

Abstract
Increased expression of glial fibrillary acidic protein (GFAP) represents astroglial activation and gliosis during neurodegeneration. However, the molecular mechanism behind increased expression of GFAP in astrocytes is poorly understood. The present study was undertaken to explore the role of nitric oxide (NO) in the expression of GFAP. Bacterial lipopolysachharides (LPSs) induced the production of NO and the expression of GFAP in mouse primary astrocytes. Either a scavenger of NO [2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (PTIO)] or an inhibitor of inducible nitric oxide synthase [l-N6-(I-iminoethyl)-lysine hydrochloride] blocked this induction of GFAP expression. Similarly, other inducers of NO production such as interferon-γ, interleukin-1β, human immunodeficiency virus type 1 gp120, fibrillar amyloid β peptides, and double-stranded RNA (polyinosinic-polycytidilic acid) also induced the expression of GFAP through NO. The role of NO in the expression of GFAP was supported further by increased expression of GFAP by S-nitroso glutathione (GSNO), an NO donor. Interestingly, inhibition of nuclear factor κB (NF-κB) suppressed LPS- but not GSNO-induced expression of GFAP, suggesting that NO does not require NF-κB to induce GFAP and that NF-κB functions upstream of NO production. However, inhibition of LPS- and GSNO-induced expression of GFAP either by NS-2028 [a specific inhibitor of guanylate cyclase (GC)] or by KT5823 [a specific inhibitor of cGMP-activated protein kinase (PKG)], and induction of GFAP expression by either 8-Br cGMP (a cell-permeable cGMP analog) or MY-5445 (a specific inhibitor of cGMP phosphodiesterase) suggests that NO induces GFAP via GC-cGMP-PKG. This study illustrates a novel biological role of NO in regulating the expression of GFAP in astrocytes through the GC-cGMP-PKG pathway that may participate in the pathogenesis of neurodegenerative disorders.
Keywords: neurotoxins, nitric oxide, astroglial activation, GFAP, guanylate cyclase, cGMP
Introduction
Astrocytes are the major glial cell population within the CNS. They play important physiological roles in brain functions. Astrocyte-neuron cross talk through the release of several neurotrophic factors is a primary event in the maintenance of CNS homeostasis. Recent evidence also suggests that astrocytes might act as immunocompetent cells within the brain (Dong and Benveniste, 2001). Astrocytes react to various neurodegenerative insults rapidly, leading to vigorous astrogliosis (Reier, 1986; Eng et al., 1992). Subsequently, activation of astrocytes has been implicated in the pathogenesis of a variety of neurodegenerative diseases, including Alzheimer's disease, inflammatory demyelinating diseases, human immunodeficiency virus (HIV)-associated dementia, acute traumatic brain injury, and prion-associated spongiform encephalopathies (Eng et al., 1994). Although activated astrocytes secrete different neurotrophic factors for neuronal survival, it is believed that rapid and severe activation augments/initiates an inflammatory response, leading to neuronal death and brain injury (Tani et al., 1996). After severe activation, astrocytes secrete various neurotoxic substances and express an enhanced level of glial fibrillary acidic protein (GFAP), which is considered a marker protein for astrogliosis (Eng et al., 1994). GFAP increases at the periphery of ischemic lesion after neurodegenerative insults (Chen et al., 1993). Senile plaques, a pathologic hallmark of Alzheimer's disease, are associated with GFAP-positive activated astrocytes (Nagele et al., 2004). It is reported that in various neuroinflammatory diseases, the increased GFAP expression corresponds to the severity of astroglial activation (Eng et al., 1992, 1994). However, the mechanism by which astroglial expression of GFAP is increased in neurodegenerative CNS remains unclear.
Because activated astrocytes also express inducible nitric oxide (NO) synthase (iNOS) to produce an excessive amount of NO, a molecule implicated virtually in all reported neurodegenerative and neuroinflammatory conditions (Murphy, 2000), we were prompted to investigate whether NO plays a role in increased expression of astroglial GFAP. Here, we report that NO is instrumental in inducing the expression of GFAP in astrocytes. Different inducers of NO production, such as lipopolysachharide (LPS), interferon-γ (IFN-γ), interleukin-1β (IL-1β), HIV-1 gp120, amyloid β (Aβ)1–42, and double-stranded RNA (dsRNA) [polyinosinic-polycytidilic acid (poly IC)], induced the expression of GFAP in mouse primary astrocytes via NO, suggesting that specific targeting of NO either by iNOS inhibitors or NO scavengers may be an important step for the attenuation of astrogliosis in neurodegenerative disorders. Furthermore, we also demonstrate that NO used the guanylate cyclase (GC)–cGMP–cGMP-activated protein kinase (PKG) signaling pathway but not nuclear factor κB (NF-κB) to induce the expression of GFAP in astrocytes.
Materials and Methods
Reagents.
Fetal bovine serum and DMEM/F-12 were obtained from Mediatech (Ormond Beach, FL), and LPS (Escherichia coli) and poly IC were purchased from Sigma (St. Louis, MO). HIV-1 coat protein gp120 was obtained from United States Biological (Cleveland, OH). NS-2028 (an inhibitor of guanylate cyclase), 8-Br cGMP, MY-5445 (an inhibitor of cGMP phosphodiesterase), KT5823 (an inhibitor of PKG), and l-N6-(I-iminoethyl)-lysine hydrochloride (l-NIL; an inhibitor of iNOS) were obtained from Biomol (Plymouth Meeting, PA). Recombinant mouse IFN-γ and IL-1β were obtained from R & D Systems (Minneapolis, MN). Human Aβ peptide 1–42 was obtained from Bachem Bioscience (King of Prussia, PA).
Preparation of fibrillar Aβ.
Fibrillar Aβ1–42 and control reverse peptide Aβ42–1 (Bachem Bioscience) were prepared by incubating freshly solubilized peptides at 50 μm in sterile distilled water at 37°C for 5 d (Pike et al., 1993).
Isolation of mouse primary astrocytes and microglia.
Astrocytes and microglia were isolated from mixed glial cultures following the procedure of Giulian and Baker (1986) as described previously (Pahan et al., 2000, 2001). Briefly, cerebra taken from 2- to 3-d-old mouse pups were chopped, triturated, passed through mesh, and trypsinized for the isolation of mixed glial cells. On day 9, the mixed glial cultures were washed three times with DMEM/F-12 and subjected to a shake at 240 rpm for 2 h at 37°C on a rotary shaker to isolate microglia. Similarly, on day 11, cells were shaken at 180 rpm for 18 h to remove oligodendroglia. Then, attached cells, primarily the astrocytes, were trypsinized, subcultured, and stimulated with LPS and other different stimuli as mentioned later.
Assay for NO synthesis.
Synthesis of NO was determined by assay of culture supernatants for nitrite, a stable reaction product of NO with molecular oxygen, using Griess reagent as described previously (Jana et al., 2001; Liu et al., 2002).
Cell viability measurement.
Mitochondrial activity was measured with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma).
Immunofluorescence analysis.
Immunofluorescence analysis was performed as described previously (Jana and Pahan, 2004). Briefly, wells containing 4–5 × 105 cells/well were fixed with 4% paraformaldehyde for 20 min followed by treatment with cold ethanol (−20°C) for 5 min and two rinses in PBS. Samples were blocked with 3% BSA in PBS–Tween 20 (PBST) for 30 min and incubated in PBST containing 1% BSA and goat anti-GFAP (1:100). After three washes in PBST (15 min each), wells were further incubated with Cy5 (Jackson ImmunoResearch, West Grove, PA). For negative controls, a set of culture wells was incubated under similar conditions without primary antibodies. The samples were mounted and observed under a Bio-Rad (Hercules, CA) MRC1024ES confocal laser-scanning microscope.
Semiquantitative reverse transcription-PCR analysis.
