(ADP-ribose) polymerase (PARP), total and phospho (Ser241) PDK1 and p110 (PI3K) from Santa Cruz Biotechnology. Anti-SHP-2 is from BD Transduction Laboratories. The total and phospho (Ser473 and Thr308) Akt antibodies were from Cell Signalling Technology. The β-actin antibody, protease inhibitor cocktail and HAL were from Sigma-Aldrich. Anti-p85 and IgG-HRP conjugates were obtained from Cedarlane Laboratories.
Cell cultures and cell viability
The PC12 rat preneuronal cell line (CRL-1721) was purchased from the American Type Culture Collection and maintained according to their specifications. Conversion of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium (MTT) to formazan crystals was used as an effective nonradioactive means of determining drug sensitivity and cell viability, as previously validated in PC12 cells.16 MTT experimental means are based on 4 to 9 individual experiments, each of which are represented by 4 to 6 replicates per test group. HAL-induced cell death was confirmed with the Live/Dead Viability/Cytotoxicity Kit (Molecular Probes, Invitrogen, Carlsbad, Calif.).
PI3K p110 lipid kinase assay
Cell extracts were immunoprecipitated for p85 (the regulatory subunit of PI3K) and washed successively with phosphate-buffered saline, 50 μmol/L LiCl /100 mM Tris-HCl (pH 7.4), and 100 mM NaCl/1 mM EDTA/10 mM Tris-HCl (pH 7.4). Lipid micelles were generated by sonicating (30 min) phosphatidylserine or phosphatidylinositol in a PI3K assay buffer (25 mM Hepes, pH 7.4, 10 mM MgCl2). Samples were then incubated with lipid micelles (5 μg phosphatidylserine + 2.5 μg phosphatidylinositol) in a PI3K assay buffer and 10 μCi of [γ-32P]ATP for 15 minutes at room temperature. The reaction was terminated by acidification. Lipid products were extracted into chloroform:methanol (1:1), washed with methanol:1 N HCl (1:1) and resolved by thin-layer chromatography (Silica Gel 60; VWR Canlab, Missisauga, Ont.). Samples were developed in 1-propanol:water:acetic acid (17.4:7.9:1) for 4 hours and were then dried and visualized with a Phosphor-Imager. Results were quantified with Quantity One software (Bio-Rad, Hercules, Calif.).
cDNA cloning and transfection
PH-mRFP1 (PH-RFP) was provided by Dr. E.R. Prossnitz (Department of Cell Biology and Physiology, University of New Mexico) with permission for the transfer of an mRFP1-based construct obtained from Dr. R.Y. Tsien (Howard Hughes Medical Institute, University of California San Diego). Given its capacity to bind PIP3s, we used this red-fluorescent protein (RFP)-tagged PH domain of Akt17 to assess PIP3 accumulation and localization. Dr. A.B. Vojtek (University of Michigan, Ann Arbor, Mich.) gave us the constitutively active, N-myristoylated full-length membrane-directed mouse Akt1 (myr-Akt) cDNA. The N-myristoylated full-length membrane-directed PDK1 (myr-PDK1) and enhanced green-fluorescent protein (eGFP)-PDK1 were from Drs. S. Kim and J. Chung (Korea Advanced Institute of Science and Technology). The yellow fluorescent protein (YFP)-p110 construct was generated with cDNA encoding the mouse p110α coding sequence (Dr. L.T. Williams, University of California San Francisco), which was PCR-amplified and subcloned into the YFP vector (Clontech Laboratories Inc., Mountain View, Calif.). The GFP-PTEN(C124S) phosphatase-dead construct was generated using human wildtype PTEN cDNA (Dr. W.R. Sellers, Dana-Farber Cancer Institute, Harvard Medical School), which was subsequently subcloned into the GFP vector (Clontech Laboratories Inc.). Site-directed mutagenesis was used to generate the PTEN Cys124-to-Ser (C124S) substitution. Human wildtype p85 and SHP-2 were used to generate the p85(Tyr688Asp) activated mutant18 and the SHP-2(C459S) phosphatase-dead mutant.19 The respective coding sequences of all constructs were confirmed by DNA sequencing. Cells were seeded in log phase and transfected with plasmid DNA (1–2 μg/well on a 24-well plate; seeded at 5 × 105 cells/well), using LipofectAmine Plus (Invitrogen, Carlsbad, Calif.). Overexpression of eGFP fluorescent protein revealed an estimated 35% transfection efficiency.
