

Abstract
Nitric oxide (NO), being a double-edged sword depending on its concentration in the microenvironment, is involved in both physiological and pathological processes of many organ systems including brain and spinal cord. It is now well-documented that once inducible nitric oxide synthase (iNOS) is expressed in CNS in a signal-dependent fashion, NO in excess of physiological thresholds is produced and this excess NO then plays a role in the pathogenesis of stroke, demyelination and other neurodegenerative diseases. Therefore, a keen interest has been generated in recent years in comprehending the regulation of this enzyme in brain cells. The present review summarizes our current understanding of signaling mechanisms leading to transcription of the iNOS gene in activated astrocytes. We attempt this comprehension with a hope to identify potential targets to intervene NO-mediated CNS disorders.
Keywords: Astrocytes, Inducible nitric oxide synthase, Inducers, Signal transduction, Inflamation
1. Signaling of iNOS in astrocytes: rationalizing the concept
Astrocytes are the major glial cells present in CNS. In counting, they greatly outnumber neurons, microglia or oligodendrocytes. More often than not, the dogma has held these cells as mere bystanders during brain pathophysiology. However, as suggested by recent research, astrocytes form the bulk of all resident brain cells with an ability to initiate and amplify immune response (Dong and Benveniste, 2001; Pekny and Nilsson, 2005). Furthermore, according to several lines of evidences, astrocytes express all three isoforms of NOS (Gibson et al., 2005), including iNOS which is expressed prolifically by these cells in response to various stimuli (see next section). Thus, volumetrically speaking, understanding signaling pathways for expression of iNOS in astrocytes is crucial in order to harness excess NO-mediated damage during brain disorders.
In addition to being quantitative heavyweights among CNS residents, the protoplasmic astrocytes of grey matter laminate the local vasculature by coating the micro-vessels with their endfeet, a property not shared by any other CNS cell-type. Due to such juxtaposition, these cells are potent regulators of local microcirculation (Mulligan and MacVicar, 2004). In light of NO being a potent vasodilator, the vasoregulatory privileges of astrocytes may spell haywire if unregulated amount of NO is produced by these cells. Taken together, these reasons advocate for enhanced neuroimmunological inquisitiveness into cell-type specific regulation of iNOS in astrocytes.
2. The cues of astroglial iNOS and their receptors
Astroglial iNOS is upregulated in response to a wide range of inducers. Broadly, these inducers can be categorized by their ability to either elicit innate or adaptive immune responses. The first group consists of products of bacterial and viral origin, while the second group is primarily composed of proinflammatory cytokines. Bacterial products like, LPS and bacterial DNA can induce iNOS in rat and mice astrocytes. However, human astrocytes do not upregulate iNOS in response to LPS. In addition to whole viruses themselves, isolated viral products, like viral nucleic acids or proteins, are strong inducers of iNOS in astrocytes. Our lab has shown that viral dsRNA induces iNOS in human astroglia (Auch et al., 2004). Similarly, various proteins of viral origin, like gp41, gp120, HIV-Tat, etc., also trigger iNOS expression.
In the other group, few cytokines, like IL-1β and IFN-γ, alone can induce iNOS in rodent astrocytes. Other cytokines, like TNF-α, usually induce iNOS in conjunction with IL-1β or IFN-γ. However, human astrocytes behave differently with respect to cytokine-mediated expression of iNOS. As for example, IFN-γ, which is a potent inducer of iNOS in rat or mouse astrocytes, fails to induce it in human astrocytes. Among different cytokines tested in our laboratory and elsewhere, it has been found that only IL-1β, alone or in combinations with other cytokines, is capable of inducing iNOS in human primary astrocytes (Hua et al., 2002; Jana et al., 2005).
Astrocytes exhibit an array of surface receptors in addition to secreting their soluble forms which may interpret signals generated by the above mentioned inducers. Most of the inducers are now known to engage specific receptors, a list of which is compiled in Table 1. However, in context of astroglial iNOS upregulation, the exact receptor has not been delineated yet for pro-inflammatory cytokines, like IL-1β. We have, however, enlisted the most probable receptors in Table 1. On the flip side, engagement of certain receptors with unknown ligands for iNOS regulation, like CD23, is also known to activate astroglial iNOS (Dugas et al., 2001).
Table 1.