Total RNA was isolated from primary astrocytes using Ultraspec-II RNA reagent (Biotecx, Houston, TX) following the protocol of the manufacturer. To remove any contaminated genomic DNA, total RNA was digested with DNase. Semiquantitative reverse transcription (RT)-PCR was performed as described previously (Dasgupta et al., 2004) using an RT-PCR kit from Clontech (Cambridge, UK). Briefly, 1 μg of total RNA was reverse transcribed using oligo (dT)12–18 as primer and Moloney murine leukemia virus reverse transcriptase (Clontech) in a 20 μl reaction mixture. The resulting cDNA was appropriately diluted, and diluted cDNA was amplified using Titanium TaqDNA polymerase and the following primers: GFAP, sense, 5′-GGCGCTCAATGCTGGCTTCA-3′; antisense, 5′-TCTGCCTCCAGCCTCAGGTT-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), sense, 5′-GGTGAAGGTCGGTGTGAACG-3′; antisense, 5′-TTGGCTCCACCCTTCAAGGTG-3′. Amplified products were electrophoresed on 1.8% agarose gels and visualized by ethidium bromide staining. The relative expression of GFAP was measured after scanning the bands with a Fluor Chem 8800 Imaging System (Alpha Innotech, San Leandro, CA).
Real-time PCR analysis for GFAP mRNA.
This was performed using the ABI-Prism7700 sequence detection system (Applied Biosystems, Foster City, CA) as described previously (Dasgupta et al., 2004). Briefly, reactions were performed in a 96-well optical reaction plated on cDNA equivalent to 50 ng of DNase-digested RNA in a volume of 25 μl, containing 12.5 μl of TaqMan Universal Master mix and optimized concentrations of carboxy fluorescein (FAM)-labeled probe and forward and reverse primers following the protocol of the manufacturer. All primers and FAM-labeled probes for mouse GFAP and GAPDH were obtained from Applied Biosystems. The mRNA expression of GFAP was normalized to the level of GAPDH mRNA. Data were processed by the ABI Sequence Detection system 1.6 software and analyzed by ANOVA.
Microinjection of LPS into the striatum of C57BL/6 mice.
Male C57BL/6 mice (8–10 week of age) were anesthetized with ketamine and xylazine and underwent cerebellar operations in a Kopf small animal stereotaxic instrument (Kopf Instruments, Tujunga, CA). Briefly, the animal was mounted in a stereotaxic frame on a heating blanket. Body temperature was maintained at 37 ± 0.5°C during the time of surgery. A midsagittal incision was made to expose the cranium, and a hole <0.5 mm in diameter was drilled with a dental drill over the cerebrum according to the following coordinates: 5 mm anterior to λ, lateral (L) 2.2 mm, ventral (V) 3.5 (see Fig. 5A). Four micrograms of LPS in the presence or absence of l-NIL (10 μg) and 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide (PTIO; 10 μg) dissolved in 3 μl of saline was injected using a 5 μl syringe (Hamilton, Reno, NV) over a period of 2 min, and the needle was held in place for another minute before withdrawing it from the skull to prevent reflux up the needle tract. Similarly, control mice received 3 μl of saline. The incision was closed with surgical staples and covered with a mixture of Bacitracin and Hurricane (20% benzocaine).
Figure 5.
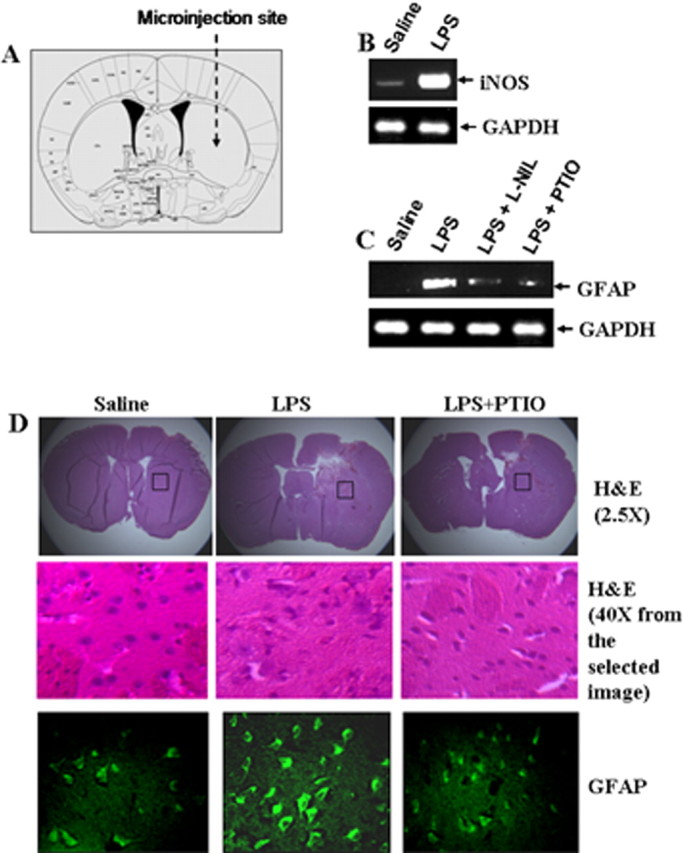
Effect of l-NIL and PTIO on LPS-mediated increase in GFAP expression in vivo in mouse striatum. A, Two micrograms of LPS in the presence or absence of l-NIL (10 μg) and PTIO (10 μg) dissolved in 3 μl of saline were stereotaxically injected into the striatum of C57BL/6 mice. B, C, After 24 h of microinjection, the expression of iNOS mRNA (B) and GFAP mRNA (C) was analyzed in the striatum by semiquantitative RT-PCR. D, Brain sections were stained for H&E and immunostained with antibodies against GFAP. Setting of the confocal microscope was strictly kept unaltered during the whole study. Figures are representative of three independent experiments.
Histological analysis.
After 24 h of microinjection, six mice from each of the following groups (saline, LPS, LPS plus l-NIL, and LPS plus PTIO) were anesthetized with a mixture of ketamine (66.6 mg/kg, i.p.) and xylazine (6.66 mg/kg, i.p.) by injection. The striatum was dissected out from each of the mice after perfusion with PBS, pH 7.4, and then with 4% (w/v) paraformaldehyde solution in PBS (Dasgupta et al., 2004). The tissues were further fixed for at least 2 h in the same fixative at room temperature followed by three PBS washes (15 min each). Routine histology was performed to obtain infiltration and morphological details of striatal tissues. Paraformaldehyde fixed tissues were embedded in paraffin, and serial sections (5 μm) were cut. Sections were stained with conventional hematoxylin–eosin (H&E) staining method.
Preparation of nuclear extract and electrophoretic mobility shift assay.
Nuclear extracts were prepared and used for electrophoretic mobility shift assay (EMSA) using 32P-end-labeled double-stranded [NF-κB, 5′-AGT TGA GGG GAC TTT CCC AGG C-3′] oligonucleotides as described previously (Pahan et al., 1998, 2001). Briefly, double-stranded oligonucleotides were radiolabeled with [γ-32P]ATP using polynucleotide T4 kinase. The labeled probe was purified with chroma spin column (BD Biosciences, San Jose, CA). Five micrograms of nuclear extract were incubated with binding buffer and nonspecific oligonucleotides for 15 min in ice before incubation with labeled probe for another 15 min. Samples were separated on a 6% polyacrylamide gel in 0.25× TBE (90 mm Tris-borate and 2 mm EDTA, pH 8.3) buffer, which were then dried and exposed to generate autoradiograms.