Immunoblot and immunoprecipitation
Standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) immunoblot conditions were used to detect expression of targeted proteins in total cell lysates (precleared; 5000 × g, 10 min, 4ºC; 20–30 μg/lane) or in immunoprecipitates (300–500 μg: precleared lysates with non-specific mouse or rabbit IgG) isolated at 4°C with antibodies diluted to the manufacturer's specifications and precipitated with protein-A/G Sepharose. Protein visualization relied on enhanced chemiluminescence. Immunoblots represent 2 to 3 determinations.
Subcellular fractionation
Cells were harvested, washed twice with 0.1 mM ice-cold phosphate-buffered saline, pelleted and then resuspended in ice-cold lysis buffer containing 20 mM Hepes-KOH (pH 7.5), 320 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol, 1 mM orthovanadate and a protease inhibitor cocktail. Cells were disrupted with a Dounce homogenizer, and the homogenates were centrifuged at 900 × g (5 min, 4°C) to isolate cell debris (P1). The cleared lysate was spun at 18 000 × g (40 min, 4°C), and the supernatant was removed and saved as the cytoplasmic fraction. The pelleted nuclei were then resuspended in 20 mM Hepes, pH 8.0, 1.5 mM MgCl2, 25% glycerol, 420 mM NaCl, 0.2 mM EDTA, 1 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulphonyl fluoride (PMSF) and incubated on ice for 20 minutes with occasional shaking. The sample was centrifuged for 10 minutes at 12 000 × g at 4°C to separate the nuclear soluble (supernatant) fraction from the nuclear insoluble (pellet) fraction, which was discarded.
Visualization of PI3K lipid products and effectors
Fluorescence confocal microscopy was used for visual localization of PI3K lipid products (using PH-RFP) and for determining the subcellular localization of overexpressed fluorophore-tagged components of the PI3K pathway, including the p110 catalytic subunit of PI3K (YFP-p110), the PIP3 substrate-trapping phosphatase PTEN (GFP-PTEN(C124S); (see Sun and colleagues20) and the PI3K effector PDK1 (GFP-PDK1). PC12 cells were grown on chamber slides and proteins were overexpressed for 24 hours. The cells were then treated with 125 μM HAL for another 24 hours. These were rinsed with warmed PBS and fixed for 30 minutes in 4% formaldehyde; they were rinsed again and then treated with ProLong Gold Antifade. For nuclear staining, cells were fixed as above and counterstained with the nuclear marker 4',6-diamidino-2-phenylindole (DAPI) for 10 minutes at room temperature. The cells were rinsed with phosphate-buffered saline (PBS) and mounted with ProLong Gold Antifade medium. Fluorescence was detected with laser-scanning confocal immunofluorescence microscopy (Olympus, FV-300, 40 × magnification). All experiments were performed 2 to 3 times, with representative scans presented. The molecular mass of fluorophores could potentially interfere with the proper localization of an expressed fusion product. As such, we included a parallel series of experiments based on epidermal growth factor treatment to confirm that proper compartmentalization of these fluorophore-tagged proteins occurred with the appropriate stimulus.
We hypothesized that deregulation of PI3K–Akt signalling is an important mechanism underlying HAL-induced toxicity in PC12 cell cultures. We assessed significance (set at p < 0.05), using analyses of variance (ANOVA), with post hoc analyses relying on Bonferroni's Multiple Comparison Test.
Results
HAL activates the PI3K complex
HAL promotes p85-associated PI3K lipid kinase activity (Fig. 1A), which correlates with an increase in the interaction between p85, the regulatory subunit of PI3K, and the docking protein Gab1 (Fig. 1B). We also found that Gab1-associated PI3K activity is similarly increased (data not shown). The appearance of a Gab1 smear/doublet in the total cell lysates of HAL-treated cells and the enhanced association of Gab1 with the SH2 domain-containing tyrosine phosphatase SHP-2 suggests that HAL-treated extracts contain phosphorylated Gab1. The association between SHP-2 and p85 is virtually unchanged (Fig. 1B).