A brief list of various receptors and inducers of astroglial iNOS. Unknown/unconfirmed elements are marked (?)
| Inducer class | Inducer | Receptor | Subsequent messengers of the signal |
|---|---|---|---|
| Viral and bacterial products | LPS | TLR4, CD14 (Galea et al., 1996) | PKCη, ERK1/2, p38, NF-κB (Chen et al., 1998; Pahan et al., 1998a,b; Bhat et al., 1998) |
| Bacterial DNA | TLR-9 (Hosoi et al., 2004) | MyD88, p38 (Hosoi et al., 2004) | |
| viral dsRNA | TLR-3 (Scumpia et al., 2005) | PKR, p38, NF-κB, C/EBP (Auch et al., 2004) | |
| Viral Tat | CCL2/MCP-1 (?) | ERK2, C/EBP (Liu et al., 2002) | |
| Cytokines | IL-1β | Type-1 IL-1βR (?) | p38, NF-κB, C/EBP, AP-1(?) (Jana et al., 2005; Da Silva et al., 1997) |
| IFNγ | IFNγR (?) | JAK2, STAT1 (Jana et al., 2005; Dell’Albani et al., 2001) | |
| Miscellaneous | LDL and HDL (Nanetti et al., 2004) | ? | ? |
| Ethanol (Blanco et al., 2004) | ? | NF-κB (Blanco et al., 2004) |
3. Signals that positively regulate iNOS expression
3.1. Role of tyrosine kinases: JAcKing it up
Broad-spectrum tyrosine kinase inhibitors (like herbimycin A) inhibit LPS- and cytokine-mediated expression of iNOS in rodent astrocytes (Kong et al., 1996; Feinstein et al., 1994) suggesting a probable involvement of tyrosine kinase(s) in the expression of astroglial iNOS. Among all the tyrosine kinases, the role of JAK in the expression astroglial iNOS has been extensively studied. JAK-STAT signaling, originally identified as the signaling pathway for IFNs, is now known to mediate signals of various cytokines, growth factors and hormones. The basic biology of JAK-mediated signal transduction is based on ligand-stimulated assembly of receptors into an active complex followed by phosphorylation of the receptor-associated JAKs (JAK1, JAK2, and JAK3) and tyrosine kinase 2 (Tyk2). Subsequently, phosphorylated JAKs lead to the phosphorylation of STATs, which in turn are released from the receptor complex and form homo- or heterodimers (Fig. 1A). These dimers translocate to the nucleus where they directly bind to the promoter region of several inflammation-associated genes including iNOS. Consistently, Xie et al. (1993) have provided evidence that the promoter region of mouse iNOS gene contains the IFN-γ activation site (GAS) and that GAS is activated by tyrosine phosphorylated STAT1α. According to Kitamura et al. (1996), the activation of JAK2 is mainly involved in IFN-γ-induced expression of iNOS in rat astrocytes. In fact, IFN-γ induces tyrosine phosphorylation of STAT1α but not STAT1β via JAK2 and that tyrosine phosphorylation of STAT1α seems to be essential for IFN-γ-induced expression of iNOS in rat astrocytes (Dell’Albani et al., 2001). These observations suggest that IFN-γ-induced activation of JAK2 phosphorylates STAT1α that in turn binds to GAS elements in the promoter of iNOS.
Fig. 1.
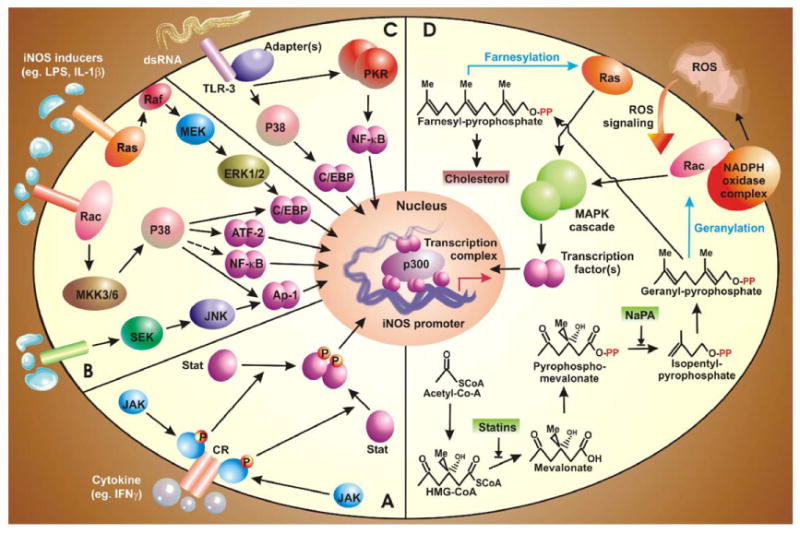
The oval of iNOS-inducing signaling pathways. Several signal transduction pathways culminate by activating transcription factors, which upon activation, translocate to the nucleus and bind with co-activators like p300 and members of basic transcription machinery to form the transcription initiation complex (TIC). This TIC facilitates the bending of iNOS promoter. Segment A: Signal transduction of dsRNA is mediated by TLR-3 which subsequently recruits NF-κB and C/EBPβ via PKR and p38-MAPK. Segment B: Cross talk of iNOS signaling and cholesterol pathway. Geranyl-pyrophosphate and farnesyl-pyrophosphate, generated from