Results
Bacterial LPSs induce the expression of GFAP in mouse primary astrocytes via NO
Astrocytes express GFAP; however, the expression of GFAP is increased during activation of astrocytes and astrogliosis. Because astroglial activation is also associated with the generation of NO, we investigated the role of nitric oxide in the expression of GFAP. Mouse primary astrocytes were stimulated with different concentrations of LPS, the prototype inducer of different immune cells including CNS astrocytes (Pahan et al., 1998, 2000). GFAP was expressed by control astrocytes; however, to monitor the increase by LPS, we used the semiquantitative approach. It is clear from Figure 1A that LPS dose dependently increased the expression of GFAP in primary astrocytes with the maximum increase observed at the highest concentration (1.0 μg/ml) of LPS tested. Assay of nitrite in culture supernatants also shows that the expression of GFAP was coordinate with the production of NO (Fig. 1B). Viability assay showed that LPS was unable to induce cell death at different concentrations tested (Fig. 1C). Subsequently, to correlate the expression of GFAP with NO production, we analyzed time-dependent expression of GFAP and production of NO in astrocytes. We observed that LPS was unable to increase the mRNA expression of GFAP within 6 h of stimulation (Fig. 1D). The increase in GFAP expression was visible at 12 h, and a higher degree of increase was observed at a later hour of stimulation with the maximum increase observed at 24 h of stimulation (Fig. 1D). Interestingly, the induction of NO production also began at 12 h and reached maximum at 24 h (Fig. 1E). Hence, the parallel induction of GFAP and NO suggests a possible involvement of NO in the increased expression of GFAP in astrocytes.
Figure 1.
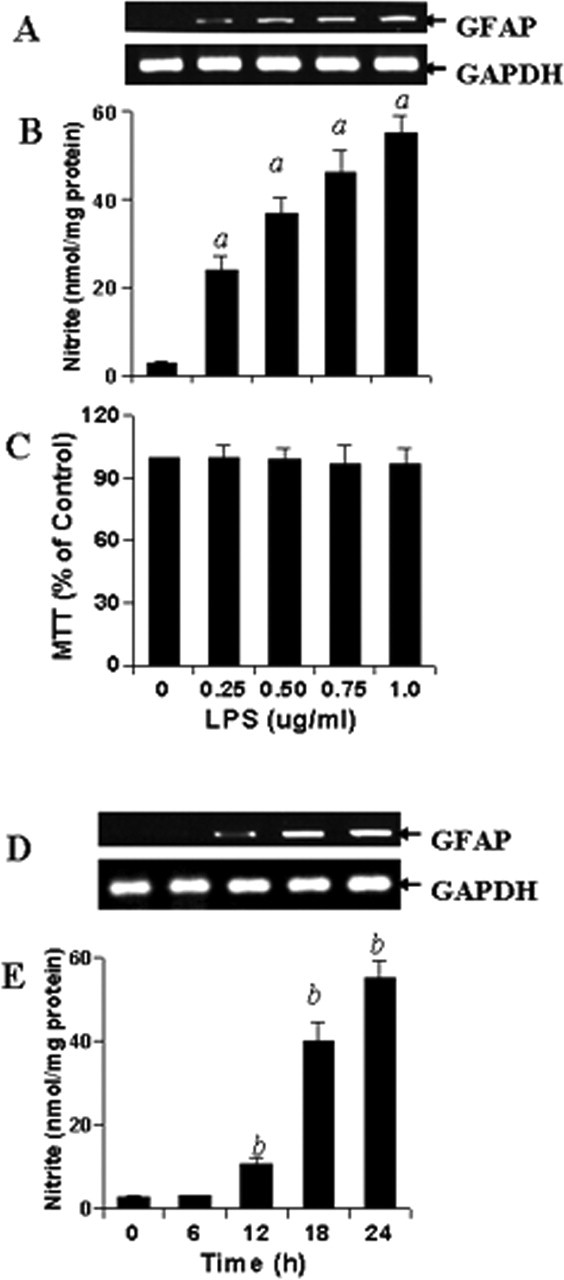
Dose- and time-dependent production of NO and increase in GFAP expression by LPS in mouse primary astrocytes. Cells were stimulated with different concentrations of LPS under serum-free condition. A–C, After 24 h of stimulation, the expression of GFAP was analyzed in cells by semiquantitative RT-PCR (A), the concentration of nitrite was measured in supernatants (B) by Griess reagent, and the viability was examined by MTT assay (C) as described in Materials and Methods. Results are mean ± SD of three different experiments. ap < 0.001 versus control (LPS, 0 μg/ml). D, E, Subsequently, cells were stimulated with 1.0 μg/ml LPS under serum-free condition for a different time period followed by analysis of GFAP mRNA expression in cells (D) and assay of nitrite in supernatants (E). Results are mean ± SD of three different experiments. bp < 0.001 versus 0 h.
Therefore, we investigated the effect of l-NIL (an inhibitor of NOS) and carboxy PTIO (a scavenger of NO) on LPS-mediated increase in GFAP mRNA expression in mouse primary astrocytes. It is clearly evident from semiquantitative RT-PCR analysis in Figure 2, A and C, and quantitative real-time PCR analysis in Figure 2, B and D, that both l-NIL and PTIO markedly inhibited LPS-mediated increase in GFAP expression. In parallel experiments, we also found decreased production of NO in LPS-stimulated cells that were also treated with l-NIL or PTIO compared with cells receiving LPS alone (Table 1). MTT results also demonstrate that both l-NIL and PTIO were unable to alter the cell viability in LPS-stimulated astrocytes (Fig. 2E). Subsequently, we investigated the effect of l-NIL and PTIO on the expression of GFAP protein in LPS-stimulated cells. Immunofluorescence analysis of GFAP in mouse primary astrocytes shows that LPS increased the expression of GFAP compared with control and that l-NIL and PTIO markedly attenuated LPS-mediated upregulation of GFAP expression (Fig. 3). Together, these studies suggest that LPS increases the expression of GFAP in mouse primary astrocytes via NO.
Figure 2.
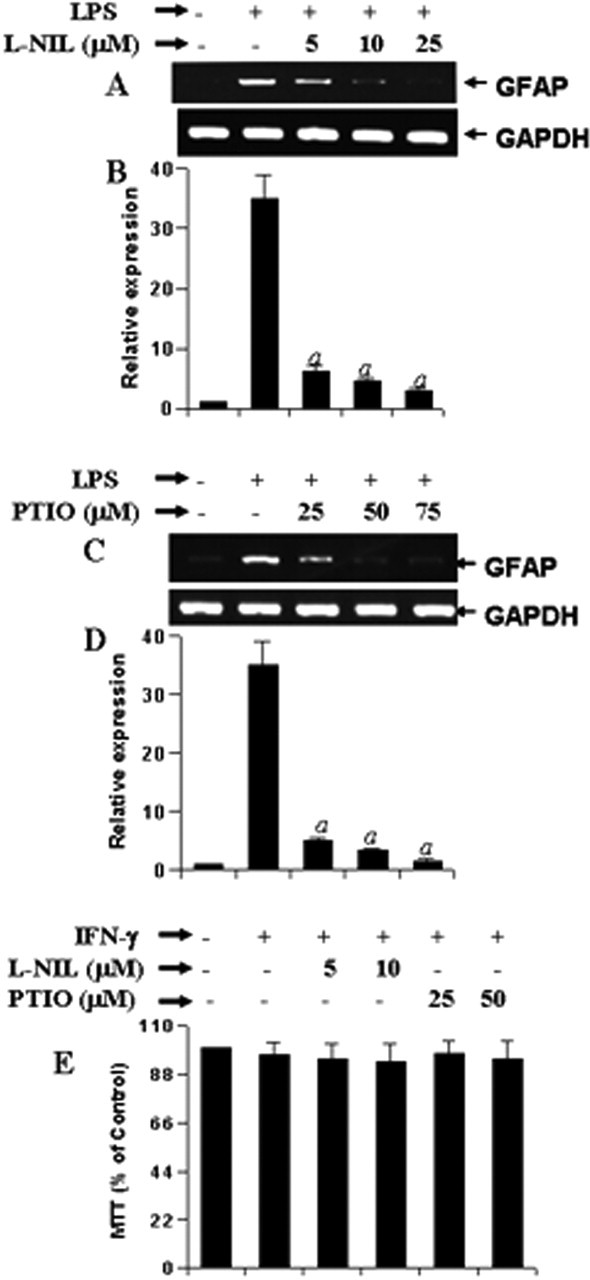
Effect of l-NIL and PTIO on LPS-mediated increase in GFAP expression in mouse primary astrocytes. Cells preincubated with different concentrations of l-NIL and PTIO for 1 h were stimulated with 1.0 μg/ml LPS. A, C, After 24 h of stimulation, the expression of GFAP mRNA was analyzed in cells by semiquantitative RT-PCR. B, D, Quantitative real-time PCR was also used to further clarify the mRNA expression of GFAP. E, MTT assay was conducted to examine cell viability. Results are mean ± SD of three different experiments. ap < 0.001 versus LPS.