Fig. 1: Haloperidol (HAL) induces PI3K activity in PC12 cells. Protein extracts from HAL-treated (125 μM, 24 h) extracts were immunoprecipitated for p85 and subjected to a p110 lipid kinase assay (A). The [γ-32P]-labelled 3'-phosphatidylinositol lipid products are indicated by PI3P on a representative autoradiogram (numbers indicate means of 2 experiments). Corresponding cell lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE), transferred to nitrocellulose and probed to determine the expression levels of p85, Gab1, Src homology-2 domain-containing protein-tyrosine phosphatase (SHP-2) (B). Levels of β-actin were used to demonstrate equal protein loading in both lanes. These same cell lysates were also immunoprecipitated for p85 or for SHP-2. The resolved immunocomplexes were probed for p85 and Gab1. A p85(Y688D) activated protein (C) or an SHP-2(C459S) dominant negative catalytically inactive protein (D) were overexpressed in PC12 cell cultures to determine their influence on HAL-induced (125 μM, 24-hour) toxicity, as assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium (MTT) reduction. Data are presented as mean, standard deviation; n = 6–8; ns: not significantly different from vehicle control; *p < 0.05 and ***p < 0.001 versus vehicle (vector) control.
HAL exerts toxicity in PC12 cell cultures (p < 0.0001, F1,27 = 327.6) (Fig. 1C) that is unaffected by overexpression (24 h) of activated p85(Y688D) (p = 0.5600, F(1,27) = 0.3482). In a separate experiment, the toxicity induced by HAL (p < 0.0001, F1,8 = 76.09) is also unaffected by overexpression of a dominant negative catalytically inactive SHP-2(C459S) (p = 0.7989, F1,8 = 0.1105) (Fig. 1D). A toxic concentration of HAL (150 μM; still in the linear dose–response range for HAL toxicity in PC12 cells16) was used in the p85(Y688D)-transfection experiment, because the experiment was intended to demonstrate a protection by the activated p85. In contrast, a moderately toxic concentration of HAL (120 μM) was used in the SHP-2(C459S) experiments, because it was expected that this catalytically inactive mutant would actually promote HAL-induced toxicity. The HAL-induced reduction in MTT conversion (Fig. 1) reflects cytotoxicity, as confirmed with a Live/Dead Viability/Toxicity kit. We observed a greater proportion of ethidium homodimer-1 (red = dead) to calcein AM (green = live) stained cells after HAL treatment, compared with vehicle-treated control subjects (Fig. 2). These cells also “round up” (i.e., they change from their normally flattened appearance and take on a spherical shape) in a manner characteristic of apoptotic cells.
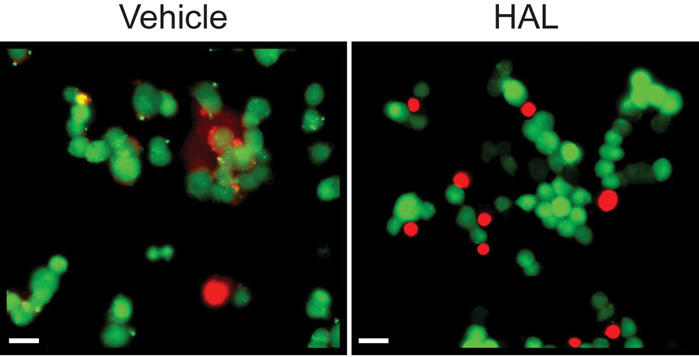
Fig. 2: Haloperidol (HAL) is toxic to PC12 cells. HAL treatment (125 μM, 24 h) results in an increase in the number of dead (labelled red) PC12 cells and induces a “rounding up” of cells, a characteristic of cells undergoing apoptosis. Bar: 20 μm.
PDK1 does not colocalize with the increased production of PIP3s in the nucleus during HAL treatment
The expression pattern of GFP alone is unaffected by either epidermal growth factor (EGF) or HAL (Fig. 3A). In unstimulated cells, the expression of PH-RFP is diffuse and throughout the cell (Fig. 3A). EGF-stimulation induces the translocation of PH-RFP to the plasma membrane, as expected. In similarly treated cells, YFP-p110 (the catalytic subunit of PI3K), GFP-PTEN(C124S) (a phosphatase-dead variant of the phosphoinositide phosphatase PTEN that behaves as a PIP3 “substrate-trap”) and GFP-PDK1 (the PIP3-activated downstream effector of PI3K) all concentrate at the plasma membrane, also as expected (Fig. 3A); this corresponds with the activation (phosphorylation) of Akt (Fig. 3B). In contrast, HAL treatment results in the concentration of the PH-RFP, YFP-p110 and GFP-PTEN(C124S) proteins in the nucleus, whereas GFP-PDK1 remains in the cytoplasmic compartment (Fig. 3A); this coincides with a loss of Akt phosphorylation (Fig. 3B). Counterstaining with the nuclear marker, DAPI, confirms the nuclear accumulation of PH-RFP in HAL-treated cultures (Fig. 3C). All cells were pictured through the plane of the nucleus.