mother carbon skeleton of isopendenyl pyrophosphate, modify membrane bound Rac and Ras, respectively, leading to their activation. Subsequently, MAP kinase pathways are employed to activate specific transcription factors. Segment C: The JAK-STAT pathway. Ligand-mediated engagement of receptor tyrosine kinases lead to their auto-activation and permits recruitment and phosphorylation of JAK. Thereafter, receptor bound JAK mediates phosphorylation, dimerization, and activation of STAT which bind to GAS elements in iNOS promoter. Segment D: MAP-kinase pathways. Activation of various receptors induces a downstream signal, which is mediated by one or more out of the three MAP-K cascades. Several transcription factors act downstream of specific MAPK. This figure indicates the central role of p38 MKP in iNOS induction.
Interestingly, inhibitors of tyrosine kinases also block LPS-induced expression of iNOS in mouse astrocytes (Kong et al., 1996), yet LPS-induced expression of iNOS does not involve the activation of JAK-STAT signaling pathway (Nishiya et al., 1995). This suggests a possible involvement of other tyrosine kinases in the process.
3.2. MAP kinases: well-mapped pathways
Several studies have now identified the involvement of all three mitogen-activated protein (MAP) kinase (i.e., ERK1/2, p38 and JNK) pathways in the regulation of iNOS. Most of these studies have utilized either inhibitors of MAP-kinases or their dominant-negative forms. Taken together, they have revealed distinct as well as overlapping role of all three MAP-kinases in astroglial iNOS machinery in response to a wide array of stimuli (Fig. 1B). In rat astrocytes, the combined treatment of LPS and IFN-γ induces p38 and ERK1/2 pathways (Bhat et al., 1998). As a matter of fact, p38 and/or MEK-ERK1/2 pathways resemble the heart of iNOS signaling in astrocytes. Treatment with a combined cytokine regimen of TNF-α and IL-1β also utilizes the MEK-ERK pathway (Marcus et al., 2003). In addition to cytokines, the same pathway has been shown to play a major role in a model of iNOS up-regulation by glucose deprivation (Yoo et al., 2005) and HIV-1 Tat (Liu et al., 2002). Similarly, p38 is required by rat astrocytes for iNOS expression in response to stimuli like Taxol (Cvetkovic et al., 2004) and activation of TGF-β-activated-kinase (TAK) (Bhat et al., 2003). Also, p38 MAP kinase pathway is a major mediator of cytokine-induced-iNOS as seen in human astrocytes in response to IL-1β and IFN-γ (our unpublished observation), IL-1β (Hua et al., 2002), and dsRNA (Auch et al., 2004). In fact, the combined treatment of TNF-α and IL-1α involves p38 but not MEK-ERK for the expression of iNOS in mouse astrocytes (Da Silva et al., 1997). Conversely, we have observed that HIV-1 Tat induces iNOS in human astroglial cells in a (MEK-ERK)- but not p38-sensitive pathway (Liu et al., 2002). In essence, these kinase cascades phosphorylate downstream signal mediators thereby enabling them to activate transcription factors. However, MAPKs may play certain non-canonical roles as well in inducing astroglial iNOS. For example, p38 has been shown to phosphorylate Ser10 of Histone3 in promoter region of pro-inflammatory genes (Saccani et al., 2002). Such phosphorylation is postulated to be the epigenetic signature for facilitated docking of pro-inflammatory transcription factors like NF-κB. Although we lack experimental evidence, we also lack reasons not to believe a similar role of p38 in astrocytes in the context of iNOS expression.