Table 1.
Induction of NO production in mouse primary astrocytes by different stimuli
| Inhibitors | Nitrite production (nmol/24 h/mg protein) |
||||||
|---|---|---|---|---|---|---|---|
| Control | LPS | IL-1β | IFN-γ | Poly IC | gp120 | Aβ1–42 | |
| None | 2.8 ± 0.3 | 50.6 ± 7.2a | 31.8 ± 4.5a | 16.8 ± 0.9a | 25.2 ± 3a | 11.2 ± 0.7a | 9.8 ± 1.2a |
| l-NIL | 2.5 ± 0.4 | 7.2 ± 1 | 5.6 ± 0.3 | 4.3 ± 0.6 | 6.2 ± 0.8 | 3.5 ± 0.5 | 4.1 ± 0.6 |
| PTIO | 0.6 ± 0.2 | 7.8 ± 0.6 | ND | ND | 5.7 ± 0.4 | 3.1 ± 0.4 | 3.5 ± 0.4 |
Cells preincubated with l-NIL and PTIO for 1 h were stimulated with LPS (1 μg/ml), IL-1β (10 ng/ml), IFN-γ (12.5 U/ml), poly IC (100 μg/ml), gp120 (200 pm), or Aβ1–42 (1 μm) under a serum-free condition. After 24 h of stimulation, the concentration of nitrite was measured in supernatants as described in Materials and Methods. Results are mean ± SD of three different experiments.
ap< 0.001 versus control.
Figure 3.
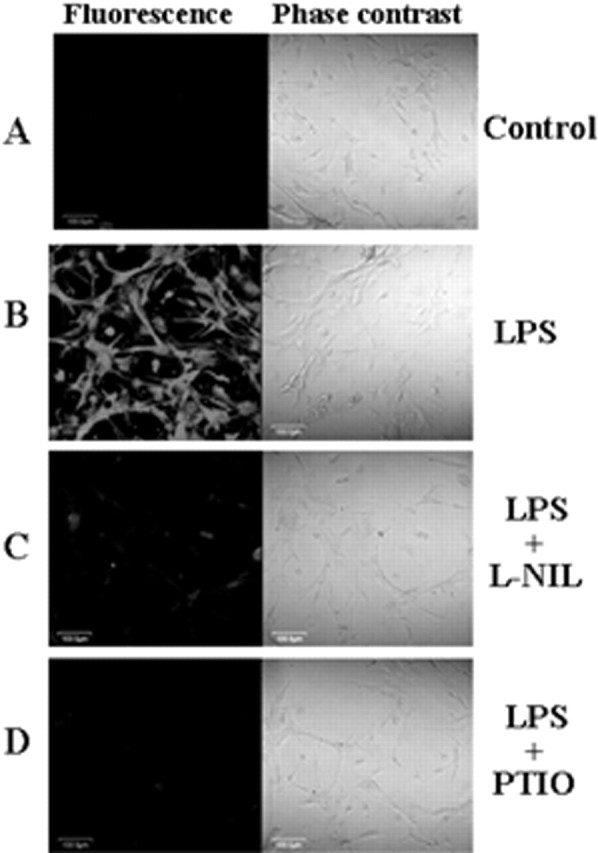
Immunofluorescence analysis of GFAP in LPS-stimulated mouse primary astrocytes. Cells preincubated with l-NIL (25 μm) and PTIO (75 μm) for 1 h were stimulated with 1.0 μg/ml LPS. After 24 h of stimulation, cells were immunostained with GFAP as described in Materials and Methods (A, control; B, LPS; C, LPS plus l-NIL; D, LPS plus PTIO). The setting of the confocal microscope was strictly kept unaltered during the entire study. Figures are representative of three independent experiments.
Involvement of NO in proinflammatory cytokine-, dsRNA-, Aβ1–42-, and HIV-1 gp120-mediated increase in GFAP expression in mouse primary astrocytes
Astrocytes are activated under various pathological conditions, such as inflammation and viral infection (Wang et al., 1993; Eng et al., 1994; Suryadevara et al., 2003; Neil et al., 2005). Because LPS increased the expression of GFAP in astrocytes via NO, we investigated whether other inducers of NO production also increased the expression of GFAP via NO. Mouse primary astrocytes were stimulated with proinflammatory cytokines (IL-1β and IFN-γ), fibrillar Aβ1–42 peptides, HIV-1 coat protein gp120 (Dawson et al., 1993), and double-stranded RNA in the form of poly IC (Auch et al., 2004) in the presence or absence of different concentrations of l-NIL and PTIO. All five stimuli (IL-1β, IFN-γ, poly IC, Aβ1–42, and gp120) induced the production of NO (Table 1) and the expression of GFAP in mouse primary astrocytes (Fig. 4). Similar to the inhibition of LPS-mediated expression of GFAP, either blocking the production of NO by l-NIL or scavenging NO by PTIO knocked down IFN-γ-, IL-1β-, gp120-, poly IC-, and Aβ1–42-mediated expression of GFAP mRNA (Fig. 4), suggesting that different neurodegenerative stimuli increase the expression of GFAP in astrocytes via NO.
Figure 4.
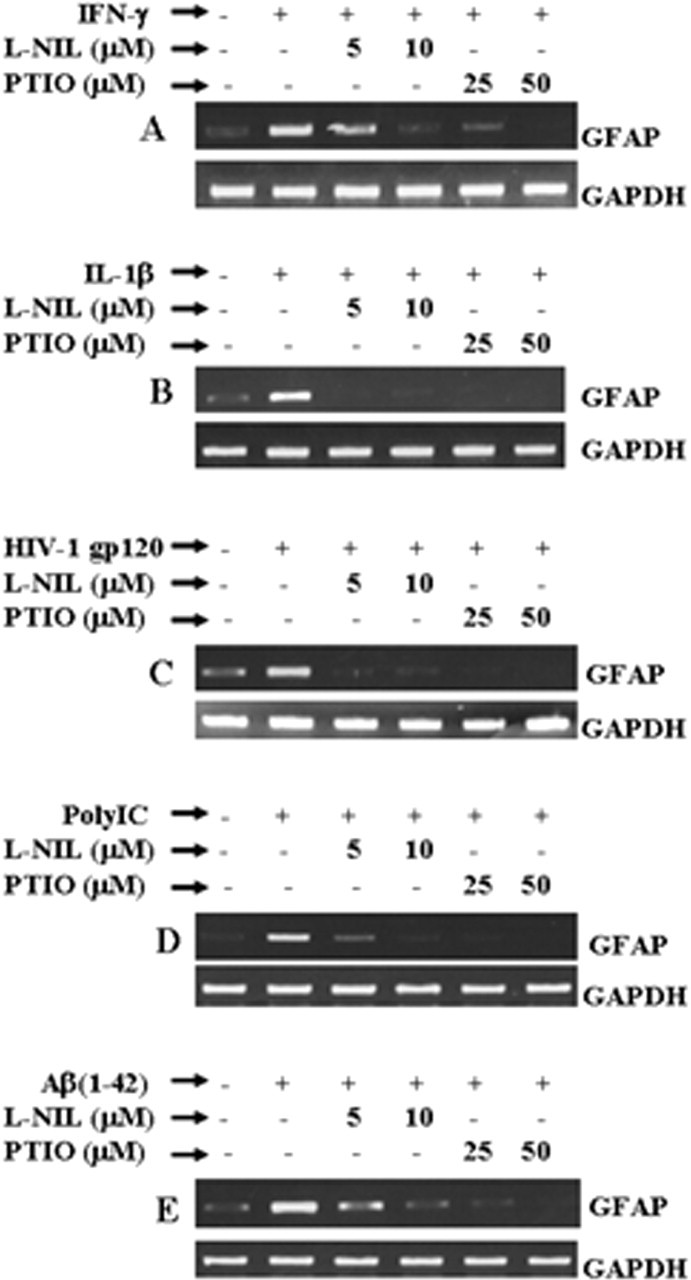
Effect of l-NIL and PTIO on IL-1β-, IFN-γ-, poly IC-, Aβ1–42-, and gp120-mediated increase in GFAP expression in mouse primary astrocytes. Cells preincubated with different concentrations of l-NIL and PTIO for 1 h were stimulated with 12.5 U/ml IFN-γ (A), 10 ng/ml IL-1β (B), 200 pm gp120 (C), 100 μg/ml poly IC (D), and 1 μm Aβ1–42 (E). After 24 h of stimulation, the expression of GFAP mRNA was analyzed in cells by semiquantitative RT-PCR. Results are representative of three independent experiments.