Fig. 3: Fluorescence localization of components of the PI3K pathway in epidermal growth factor (EGF)- and haloperidol (HAL)-treated PC12 cells. (A) The localization of overexpressed proteins was monitored with confocal microscopy in EGF- (10 ng/ml; 10 min) or HAL- (125 μM, 24 h) treated PC12 cell cultures. The localization of these proteins to the plasma membrane (indicated by the arrows; centre column) is evident in EGF-treated cultures, whereas a pool of these same proteins (with the exception of PDK1) relocalizes to the nucleus (indicated by arrows; right column) in HAL-treated cultures. All micrographs are depicted through the plane of the nucleus. (B) The phosphorylation of Ser473-Akt is enhanced in EGF-treated PC12 cells, but not in HAL-treated cells. β-Actin demonstrates equal protein loading. (C) HAL treatment results in the concentration of the PH-RFP construct in the nucleus as demonstrated by counterstaining with the nuclear marker, 4',6-diamidino-2-phenylindole. Bar: 5 μm.
The distribution of the regulatory subunit of PI3K, p85 and of the p110/PI3K catalytic subunit are increased, albeit modestly, in the nuclear fraction after HAL treatment (Fig. 4). The phosphorylation of Thr308-Akt (which is predominantly nuclear) and Ser473-Akt (which is predominantly cytosolic) are both decreased by HAL. This latter observation indicates the existence of distinct pools of Akt in PC12 cells. The levels of Ser241-PDK1 (also predominantly cytosolic) appear to increase, while the expression of total PDK1 is essentially unchanged after HAL treatment (Fig. 4).
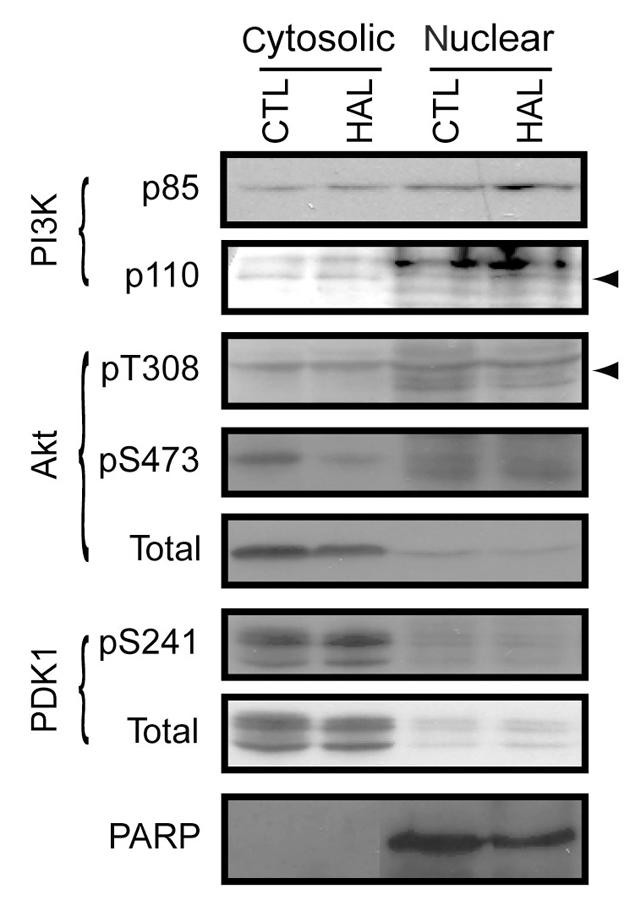
Fig. 4: Haloperidol (HAL) induces the nuclear translocation of p110/PI3K and dephosphorylates Akt in specific subcellular compartments. PC12 cells were treated with HAL (125 μM, 24 h). The cells were then fractionated, and the nuclear and cytosolic fractions were examined for the distribution of p85, p110 (indicate by arrow), phosphoThr308- (indicated by arrow) and phosphoSer473-Akt, phosphoSer241-protein kinase-1, and poly(ADP-ribose) polymerase (which demonstrates fraction purity). CTL = control; PARP = poly(ADP-ribose) polymerase.