More often than not, p38 MAPkinase pathway functions in parallel with JNK pathway. In rat astrocytes, dual activity of JNK and p38 has been observed in several cases (Cvetkovic et al., 2004; Bhat et al., 2003). In human astrocytes, similar trend is also reported for iNOS induction by IL-1β (Hua et al., 2002). However, when co-administered with IFN-γ the combination (IL-1β + IFN-γ), triggers iNOS in a p38-sensitive (our unpublished observation) but JNK-insensitive way (Jana et al., 2005). Taken together, this mélange of diverse observations indicates that signal transduction for astroglial expression of iNOS may involve all three MAP-kinase depending mainly upon the inducing stimuli (Fig. 1B).
3.3. A picky kinase: regulation by PKR
Glial cells produce iNOS when treated with viral dsRNA or its synthetic mimick, the polyinosinic-polycytidylic acid (Poly-IC) (Auch et al., 2004; Scumpia et al., 2005). This signaling involves a unique 68-kDa serine-threonine kinase called the dsRNA-dependent protein kinase (PKR), which cans autophosphorylate itself by dimerization (Fig. 1C). Involvement of PKR in induction of astroglial iNOS in response to poly-IC is proved by attenuation of iNOS expression in cells expressing either ΔPKR (a dominant negative mutant of PKR) or K3L (the Vaccinia viral inhibitor of PKR) (Auch et al., 2004). Several studies in various cell-types have shown that PKR can activate transcription factor NF-κB. Indeed, in human astrocytes, both ΔPKR and pMT-K3L inhibited dsRNA-induced iNOS expression by interfering with activity of NF-κB (Auch et al., 2004). Similar observations have also been recorded when PKR activity was blocked with 2-aminopurine (Scumpia et al., 2005).
3.4. Indulging in ROS: regulation by reactive oxygen species
ROS are multi-potent diffusible molecules capable of carrying out several signal transduction processes in response to several extracellular stimuli. Consistent with their versatile cellular functions, ROS have been also shown to regulate the iNOS expression in different cells including astroglial cells. Antioxidants, like N-acetyl cysteine (NAC), pyrrolidine dithiocarbamate (PDTC) and lycopene are potent inhibitors of production of NO and the expression of iNOS in rat primary astrocytes and C6 glial cells (Pahan et al., 1998c; Pannu et al., 2004), thereby proclaiming a role of ROS in iNOS induction.
Recent studies have identified the ROS-producing molecule in activated glial cells. Pawate et al. (2004) have defined a critical role of NADPH oxidase in (LPS + IFN-γ)-induced expression of iNOS in glial cells. They have shown that stimulation of rat primary astrocytes leads to rapid activation of NADPH oxidase and release of ROS followed by expression of iNOS. Consistently, attenuated expression of iNOS is observed in primary astrocytes derived from gp91Phox-deficient mice (Pawate et al., 2004). Furthermore, inhibition of (LPS + IFN-γ)-induced expression of iNOS and production of NO by exogenous addition of catalase but not superoxide dismutase suggests that H2O2 but not O2− is actually required for astroglial expression of iNOS. Because activation of NF-κB is involved in the expression of iNOS and antioxidants (like PDTC and NAC) suppress the activation of NF-κB in glial cells (Pahan et al., 1998c; Marcus et al., 2003), ROS are believed to regulate iNOS expression via NF-κB. However, the involvement of other transcription factors in ROS-mediated induction of iNOS in glial cells cannot be ruled out as many such factors are redox-sensitive in other cell-types.
3.5. Greasy roads: lipid pathways
3.5.1. Ceramide: Jekyll and Hyde
Ceramide is a major cellular sphingolipid with sphingosine as its basic structure. Several extracellular stimuli have been shown to couple the sphingomyelin-ceramide pathway that eventually leads to important biochemical and cellular responses including, growth arrest and apoptosis (55, 74). This pathway is initiated by the activation of neutral sphingomyelinase (NSMase) that hydrolyzes membrane sphingomyelin to ceramide and phosphocholine. Does ceramide play any role in induction of iNOS in astrocytes? In a study attempted to provide answer to this question, it was found that although sphingomyelinase (SMase) increases the cellular levels of ceramide, either SMase or cell-permeable ceramide analog alone was unable to induce iNOS in rat primary astrocytes (Pahan et al., 1998a). However, incubation of LPS or cytokine-stimulated rat astrocytes with SMase, cell-permeable ceramide analogs (C2- or C6-ceramide) or inhibitor of ceramidase (N-oleoyl ethanolamine) led to a time- and dose-dependent increase in the expression of iNOS in rat astrocytes and C6 glial cells (Pahan et al., 1998a). Thus, although being an ineffective iNOS inducer by itself, this second messenger takes on a different role of augmenting LPS- or cytokine-induced signals. This shift in functionality of ceramide is dependent on potentiation of NF-κB (Pahan et al., 1998a). Furthermore, inhibition of iNOS expression in (LPS + ceramide)-activated rat astrocytes by antioxidants and inhibitors of farnesyltransferase and MEK suggests possible involvement of redox-sensitive Ras-ERK pathway in the activation of NF-κB and the expression of iNOS (Pahan et al., 1998a). Consistently, a recent study from the Singh laboratory in South Carolina (Pannu et al., 2004) has identified lactosylceramide (LacCer) as an important mediator of expression of iNOS in (LPS + IFN-γ)-activated primary rat astrocytes. This study provides evidence that LPS/IFN-γ induces the production of LacCer that in turn activates Ras-ERK pathways via redox-sensitive pathway. This Ras-ERK pathway couples to the activation of NF-κB thereby inducing iNOS in astrocytes.