Involvement of NO in LPS-mediated increase in GFAP expression in vivo in mouse striatum
Using various approaches, the studies presented above have shown that different neurotoxins increase the expression of GFAP in primary astrocytes via NO. However, these studies do not indicate whether NO may have the capacity to influence the expression of GFAP in vivo in the CNS. Therefore, we were prompted to investigate whether NO is also involved in the upregulation of GFAP expression in vivo in the striatum. As expected, microinjection of LPS but not saline into the striatum of 8- to 10-week-old male C57BL/6 mice (Fig. 5A) induced the expression of iNOS (Fig. 5B) and GFAP (Fig. 5C). Subsequently, to analyze the role of NO in LPS-mediated expression of GFAP, either l-NIL or PTIO was microinjected together with LPS. As revealed by semiquantitative RT-PCR, both l-NIL and PTIO knocked down LPS-mediated increase in GFAP expression in vivo in the striatum (Fig. 5C). This has been further supported by immunofluorescence analysis. Increased GFAP staining was observed in LPS-microinjected brain sections compared with that in saline-microinjected sections (Fig. 5D, third panel). However, comicroinjection of PTIO markedly inhibited LPS-mediated increase in GFAP expression (Fig. 5D, third panel). H&E analysis (Fig. 5D, first and second panels) also reveals that the decreased GFAP staining after PTIO comicroinjection is not attributable to the health of the tissues. These results demonstrate the requirement of NO in LPS-induced astrogliosis in vivo in the brain.
S-nitroso glutathione alone increases the expression of GFAP in mouse primary astrocytes
Because LPS and other inducers of iNOS increased the expression of GFAP via NO, we investigated whether NO alone was capable of increasing the expression of GFAP in astrocytes. Therefore, we examined the effect of S-nitroso glutathione (GSNO), a NO donor, on the mRNA expression of GFAP in mouse primary astrocytes. As evident from the nitrite assay in Figure 6C, GSNO was capable of releasing NO into the media. NO produced from 100 μm GSNO was almost equivalent to that produced by 1 μg/ml LPS (Table 1). In contrast, NO produced by 50 μm GSNO was comparable with that produced by IL-1β and poly IC (Table 1). Semiquantitative RT-PCR analysis (Fig. 6A) and quantitative real-time PCR analysis (Fig. 6B) demonstrate that GSNO alone dose-dependently increased the expression of GFAP mRNA with maximum induction observed at 200 or 500 μm. These results suggest that NO alone is sufficient to stimulate the expression of GFAP in astrocytes.
Figure 6.
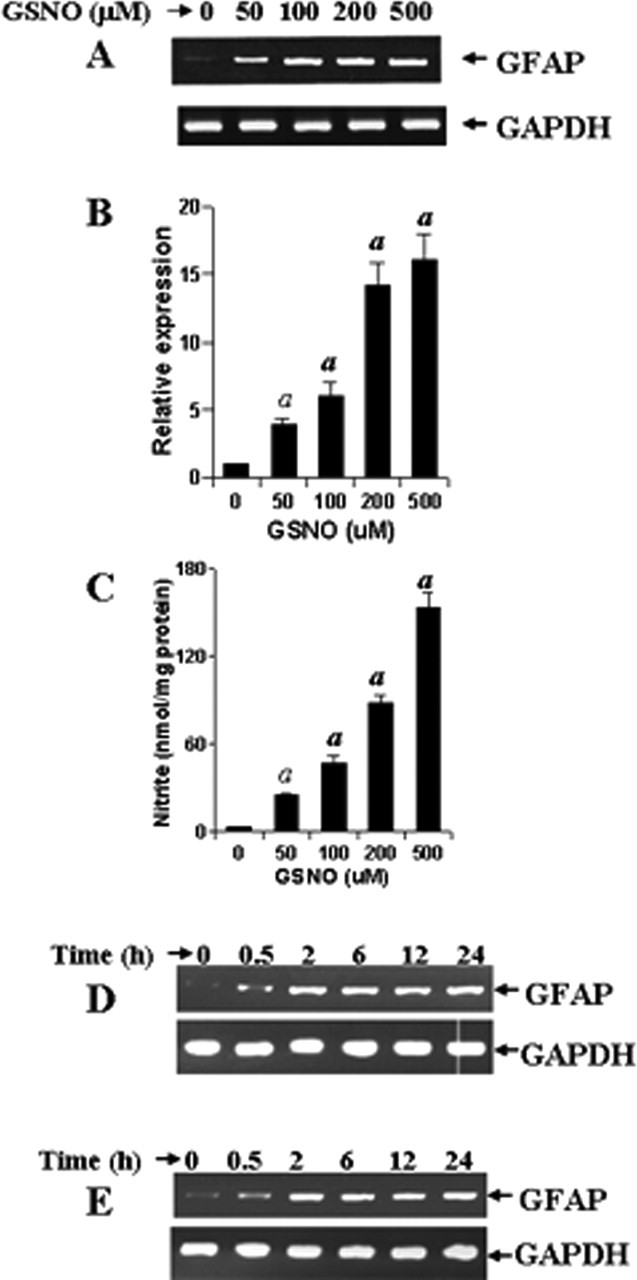
GSNO increases the expression of GFAP in mouse primary astrocytes. Cells were stimulated with different concentrations of GSNO under serum-free condition. A, B, After 24 h of stimulation, the expression of GFAP was analyzed in cells by semiquantitative RT-PCR (A) and quantitative real-time PCR (B). C, The release of nitrite from GSNO was also assayed by Griess reagent. Results are mean ± SD of three different experiments. ap < 0.001 versus control. D, E, Mouse primary astrocytes (D) and a mixture of primary astrocytes and microglia at a ratio of 50:1 (E) were stimulated by GSNO (200 μm) for different time periods under serum-free condition. The expression of GFAP was analyzed in cells by semiquantitative RT-PCR. Figures are representative of three independent experiments.
Do microglia influence an NO-mediated increase in GFAP expression in mouse primary astrocytes?
In addition to astrocytes, there is another population of cells in the brain that also act as mediators of inflammation, the microglia. Although astrocytes comprise ∼55–60% of total brain cells, microglia comprise only 2–5% in healthy humans (Eng and Ghirnikar, 1994; Gonzalez-Scarano and Baltuch, 1999). We investigated whether NO-mediated upregulation of GFAP is a direct effect or indirect effect of microglia. Therefore, we isolated mouse primary astrocytes (<95% pure) and microglia (<95% pure) and mixed them together at a ratio of 50:1. Then, pure primary astrocytes and the astrocyte:microglia coculture were stimulated by 200 μm GSNO. Time course experiments for GSNO-mediated increase in GFAP expression in pure astrocytes (Fig. 6D) and astrocyte:microglia coculture (Fig. 6E) demonstrate that GSNO was capable of increasing the expression of CD11b within 2 h of stimulation in both instances, suggesting that NO increases the expression of GFAP in astrocytes independent of microglia. In addition, whereas LPS-mediated upregulation of GFAP was visible after 18 or 24 h of stimulation (Fig. 1D), GSNO was capable of increasing the expression of GFAP in primary astrocytes within 2 h of stimulation (Fig. 6D), confirming that NO functions downstream to LPS in regulating the expression of GFAP in astrocytes.