Overexpression of myristoylated (membrane-directed), constitutively active Akt (myr-Akt) partially reverses (p = 0.0274, F2,15 = 4.804) the cytotoxicity induced by HAL (p < 0.0001, F1,26 = 121.6) in PC12 cells, whereas overexpression of myristoylated (membrane-directed) PDK1 (myr-PDK1) does not (Fig. 5A). In corresponding cell lysates, myr-Akt prevents HAL-induced Akt dephosphorylation, whereas myr-PDK1 does not (Fig. 5B).

Fig. 5: Membrane-directed, constitutively active Akt diminishes the effects of haloperidol (HAL). (A) Constitutively active myristoylated (myr; membrane-directed) Akt (myr-Akt) and PDK1 (myr-PDK1) were overexpressed (24 h) in PC12 cell cultures. Their effect on the cytotoxicity of 125 mM of HAL was monitored by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium (MTT) reduction. Data are presented as mean, standard deviation; n ≥ 6: ***p < 0.001 versus vehicle control; #p < 0.05 relative to vector (vec)/HAL. (B) Protein extracts from similarly treated cells were used to demonstrate overexpression of the respective myristoylated-proteins and to monitor the phosphorylation status of Thr308- and Ser473-Akt residues. PDK1 = phosphoinositide-dependent protein kinase1; endo = endogenous.
Discussion
We and others21 have observed that HAL can inhibit Akt signalling in neuronal cell cultures. We aimed to determine which step of the PI3K–PDK1–Akt pathway was perturbed in HAL-treated PC12 cell cultures. It is well documented that the brain concentrations of antipsychotic drugs, particularly lipophilic compounds such as HAL, in both the laboratory and the clinical setting are 20 to 100 times higher than in corresponding plasma (e.g., it can reach 10 μM in brain; see Kornhuber et al22 and references therein). Our use of μM concentrations of HAL, therefore, is not unreasonable and is in keeping with concentrations required elsewhere to examine the molecular effects of HAL in the experimental setting.16,23–26
HAL treatment of PC12 cells increases p85-associated p110 lipid kinase activity and causes the expected changes in the associated p85, Gab1 and SHP-2 positive regulatory complex.6,27,28 However, the concurrent dephosphorylation of Akt ultimately supersedes any benefit to the cell. Interestingly, treatment with HAL can exert positive effects on Akt function in vivo,29,30 although we and others21 have demonstrated that HAL can also negatively regulate Akt, specifically in primary neuronal cultures and PC12 cells (present study). Antagonism of the dopamine D2 receptor may contribute to these effects in some models,21,29,30 although this is not the case for the cytotoxicity observed in HAL-treated PC12 cells, which is mediated by the proapoptotic σ2 receptor in a p53- and Bax-independent manner.16 HAL-mediated antagonism of the σ1 receptor may also contribute to the regulation of Akt function via a calcium-independent event.31
The generation of PIP3s by PI3K at the plasma membrane is essential for the recruitment and activation of PH domain-containing proteins, including PDK1 and Akt. These proteins, as well as PI3K, are also expressed in the nucleus, which, in combination with a nuclear phospholipid cycle that is independent of the cytosolic cycle, suggests a functional PI3K–PDK1–Akt pathway in this subcellular compartment (for reviews see Martelli et al9 and Lian and Di Cristofano32). Nuclear PDK1, however, may not target Akt in this compartment. It has been postulated that PDK1 nuclear translocation could be a means of sequestering PDK1 from activating cytosolic Akt signalling pathways.33 In keeping with this unique means of regulating Akt signalling, we demonstrate that PDK1 does not relocalize to the nucleus along with p110/PI3K in HAL-treated PC12 cells. The nuclear translocation of p110 was more evident with the fluorophore-tagged p110 protein than with our immunoblot/subcellular fractionation studies, possibly reflecting the extremely low levels of endogenous protein and limitations of the p110 antibody. The retention of PDK1 in the cytoplasm effectively sequesters it from newly generated nuclear PI3K lipid products, and this “inactive” PDK1 is reflected in the reduced phosphorylation of both cytosolic Thr308-Akt and nuclear Ser473–Akt. The immediate effect of diminished Akt function in HAL-treated PC12 cells includes, for example, the dephosphorylation of Ser9–GSK–3β (Zhang and Mousseau, unpublished observations), the cleavage of caspase-916 and the dephosphorylation of nuclear IkBa, an inhibitor of NF-kB–Rel proteins (Wei and Mousseau, unpublished observations). Further, HAL-induced apoptosis is demonstrated by Annexin-V staining and the processing of PARP (see Wei et al16) and caspase-3 activation/DNA laddering and loss of microtubule-associated protein-2 expression.21