3.5.2. Isoprenoids and iNOS: regulation by mevalonate pathway
Isoprenoids are a major class of non-saponifiable lipids which is polymeric derivative of C5 compound isopentenylpyr-ophosphate (IPP). Isoprenoids like farnesyl- and geranylgeranyl-pyrophosphate, are biosynthesized in animals from acetyl CoA via the mevalonate pathway (Goldstein and Brown, 1990), as is depicted in Fig. 1D. These isoprenoids covalently modify and thus modulate the biological activity of important signal transduction components like small G-proteins Ras, Rac and Rho (Maltese, 1990). Upon isoprenylation, these G proteins become membrane-bound and transduce several intracellular signaling pathways that lead to iNOS expression. The experimental evidence for this statement has been provided by a pioneering study by Pahan et al. (1997b), where lovastatin, a blocker of mevalonate pathway (Fig. 1D) and also a FDA-approved drug for hypercholesterolemia, inhibits the activation of NF-κB and the expression of iNOS in LPS-stimulated rat primary astrocytes. Furthermore, similar to lovastatin, sodium phenylacetate (NaPA) also inhibits the activation of NF-κB and the expression of iNOS in glial cells (Pahan et al., 1997b). As lovastatin inhibits HMG-CoA reductase, exogenous mevalonate and farnesyl pyrophosphate (FPP) are capable of reversing the inhibitory effect of lovastatin on iNOS expression and NF-κB activation. On the contrary, NaPA inhibits mevalonate pyrophosphate decarboxylase downstream of mevalonate synthesis, therefore FPP but not mevalonate, blocks the inhibitory effect of NaPA (Fig. 1D). However, addition of ubiquinone and cholesterol to rat astrocytes fails to reverse the inhibitory effect of lovastatin and NaPA. These results suggest that depletion of farnesyl pyrophosphate, rather than end products of mevalonate pathway, is responsible for the observed inhibitory effect of lovastatin and NaPA on the expression of iNOS (Fig. 1D).
Suppression of LPS-induced activation of NF-κB and expression of iNOS in rat astrocytes by farnesyltransferase inhibitors (Pahan et al., 1997b) suggests an important role of farnesylation reaction in the regulation of iNOS gene. Consistent to a role of farnesylation in the activation of p21Ras, a dominant-negative mutant of p21Ras (S17N) also attenuated activation of NF-κB and expression of iNOS in rat and human primary astrocytes (Pahan et al., 1997b). Taken together, there is evidence to suggest that mevalonate metabolites regulate the expression of iNOS in astrocytes via modulating isoprenylation of small G-proteins like Ras.
4. Signals that negatively regulate iNOS expression
4.1. The camp in glial iNOS regulation: unique regulation by cAMP
Cyclic AMP (cAMP), one of the most important cellular second messengers, has been shown to regulate the induction of iNOS in various cell-types. Such as, cAMP induces the expression of iNOS in LPS- and cytokine-stimulated glomerular mesangial cells (Muhl et al., 1994), vascular smooth muscle cells (Koide et al., 1993), cardiac myocytes (Ikeda et al., 1996), murine 3T3 fibroblasts (Kleinert et al., 1996) and rat peritoneal macrophages (Sowa and Przewlocki, 1994). In these studies, the increase in cAMP level resulting due to inhibition of phosphodiesterase (an enzyme that degrades cAMP), or due to an exogenous input of cAMP derivatives or compounds that enhance intracellular levels of cAMP, induce and/or stimulate iNOS expression. However, the script is altered when it comes to astrocytes. Surprisingly, cAMP has been found to inhibit the expression of iNOS and the production of NO in LPS- or cytokine-stimulated rat primary astrocytes or C6 glial cells (Pahan et al., 1997a). Compounds that increase cAMP and activate protein kinase A (PKA) [forskolin, 8-bromo-cAMP, and (Sp)-cAMP] were found to inhibit LPS- and cytokine-mediated production of NO as well as the expression of iNOS, whereas compounds that decrease cAMP and PKA activity [H-89 and (Rp)-cAMP] stimulated the production of NO and the expression of iNOS in rat primary astrocytes (Pahan et al., 1997a).