Is NF-κB involved in an NO-mediated increase in GFAP expression in astrocytes?
Subsequently, we investigated the mechanism by which NO increased the expression of GFAP in astrocytes. Because the expression of many inducible genes in activated astrocytes depends on the activation of NF-κB, we investigated whether NO increases the expression of GFAP in astrocytes via NF-κB. NEMO (NF-κB essential modifier)-binding domain (NBD) peptides specifically inhibit the induction of NF-κB activation without altering the basal activation of NF-κB (May et al., 2000). We examined the effect of NBD peptides on LPS- and GSNO-mediated increase in GFAP expression. LPS induced the DNA-binding activity of NF-κB (Fig. 7A), and this DNA binding was competed out by excess unlabeled oligonucleotides (data not shown). As expected, NBD peptides dose-dependently inhibited LPS-induced activation of NF-κB (Fig. 7A) and production of NO (Fig. 7B). Because NO is involved in the increase in GFAP expression, NBD peptides suppressed the expression of GFAP in LPS-stimulated cells in a dose-dependent manner (Fig. 7C). Similarly, NBD peptides also suppressed IL-1β-, IFN-γ-, poly IC-, Aβ1–42-, and gp120-mediated increase in GFAP expression in astrocytes (data not shown). In contrast, under similar condition, NBD peptides had no effect on GSNO-mediated increase in GFAP expression (Fig. 7D). These studies suggest that the activation of NF-κB is involved upstream of NO production and that NO increases the expression of GFAP in astrocytes independent of NF-κB activation.
Figure 7.
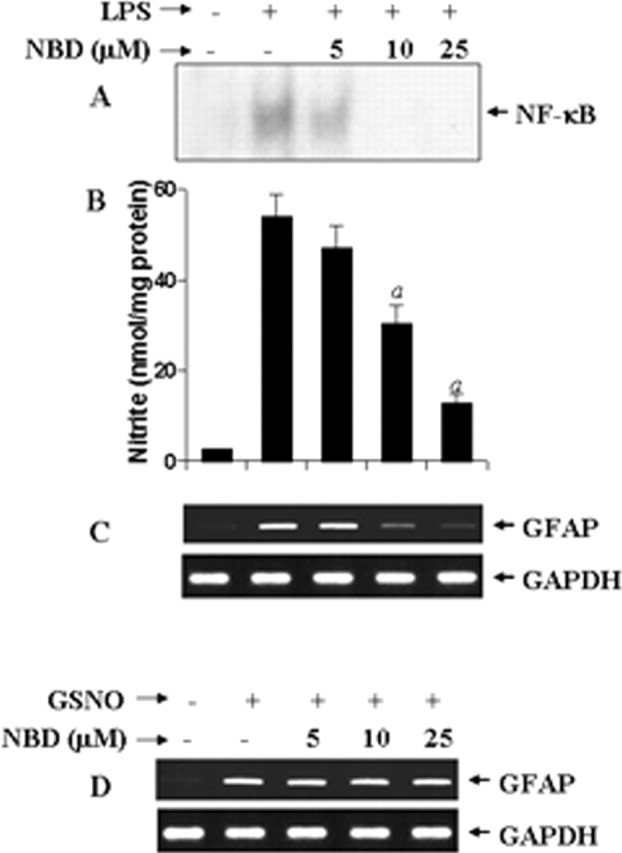
Effect of NBD peptides on LPS- and GSNO-mediated increase in GFAP expression in mouse primary astrocytes. Cells preincubated with different concentrations of NBD peptides for 1 h were stimulated with 1 μg/ml LPS. A, After 1 h of stimulation, DNA-binding activity of NF-κB was monitored by EMSA as described in Materials and Methods. B, C, After 24 h of stimulation, nitrite concentrations (B) were measured in supernatants by Griess reagent, and cells were analyzed for GFAP mRNA by semiquantitative RT-PCR (C). Results are mean ± SD of three different experiments. ap < 0.001 versus LPS. Cells preincubated with different concentrations of NBD peptides for 1 h were stimulated with 200 μm GSNO. D, After 24 h of stimulation, cells were analyzed for GFAP mRNA by semiquantitative RT-PCR.
Involvement of GC in LPS- and GSNO-mediated expression of GFAP in astrocytes
Because GC is intimately coupled to NO-induced downstream signaling events (Hawkins et al., 1998; Bredt, 2003), we attempted to explore the role of GC in LPS- and GSNO-mediated expression of GFAP in astrocytes. We examined the effect of NS-2028, a specific inhibitor of GC (Stoner and Kellum, 2001), on LPS- and GSNO-mediated expression of GFAP. As evident from semiquantitative RT-PCR analysis (Fig. 8A,C) and real-time PCR analysis (Fig. 8B,D), NS-2028 markedly inhibited LPS- as well as GSNO-mediated expression of GFAP, suggesting that NO itself and inducer of NO production increase the expression of GFAP via GC.
Figure 8.

Effect of NS-2028 on LPS- and GSNO-mediated increase in GFAP expression in mouse primary astrocytes. Cells preincubated with different concentrations of NS-2028 (NS) for 1 h were stimulated with either 1.0 μg/ml LPS (A, B) or 200 μm GSNO (C, D) under serum-free conditions. After 24 h of stimulation, the expression of GFAP was analyzed in cells by semiquantitative RT-PCR (A, C) and quantitative real-time PCR (B, D). Results are mean ± SD of three different experiments. ap < 0.001 versus LPS. bp < 0.001 versus GSNO.
Is cGMP alone capable of inducing the expression of GFAP in astrocytes?
Because GC catalyzes the formation of cGMP from GTP, we investigated whether cGMP alone is sufficient to induce the expression of GFAP in primary astrocytes. The level of cGMP in a cell could be increased by either a cell-permeable cGMP derivative (8-Br-cGMP) or an inhibitor of cGMP phosphodiesterase (MY-5445). Therefore, the effect of 8-Br-cGMP and MY-5445 on the expression of GFAP was tested in astrocytes. Interestingly, both 8-Br-cGMP (Fig. 9A,B) and MY-5445 (Fig. 9C,D) markedly increased the expression of GFAP, suggesting that cGMP alone is capable of increasing the expression of GFAP in astrocytes and that LPS induces the expression of GFAP in astrocytes via NO-GC-cGMP pathway.
Figure 9.

8-Br-cGMP and MY-5445 increase the expression of GFAP in mouse primary astrocytes. Cells were stimulated with either different concentrations of 8-Br-cGMP (A, B) or MY-5445 (C, D) under serum-free condition. After 24 h of stimulation, the expression of GFAP was analyzed in cells by semiquantitative RT-PCR. Results are mean ± SD of three different experiments. ap < 0.001 versus control.
Role of PKG in LPS- and GSNO-mediated expression of GFAP in astrocytes
Because cGMP transduces many of its downstream signaling pathways via PKG (Monfort et al., 2002), we were prompted to investigate whether PKG was involved in LPS- and NO-mediated expression of GFAP in astrocytes. A specific cell-permeable inhibitor, KT5823 (Jouvert et al., 2004), was used to inhibit the activity of PKG in mouse primary astrocytes. It is clearly evident from Figure 10, A and B, that KT5823 dose-dependently inhibited LPS-mediated expression of GFAP mRNA with maximum inhibition observed at 25 μm. Similarly, semiquantitative RT-PCR and quantitative real-time PCR analyses also revealed marked inhibition of GSNO-mediated expression of GFAP mRNA by KT5823 in mouse primary astrocytes (Fig. 10C,D). These results suggest that LPS increases the expression of GFAP in astrocytes via NO-GC-cGMP-PKG pathway.
Figure 10.