Proteins that contain both nuclear localization signals (NLSs) and nuclear export signals (NESs) are amenable to nuclear-cytoplasmic shuttling. PDK1 does not have an NLS but is rapidly phosphorylated on Ser396, which is adjacent to an NES. Although it is unclear how phosphorylated Ser396 could enhance PDK1 nuclear import, a more suitable explanation is that its potential for interfering with nuclear export leads to the observed nuclear PDK1 accumulation.34 These authors34 also suggest that tyrosine phosphorylation of PDK1 could affect its localization and function. Whatever the mechanism, the phosphorylation of PDK1 on Ser396 depends on its localization at the plasma membrane.34 In the current model, the relocalization of PI3K to the nucleus would preclude any phosphorylation of Ser396-PDK1 at the plasma membrane and consequently interfere with the potential for nuclear translocation/accumulation of PDK1. We do observe 2 pools of PDK1, 1 cytoplasmic and 1 nuclear. We are uncertain at this time as to why nuclear PDK1 is not activated when there is an increase in local PIP3 production. Perhaps these supraphysiological levels of PIP3 are affecting the affinity of the PH domain in PDK1 by a mechanism similar to that proposed for the regulation of Akt function in PTEN-null Drosophila.35 Our current data confirm36 that the phosphorylation of Ser241-PDK1, which is often used to confirm activated PDK1, does not necessarily coincide with the phosphorylation status of its downstream target, Akt.
The potential antiproliferative effect of HAL has been linked to σ receptor activation in cancer cell lines.37 Interestingly, we have also linked HAL-induced apoptosis and the associated Akt-sensitive mitochondrial localization of proapoptotic Bcl-xS to the σ2 receptor system in PC12 cells and not to the antagonism of dopamine D2 receptors, as originally anticipated.16 We continue to investigate whether this mechanism contributes to the cytotoxicity associated with all antipsychotics or σ2 receptor ligands.
We have now linked HAL treatment of PC12 cells to the translocation of pivotal components of the PI3K signalling to the nucleus. Despite the activation of PI3K in this subcellular compartment, the fact that PDK1 is not concurrently translocated to this compartment appears to disrupt PI3K/Akt signalling and the associated prosurvival mechanisms in these cells. The actual mechanism that results in retention of PDK1 in the cytosol remains unclear, yet it is clear that this event correlates with HAL-induced toxicity. Although the toxicity and antiproliferative properties of HAL, which appear to rely predominantly on σ receptor activation,16,37 could limit the use of HAL in the clinical setting as an antipsychotic, this same “negative” profile could benefit the proposed use of HAL as an anticancer therapeutic modality,15,38–40 where toxicity is desirable.
Acknowledgments
This work was supported in part by a Saskatchewan Health Research Foundation (SHRF) operating grant and a Canadian Institutes of Health Research (CIHR)–SHRF New Investigator Award (to DDM), by a University Graduate Scholarship (to CFS) and by a Fellowship from the CIHR/Rx&D Research Program and AstraZeneca/Alzheimer Society of Canada (to ZW).
Footnotes
Contributors: Ms. Dai and Dr. Mousseau designed the study. Ms. Dai and Ms. Zhang and Drs. Wei, Sephton and Anderson acquired the data, which Ms. Dai and Ms. Zhang and Drs. Wei, Sephton and Mousseau analyzed. Drs. Wei, Anderson and Mousseau wrote the article, which Ms. Dai and Ms. Zhang and Drs. Wei, Sephton and Anderson revised. All authors gave final approval for the article to be published.
Competing interests: None declared.