How does cAMP mediate its signal? Treatment of rat astrocytes with cAMP synthesis activator Forskolin (FSK) leads to activation of PKA and subsequent inactivation of transcription factor NF-κB (Pahan et al., 1997a) (Fig. 2, right panel). This is in sharp contrast with macrophages, where FSK activates PKA to activate NF-κB (Pahan et al., 1997a). Furthermore, one recent study with rat primary astrocytes and C6 glial cells shows that excess cAMP, generated by treating cells with FSK, inhibits iNOS expression by interfering with activation of p38 MAP kinase (Won et al., 2004). FSK inhibits LPS/interferon-gamma (IFN)-induced iNOS transcription by blocking the p38-MAPK/ATF-2 pathway in glial cells (Fig. 2, right panel). Apart from ATF-2, p38 is also involved in the activation of AP-1, C/EBPβ, and NF-κB, the transcription factors that positively regulate the transcription of iNOS gene in astrocytes. Therefore, cAMP is expected to inhibit astroglial expression of iNOS via down-regulation of p38-coupled proinflammatory transcription factors (Bhat et al., 2002). In addition, the role of cAMP-response element binding protein (CREB) in cAMP-mediated down-regulation of astroglial iNOS cannot be over-ruled. Because cAMP is capable of activating CREB via PKA-mediated phosphorylation and the promoter of iNOS contains CRE, it is possible that cAMP-PKA may down-regulate the expression of iNOS via CREB (Bhat et al., 2002). Therefore, it will be interesting to explore the role of CREB, if any, in expression of astroglial iNOS.
Fig. 2.
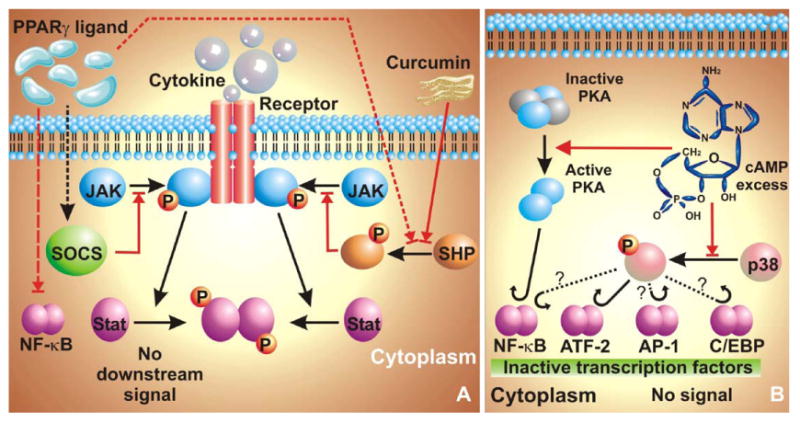
Blocks of iNOS-blocking signaling pathways. The central idea of blocking iNOS transactivation involves interfering with activation of transcription factors. (A) Blocking of JAK-STAT pathway. SHP-1 and SHP-2 can block recruitment of JAK to the receptor tyrosine kinase and thus prevent activation of the JAK-STAT pathway. This scheme is utilized by Curcumin {diferuloylmethane,1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione} and PPARγ ligands, as they can activate SHP. PPARγ ligands can also upregulate SOCS expression, which subsequently blocks recruitment of JAK to the RTK. PPARγ ligands furthermore, block activation of NF-κB. (B) Dampening by cAMP. Presence of excess cAMP in astrocytes blocks activation of p38-MAPK thereby attenuating signals downstream of it. It also binds with PKA and subsequently activates it, which then somehow blocks activation of NF-κB.