Effect of KT5823 on LPS- and GSNO-mediated increase in GFAP expression in mouse primary astrocytes. Cells preincubated with different concentrations of KT5823 for 1 h were stimulated with either 1.0 μg/ml LPS (A, B) or 200 μm GSNO (C, D) under serum-free condition. After 24 h of stimulation, the expression of GFAP was analyzed in cells by semiquantitative RT-PCR (A, C) and quantitative real-time PCR (B, D). Results are mean ± SD of three different experiments. ap < 0.001 versus LPS. bp < 0.05 versus GSNO. cp < 0.001 versus GSNO.
Discussion
Astrocytes, constituting the majority of resident glial cells, are present in almost 10 times more in number than neurons in normal adult brain. Increasing evidence suggests that astrocytes play an active role in CNS function by influencing or even directing the neuronal activities (Dong and Benveniste, 2001). GFAP is the principal intermediate filament (IF) protein of mature astrocytes in the CNS. Because vimentin is unable to assemble into IFs on its own, the absence of GFAP leads to an absence of IFs in nonreactive astrocytes in vivo. Therefore, GFAP is probably involved in controlling the shape and movement of astrocytes. Although GFAP is not required for the morphogenesis of the CNS (Pekny et al., 1999), this protein does play other important roles. For example, according to McCall et al. (1996), GFAP−/− mice display enhanced long-term potentiation of both population spike amplitude and EPSP slope in the hippocampus after tetanic stimulation. The study by Shibuki et al. (1996) has also indicated that GFAP-negative mice are deficient in long-term depression at distinct sites in the brain as well as in eyeblink conditioning. Therefore, it has been suggested that GFAP is important for astrocyte-neuronal interactions and that GFAP-mediated astrocytic processes play a vital role in modulating synaptic efficacy in the CNS. In addition, a study by Liedtke et al. (1996) also demonstrates that GFAP expression is essential for normal white matter architecture and blood–brain barrier integrity in aged mice, and its absence may be responsible for a late onset of CNS demyelination.
Because most of the biomolecules in our body possess a physiological as well as a pathophysiological role, in addition to having many physiological roles as mentioned above, GFAP may have CNS-damaging roles as well. After any degenerative injury or insult, astrocytes react rapidly by producing more GFAP, leading to vigorous astrogliosis (Yu et al., 1993; Sriram et al., 2004). Therefore, common pathological hallmarks of several neurodegenerative diseases (e.g., Alzheimer's disease, Parkinson's disease, and HIV-dementia) include the loss of invaluable neurons associated with or followed by massive activation of astrocytes. Although activation of astrocytes may have important beneficial effects in the recovery of injured CNS by actively monitoring and controlling the extracellular water, pH, and ion homeostasis, once astrocytes are activated in neurodegenerating microenvironment, it goes beyond control and eventually detrimental effects outclass beneficial effects (Yu et al., 1993). Therefore, understanding mechanisms that regulate the expression of GFAP in astrocytes is an important area of investigation, because that may provide a tool to control both physiological and pathophysiological processes in the CNS. Several lines of evidence presented in this manuscript clearly demonstrate that NO plays a key role in regulating the expression of GFAP in astrocytes. First, in LPS-stimulated astrocytes, the increase of GFAP expression began after the production of NO. Second, LPS and other inducers of iNOS (IFN-γ, IL-1β, HIV-1 gp120, and poly IC) were unable to increase the expression of GFAP in astrocytes, where either NO was scavenged by PTIO or NO production was inhibited by l-NIL. Similarly, microinjection of LPS into the striatum caused marked upregulation of GFAP. Consistent with the effect observed in isolated astrocytes, both l-NIL and PTIO strongly inhibited LPS-mediated increase in striatal GFAP expression in vivo. Third, GSNO, an NO donor, alone increased the expression of GFAP in astrocytes. Astrocytes in the healthy brain usually do not express iNOS, but after ischemic, traumatic, neurotoxic, or inflammatory damage, activated astrocytes express iNOS and produce an excessive amount of NO in mouse, rat, and human (Pahan et al., 1997; Akama et al., 1998; Liu et al., 2002). Our results clearly indicate that the upregulation of GFAP expression in reactive astrocytes follows NO production. Recently, Hwang et al. (2006) have shown that ionizing radiation induces astrocyte gliosis through microglial activation. In contrast, we found that NO-mediated increase in GFAP expression in astrocytes does not depend on microglia. Therefore, it is possible that ionizing radiation is capable of inducing the production of NO in microglia but not astrocytes. In fact, microglia respond to any insult and injury faster than astrocytes (Gonzalez-Scarano and Baltuch, 1999). However, once NO is produced from microglia, it can increase the expression of GFAP in astrocytes independent of microglia.
The signaling mechanisms for the increase in GFAP expression in astrocytes are not known. However, activation of NF-κB plays an important role in the expression of many proinflammatory molecules in astrocytes [such as, LPS induces the expression of iNOS and proinflammatory cytokines (tumor necrosis factor-α, IL-1β, and IL-6) in astrocytes via activation of NF-κB (Pahan et al., 1997, 1998, 2000)]. Different neurotoxins such as Tat and amyloid-β peptides also induce iNOS via NF-κB (Akama et al., 1998; Liu et al., 2002). Consistently, we also observed that LPS induced the production of NO in mouse astrocytes via an NF-κB-dependent mechanism. Because NO is playing a crucial role in increasing the expression of GFAP, NBD peptides (NF-κB inhibitor) also suppressed LPS- and other stimuli-mediated increase in GFAP expression in astrocytes. However, NBD peptides were unable to inhibit a GSNO-mediated increase in GFAP expression, suggesting that once NO is produced, it does not require NF-κB to increase the expression of GFAP (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Among many downstream targets of NO, activation of GC represents the most important one attributable to its involvement in many physiological processes, including vasodilation, modulation of synaptic transmission, and inhibition of platelet aggregation (Hawkins et al., 1998; Bredt et al., 2003). We were prompted to explore the role of GC in astroglial expression of GFAP. Interestingly, our results demonstrate that NO uses GC for the induction of GFAP in astrocytes (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Our conclusion is based on the following. First, LPS as well as GSNO, an NO donor, was unable to increase the expression of GFAP in astrocytes that were treated with NS-2028, an inhibitor of GC. Second, cell-permeable cGMP analog (8-Br cGMP) and MY-5445, an inhibitor of cGMP phosphodiesterase, were also capable of increasing the expression of GFAP in primary astrocytes. Third, cGMP is known to execute its function via PKG. Consistently, KT5823, a specific inhibitor of PKG, attenuated LPS- as well as GSNO-mediated expression of GFAP. Because the activation of astrocytes is involved in the pathophysiology of several neurodegenerative disorders, our results suggest that NO-coupled GC-cGMP-PKG signaling pathway may also be an important player under neurodegenerative conditions.
NO, a short-lived and diffusible free radical, plays many roles as a signaling and effector molecule in diverse biological systems; it is a neuronal messenger and is involved in vasodilation as well as in antimicrobial and antitumor activities (Nathan, 1992). In contrast, NO has also been implicated in several CNS disorders, including inflammatory, infectious, traumatic, and degenerative diseases (Merrill et al., 1993; Brosnan et al., 1994; Mitrovic et al., 1994; Samdani et al., 1997; Akama et al., 1998). Here, we present another example in which NO may play an important role in physiology and pathophysiology of astrocytes via regulation of GFAP.
Footnotes
This work was supported by National Institutes of Health Grants NS39940 and NS48923, National Multiple Sclerosis Society Grant RG3422A1/1, and the Michael J. Fox Foundation for Parkinson's Research. We thank Avik Roy for his help in animal studies and Dr. You Zhou (University of Nebraska at Lincoln) for his help in confocal microscopy.