Correspondence to: Dr. Darrell D. Mousseau, Cell Signalling Laboratory, Neuropsychiatry Research Unit, University of Saskatchewan, 103 Wiggins Rd., Saskatoon SK S7N 5E4; fax 306 966-8830; Darrell.Mousseau@usask.ca
References
- 1.Brunet A, Datta SR, Greenberg ME. Transcription-dependent and independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 2001;11:297-305. [DOI] [PubMed]
- 2.Mora A, Komander D, van Aalten DM, et al. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol 2004; 15:161-70. [DOI] [PubMed]
- 3.Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J 2000;346:561-76. [PMC free article] [PubMed]
- 4.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev 1999;13:2905-27. [DOI] [PubMed]
- 5.Gericke A, Munson M, Ross AH. Regulation of the PTEN phosphatase. Gene 2006;374:1-9. [DOI] [PubMed]
- 6.Wu CJ, O'Rourke DM, Feng GS, et al. The tyrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-kinase/Akt activation by growth factors. Oncogene 2001;20:6018-25. [DOI] [PubMed]
- 7.Mood K, Saucier C, Bong YS, et al. Gab1 is required for cell-cycle transition, cell proliferation, and transformation induced by an oncogenic met receptor. Mol Biol Cell 2006;17:3717-28. [DOI] [PMC free article] [PubMed]
- 8.Gardai SJ, Hildeman DA, Frankel SK, et al. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem 2004;279:21085-95. [DOI] [PubMed]
- 9.Martelli AM, Faenza I, Billi AM, et al. Intranuclear 3′-phosphoinositide metabolism and Akt signaling: new mechanisms for tumorigenesis and protection against apoptosis? Cell Signal 2006; 18:1101-7. [DOI] [PubMed]
- 10.Yokogawa T, Nagata S, Nishio Y, et al. Evidence that 3′-phosphorylated polyphosphoinositides are generated at the nuclear surface: use of immunostaining technique with monoclonal antibodies specific for PI 3,4-P(2). FEBS Lett 2000;473:222-6. [DOI] [PubMed]
- 11.Gozani O, Karuman P, Jones DR, et al. The PHD finger of the chromatin-associated protein ING2 functions as a nuclear phosphoinositide receptor. Cell 2003;114:99-111. [DOI] [PubMed]
- 12.Mejillano M, Yamamoto M, Rozelle AL, et al. Regulation of apoptosis by phosphatidylinositol 4,5-bisphosphate inhibition of caspases, and caspase inactivation of phosphatidylinositol phosphate 5-kinases. J Biol Chem 2001;276:1865-72. [DOI] [PubMed]
- 13.Ahn JY, Rong R, Liu X, et al. PIKE/nuclear PI 3-kinase signaling mediates the antiapoptotic actions of NGF in the nucleus. EMBO J 2004;23:3995-4006. [DOI] [PMC free article] [PubMed]
- 14.Post A, Rucker M, Ohl F, et al. Mechanisms underlying the protective potential of a-tocopherol (vitamin E) against haloperidol-associated neurotoxicity. Neuropsychopharmacology 2002;26:397-407. [DOI] [PubMed]
- 15.Crawford KW, Bowen WD. σ-2 receptor agonists activate a novel apoptotic pathway and potentiate antineoplastic drugs in breast tumor cell lines. Cancer Res 2002;62:313-22. [PubMed]
- 16.Wei Z, Mousseau DD, Dai Y, et al. Haloperidol induces apoptosis via the σ2 receptor system and Bcl-XS. Pharmacogenomics J 2006;6:279-88. [DOI] [PubMed]
- 17.Revankar CM, Cimino DF, Sklar LA, et al. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005;307:1625-30. [DOI] [PubMed]
- 18.Cuevas BD, Lu Y, Mao M, et al. Tyrosine phosphorylation of p85 relieves its inhibitory activity on phosphatidylinositol 3-kinase. J Biol Chem 2001;276:27455-61. [DOI] [PubMed]
- 19.Arkinstall S, Gillieron C, Vial-Knecht E, et al. Negative regulatory function for the protein tyrosine phosphatase PTP2C revealed by reconstruction of platelet-derived growth factor receptor signalling in Schizosaccharomyces pombe. FEBS Lett 1998;422:321-7. [DOI] [PubMed]
- 20.Sun H, Charles CH, Lau LF, et al. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell 1993;75:487-93. [DOI] [PubMed]
- 21.Ukai W, Ozawa H, Tateno M, et al. Neurotoxic potential of haloperidol in comparison with risperidone: implication of Akt-mediated signal changes by haloperidol. J Neural Transm 2004;111:667-81. [DOI] [PubMed]