4.2. Legend of ligands (of nuclear hormone receptors)
Ligands of nuclear receptors (NRL) recruit coactivators to the DNA-bound NR thus facilitating transactivation of target genes. Quite interestingly, this script is not followed during regulation of iNOS. Several observations have now confirmed that NRLs repress iNOS transcription independent of nuclear receptor itself. For example, gemfibrozil, a well-known ligand for peroxisome proliferator-activated receptor-alpha (PPAR-α), inhibits cytokine-induced iNOS expression in human astrocytes independent of PPAR-α (Pahan et al., 2002). Gemfibrozil induces peroxisome proliferator-responsive element (PPRE)-dependent luciferase activity, which is inhibited by the expression of ΔPPAR-α, the dominant-negative mutant of human PPAR-α. However, ΔPPAR-α does not reverse gemfibrozil-mediated inhibition of iNOS suggesting that gemfibrozil inhibits iNOS independent of PPAR-α. Similarly, 15-deoxy-12, 14-PGJ2 (15d-PGJ2), a ligand for PPAR-γ, attenuates (LPS + IFN-γ)-induced expression of iNOS in rat primary astrocytes independent of the PPAR-γ itself (Giri et al., 2004). Recently, ligands for other NR such as RAR and RXR have been also shown to suppress the expression of iNOS independent of NR in macrophages (Saura et al., 1999).
How can iNOS repression be contingent on NRLs but not on NRs? Although details are yet not well-understood, yet we have few scattered pieces to generate a broad based answer. Gemfibrozil, the PPAR-αligand, strongly inhibited (IL-1β + IFN-γ)-induced activation of NF-κB, AP-1, and C/EBPβ but not that of STAT-GAS in human astroglial cells (Pahan et al., 2002). As a matter of fact, 15d-PGJ2, the PPAR-γ ligand, inhibits the NF-κB pathway at multiple points (Giri et al., 2004). Blocking of NF-κB and other TFs is an effective way of down-regulating signaling in a short period of time. Additionally, few other mechanisms have also been offered to explain the blocking effect of NRLs on pro-inflammatory TFs. As revealed in Fig. 2 (left panel), PPAR-γ ligands 15d-PGJ2 and rosiglitazone, reduce phosphorylation of JAK-STAT pathway in activated rat astroglia and microglia thereby leading to the suppression of JAK-STAT-dependent inflammatory responses (Park et al., 2003). This blockage is not contingent on PPAR-γ SHP-2 is also involved in the anti-inflammatory action of NRLs. NRL treatment was shown to phosphorylate SHP2 within minutes (Park et al., 2003). As phosphorylated SHP2 dephosphorylates JAK, this creates yet another avenue of blocking the JAK-STAT pathway (Fig. 2, left panel).
4.3. Shock JAcKs: saga of SOCS
In order to counter pro-inflammatory signaling pathways of various cytokines, cells employ a family of proteins called suppressors of cytokine signaling (SOCS). Due to their ability to regulate and subdue a pro-inflammatory signal, these proteins are now considered important regulators of normal immune physiology and immune disease (Leroith and Nissley, 2005). In general, SOCS are present in cells at very low levels. However, SOCS are rapidly transcribed upon exposure of cells to certain stimuli. Subsequently several studies indicate that SOCS can negatively regulate the response of immune cells either by inhibiting the activity of JAK or by competing with signaling molecules for binding to the phosphorylated receptor (Alexander and Hilton, 2004). According to Park et al. (2003), 15d-PGJ2 and rosiglitazone, activators of PPAR-γ, induce the transcription of SOCS1 and SOCS3 to inhibit the activity of JAK1 and JAK2 in rat primary astrocytes (Fig. 2, left panel). Both SOCS1 and SOCS3 are capable of binding JAKs to suppress their tyrosine kinase activity. Therefore, 15d-PGJ2 and rosiglitazone reduce the phosphorylation of STAT1 and STAT3 and attenuate the iNOS inducing signal in activated glial cells. These results suggest that up-regulation of SOCS may represent a critical step for suppressing expression of iNOS in glial cells via negative regulation of the JAK-STAT pathway.