References
- Akama KT, Albanese C, Pestell RG, Van Eldik LJ (1998). Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFkappaB-dependent mechanism. Proc Natl Acad Sci USA 95:5795–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auch CJ, Saha RN, Sheikh FG, Liu X, Jacobs BL, Pahan K (2004). Role of protein kinase R in double-stranded RNA-induced expression of nitric oxide synthase in human astroglia. FEBS Lett 563:223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS (2003). Nitric oxide signaling in brain: potentiating the gain with YC-1. Mol Pharmacol 63:1206–1208. [DOI] [PubMed] [Google Scholar]
- Brosnan CF, Battistini L, Raine CS, Dickson DW, Casadevall A, Lee SC (1994). Reactive nitrogen intermediates in human neuropathology: an overview. Dev Neurosci 16:152–161. [DOI] [PubMed] [Google Scholar]
- Chen H, Chopp M, Schultz L, Bodzin G, Garcia JH (1993). Sequential neuronal and astrocytic changes after transient middle cerebral artery occlusion in the rat. J Neurol Sci 188:109–116. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Jana M, Zhou Y, Fung YK, Ghosh S, Pahan K (2004). Antineuroinflammatory effect of NF-kappaB essential modifier-binding domain peptides in the adoptive transfer model of experimental allergic encephalomyelitis. J Immunol 173:1344–1354. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, Uhl GR, Snyder SH (1993). Human immunodeficiency virus type 1 coat protein neurotoxicity mediated by nitric oxide in primary cortical cultures. Proc Natl Acad Sci USA 90:3256–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN (2001). Immune function of astrocytes. Glia 36:180–190. [DOI] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS (1994). GFAP and astrogliosis. Brain Pathol 4:229–237. [DOI] [PubMed] [Google Scholar]
- Eng LF, Yu AC, Lee YL (1992). Astrocytic response to injury. Prog Brain Res 94:353–365. [DOI] [PubMed] [Google Scholar]
- Giulian D, Baker TJ (1986). Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci 6:2163–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Baltuch G (1999). Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci 22:219–240. [DOI] [PubMed] [Google Scholar]
- Hawkins RD, Son H, Arancio O (1998). Nitric oxide as a retrograde messenger during long-term potentiation in hippocampus. Prog Brain Res 118:155–172. [DOI] [PubMed] [Google Scholar]
- Hwang SY, Jung JS, Kim TH, Lim SJ, Oh ES, Kim JY, Ji KA, Joe EH, Cho KH, Han IO (2006). Ionizing radiation induces astrocyte gliosis through microglia activation. Neurobiol Dis 21:457–467. [DOI] [PubMed] [Google Scholar]
- Jana A, Pahan K (2004). Human immunodeficiency virus type 1 gp120 induces apoptosis in human primary neurons through redox-regulated activation of neutral sphingomyelinase. J Neurosci 24:9531–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana M, Liu X, Koka S, Ghosh S, Petro TM, Pahan K (2001). Ligation of CD40 stimulates the induction of nitric-oxide synthase in microglial cells. J Biol Chem 276:44527–44533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvert P, Revel MO, Lazaris A, Aunis D, Langley K, Zwiller J (2004). Activation of the cGMP pathway in dopaminergic structures reduces cocaine-induced EGR-1 expression and locomotor activity. J Neurosci 24:10716–10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R, Raine CS (1996). GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron 17:607–615. [DOI] [PubMed] [Google Scholar]
- Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K (2002). Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J Biol Chem 277:39312–39319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May MJ, D'Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S (2000). Selective inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the IκB kinase complex. Science 289:1550–1554. [DOI] [PubMed] [Google Scholar]
- McCall MA, Gregg RG, Behringer RR, Brenner M, Delaney CL, Calbreath EJ, Zhang CL, Pearce RA, Chiu SY, Messing A (1996). Targeted deletion in astrocyte intermediate filament (GFAP) alters neuronal physiology. Proc Natl Acad Sci USA 93:6361–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE (1993). Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J Immunol 151:2132–2141. [PubMed] [Google Scholar]
- Mitrovic B, Ignarro LJ, Montestruque S, Smoll A, Merrill JE (1994). Nitric oxide as a potential pathological mechanism in demyelination: its differential effects on primary glial cells in vitro. Neuroscience 61:575–585. [DOI] [PubMed] [Google Scholar]
- Monfort P, Munoz MD, Kosenko E, Felipo V (2002). Long-term potentiation in hippocampus involves sequential activation of soluble guanylate cyclase, cGMP-dependent protein kinase, and cGMP-degrading phosphodiesterase. J Neurosci 22:10116–10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S (2000). Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia 29:1–13. [DOI] [PubMed] [Google Scholar]
- Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC, Wegiel J (2004). Contribution of glial cells to the development of amyloid plaques in Alzheimer's disease. Neurobiol Aging 25:663–674. [DOI] [PubMed] [Google Scholar]
- Nathan C (1992). Nitric oxide as a secretory product of mammalian cells. FASEB J 6:3051–3064. [PubMed] [Google Scholar]
- Neil SJ, Aasa-Chapman MM, Clapham PR, Nibbs RJ, McKnight A, Weiss RA (2005). The promiscuous CC chemokine receptor D6 is a functional coreceptor for primary isolates of human immunodeficiency virus type 1 (HIV-1) and HIV-2 on astrocytes. J Virol 79:9618–9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Namboodiri AMS, Singh I (1997). Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J Clin Invest 100:2671–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Khan M, Namboodiri AM, Singh I (1998). Sphingomyelinase and ceramide stimulate the expression of inducible nitric-oxide synthase in rat primary astrocytes. J Biol Chem 273:2591–2600. [DOI] [PubMed] [Google Scholar]
- Pahan K, Liu X, McKinney MJ, Wood C, Sheikh FG, Raymond JR (2000). Expression of a dominant-negative mutant of p21(ras) inhibits induction of nitric oxide synthase and activation of nuclear factor-kappaB in primary astrocytes. J Neurochem 74:2288–2295. [DOI] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Liu X, Hilger S, McKinney MJ, Petro TM (2001). Interleukin-12 p40 induces nitric oxide synthase and activates NF-κB in microglial cells. J Biol Chem 276:7899–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Johansson CB, Eliasson C, Stakeberg J, Wallén A, Perlmann T, Lendahl U, Betsholtz C, Berthold CH, Frisén J (1999). Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J Cell Biol 145:503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW (1993). Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci 13:1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reier PJ (1986). Gliosis following CNS injury: the anatomy of astrocytic scars and their influences on axonal elongation. Astrocytes 3:263–324. [Google Scholar]
- Samdani AF, Dawson TM, Dawson VL (1997). Nitric oxide synthase in models of focal ischemia. Stroke 28:1283–1288. [DOI] [PubMed] [Google Scholar]
- Shibuki K, Gomi H, Chen L, Bao S, Kim JJ, Wakatsuki H, Fujisaki K, Fujimoto K, Katoh A, Ikeda T, Chen C, Thomson RF, Itohara S (1996). Deficient cerebellar long-term depression, impaired eyeblink conditioning, and normal motor coordination in GFAP mutant mice. Neuron 16:587–599. [DOI] [PubMed] [Google Scholar]
- Sriram K, Benkovic SA, Hebert MA, Miller DB, O'Callaghan JP (2004). Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: key signaling pathway for astrogliosis in vivo? J Biol Chem 279:19936–19947. [DOI] [PubMed] [Google Scholar]
- Stoner MC, Kellum JM (2001). Both serotonin and a nitric-oxide donor cause chloride secretion in rat colonocytes by stimulating cGMP. Surgery 130:236–241. [DOI] [PubMed] [Google Scholar]
- Suryadevara R, Holter S, Borgmann K, Persidsky R, Labenz-Zink C, Persidsky Y, Gendelman HE, Wu L, Ghorpade A (2003). Regulation of tissue inhibitor of metalloproteinase-1 by astrocytes: links to HIV-1 dementia. Glia 44:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani M, Glabinski AR, Tuohy VK, Stoler MH, Estes ML, Ransohoff RM (1996). In situ hybridization analysis of glial fibrillary acidic protein mRNA reveals evidence of biphasic astrocyte activation during acute experimental autoimmune encephalomyelitis. Am J Pathol 148:889–896. [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, Rothstein JD, Yu AC, Lee YL, Eng LF (1993). Astrogliosis in culture: I. The model and the effect of antisense oligonucleotides on glial fibrillary acidic protein synthesis. J Neurosci Res 34:295–303. [DOI] [PubMed] [Google Scholar]