- 22.Kornhuber J., Wiltfang J., Riederer P., et al. Neuroleptic drugs in the human brain: clinical impact of persistence and region-specific distribution. Eur Arch Psychiatry Clin Neurosci 2006;256:274-80. [DOI] [PubMed]
- 23.Vilner BJ, de Costa BR, Bowen WD. Cytotoxic effects of σ ligands: σ receptor-mediated alterations in cellular morphology and viability. J Neurosci 1995;15:117-34. [DOI] [PMC free article] [PubMed]
- 24.Brent PJ, Pang G, Little G, et al. The sigma receptor ligand, reduced haloperidol, induces apoptosis and increases intracellular-free calcium levels [Ca2+]i in colon and mammary adenocarcinoma cells. Biochem Biophys Res Commun 1996;219:219-26. [DOI] [PubMed]
- 25.Gil-Ad I, Shtaif B, Shiloh R, et al. Evaluation of the neurotoxic activity of typical and atypical neuroleptics: relevance to iatrogenic extrapyramidal symptoms. Cell Mol Neurobiol 2001;21:705-16. [DOI] [PMC free article] [PubMed]
- 26.Wei Z, Mousseau DD, Richardson JS, et al. Atypical antipsychotics attenuate neurotoxicity of β-amyloid(25-35) by modulating Bax and Bcl-X(l/s) expression and localization. J Neurosci Res 2003;74:942-7. [DOI] [PubMed]
- 27.Yu CF, Liu ZX, Cantley LG. ERK negatively regulates the epidermal growth factor-mediated interaction of Gab1 and the phosphatidylinositol 3-kinase. J Biol Chem 2002;277:19382-8. [DOI] [PubMed]
- 28.Holgado-Madruga M, Wong AJ. Gab1 is an integrator of cell death versus cell survival signals in oxidative stress. Mol Cell Biol 2003;23: 4471-84. [DOI] [PMC free article] [PubMed]
- 29.Emamian ES, Hall D, Birmbaum MJ, et al. Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat Genet 2004;36:131-7. [DOI] [PubMed]
- 30.Alimohamad H, Sutton L, Mouyal J, et al. The effects of antipsychotics on β-catenin, glycogen synthase kinase-3 and dishevelled in the ventral midbrain of rats. J Neurochem 2005;95:513-25. [DOI] [PubMed]
- 31.Spruce BA, Campbell LA, McTavish N, et al. Small molecule antagonists of the sigma-1 receptor cause selective release of the death program in tumor and self-reliant cells and inhibit tumor growth in vitro and in vivo. Cancer Res 2004;64:4875-86. [DOI] [PubMed]
- 32.Lian Z, Di Cristofano A. Class reunion: PTEN joins the nuclear crew. Oncogene 2005;24:7394-400. [DOI] [PubMed]
- 33.Lim MA, Kikani CK, Wick MJ, et al. Nuclear translocation of 3′-phosphoinositide-dependent protein kinase 1 (PDK-1): a potential regulatory mechanism for PDK-1 function. Proc Natl Acad Sci U S A 2003;100:14006-11. [DOI] [PMC free article] [PubMed]
- 34.Scheid MP, Parsons M, Woodgett JR. Phosphoinositide-dependent phosphorylation of PDK1 regulates nuclear translocation. Mol Cell Biol 2005;25:2347-63. [DOI] [PMC free article] [PubMed]
- 35.Stocker H, Andjelkovic M, Oldham S, et al. Living with lethal PIP3 levels: viability of flies lacking PTEN restored by a PH domain mutation in Akt/PKB. Science 2002;295:2088-91. [DOI] [PubMed]
- 36.An WL, Bjorkdahl C, Liu R, et al. Mechanism of zinc-induced phosphorylation of p70 S6 kinase and glycogen synthase kinase 3β in SH-SY5Y neuroblastoma cells. J Neurochem 2005;92:1104-15. [DOI] [PubMed]
- 37.Nordenberg J, Perlmutter I, Lavie G, et al. Anti-proliferative activity of haloperidol in B16 mouse and human SK-MEL-28 melanoma cell lines. Int J Oncol 2005;27:1097-103. [PubMed]
- 38.Gil-Ad I, Shtaif B, Levkovitz Y, et al. Characterization of phenothiazine-induced apoptosis in neuroblastoma and glioma cell lines: clinical relevance and possible application for brain-derived tumors. J Mol Neurosci 2004;22:189-98. [DOI] [PubMed]
- 39.Wang B, Rouzier R, Albarracin CT, et al. Expression of σ1 receptor in human breast cancer. Breast Cancer Res Treat 2004;87:205-14. [DOI] [PubMed]
- 40.Mukherjee A, Prasad TK, Rao NM, et al. Haloperidol-associated stealth liposomes: a potent carrier for delivering genes to human breast cancer cells. J Biol Chem 2005;280:15619-27. [DOI] [PubMed]