4.4. Splitting phosphoric acid Mon(oe)sters: the case of phosphatases
4.4.1. Regulation by phosphoserine/threonine protein phosphatase
Phosphoserine threonine protein phosphatases (PP1, PP2A, PP2B, and PP2C) are specific dephosphorylating agents of phosphoserine and phosphothreonine residues. Since iNOS upregulating signals involve phosphorylation by serine/threonine kinases, it is quite logical to assume that signals downregulating iNOS may involve serine/threonine protein phosphatases. The assumption was put to test in rat primary astrocytes, where compounds that inhibit PP 1 and PP 2A (calyculin A, microcystin, okadaic acid, and cantharidin) were utilized to investigate any alteration in iNOS regulation (Pahan et al., 1998b). Treatment with these compounds was found to stimulate the LPS- and cytokine-mediated expression of iNOS and production of NO in rat primary astrocytes and C6 glial cells suggesting that protein phosphatases are indeed involved in negative iNOS regulation in astrocytes (Pahan et al., 1998b). Because the activation of NF-κB is necessary for the induction of iNOS, the effect of okadaic acid on the LPS-mediated activation of NF-κB was tested in rat primary astrocytes. Interestingly, okadaic acid stimulated the LPS-mediated DNA binding as well as transcriptional activity of NF-κB suggesting that the stimulation of iNOS expression in astrocytes by inhibitors of PP1/2A is possibly due to an enhanced activity of NF-κB.
4.4.2. Regulation by phosphotyrosine protein phosphatase
Consistent to iNOS regulation by tyrosine kinases (see above), it has been proposed that tyrosine phosphatase(s) also play an important role in controlling the expression of iNOS. SH2 domain-containing tyrosine phosphatases (SHP)-1 and SHP-2 have two SH2 domains at the N terminus and a phosphatase catalytic domain at the C terminus capable of dephosphorylating tyrosine phosphorylated JAKs, receptors or other cellular proteins (Qu, 2002; Zhang et al., 2000) (Fig. 2, left panel). Therefore, SHP-1 and SHP-2 play important roles in the control of proinflammatory cytokine signaling. A study, showing greater iNOS expression in SHP-1 deficient astrocytes, supports the suggested role of SHPs in regulating iNOS (90). Furthermore, SHP-2 has also been proposed to be involved in anti-inflammatory role of PPARγ ligands 15d-PGJ2 and rosiglitazone (Park et al., 2003). Interestingly, this proposal comes in the light of current understanding whereby SHP-2 and SHP-1 are believed to mediate an opposite effect on cell activation (Poole and Jones, 2005).
4.5. Containing iNOS: regulation by phospho(iNOS)itol-3kinase
Phosphoinositol-3kinase (PI-3K) is a lipid kinase that phosphorylates 3′-OH position of the inositol ring of inositol phospholipids, producing PtdIns(3)P, PtdIns(3,4)P2, PtdIns(3,4,5)P3. The prototypical class-I PI-3K comprises a p110 catalytic subunit and a p85 regulatory subunit (three mammalian subtypes; α, β, and γ). Although this kinase activity is known to initiate several positive cellular responses (for more information, see Wymann and Marone, 2005), inhibition of PI-3K pathway induces/stimulates LPS or cytokine-induced iNOS expression in glial cells. Treatment of C6 glial cells and rat primary astrocytes with specific inhibitors of PI-3K (Wortmannin and LY294002) encourages expression of iNOS in response to LPS- or cytokine-stimulation (Pahan et al., 1999). Similar effects are seen by over-expression of dominant-negative mutant of adapter p85α in these cells (Pahan et al., 1999). Alternatively, expression of a catalytically active p110 subunit of PI 3-kinase but not that of a kinase-deficient mutant of p110 induced an increase in PI 3-kinase activity and inhibits cytokine-induced production of NO and expression of iNOS (Pahan et al., 2000). Although, the exact mechanism of PI-3K-mediated containment of iNOS expression is yet elusive, it is however known that these inhibitory effects are not dependent on MAP-kinase pathways or activation of NF-κB (Pahan et al., 1999, 2000).
5. In the nux: nuclear events
Basic iNOS regulatory events in the nucleus are greatly juxtaposed to the promoter of iNOS, where a number of signal-recruited transcription factors (TFs) converge in concert with co-regulatory elements to initiate transcription of this gene. This simplification however, overly understates the complexity of iNOS promoter regulation. The iNOS promoter has been found to possess extensive species-specific properties. Here are the facts. While a 1.7-kb 5′-UTR stretch of mouse iNOS gene exhibits full inducibility to a mixture of LPS and IFN-γ (Xie et al., 1993), a 3.2-kb 5′-UTR stretch is required for full inducibility of iNOS in rats (Zhang et al., 1998). In human, a 7.2-kb segment confers partial inducibility of iNOS in response to cytokines (Taylor et al., 1998). For higher level of activation of iNOS in human, a promoter segment of 8.3 or 16 kb is required (Kristof et al., 2001; Taylor and Geller, 2000). We, in our laboratory, have observed differential activation of a 7.2 and 8.8-kb human iNOS promoter in astrocytes. Treatment of U373MG astroglial cells or primary astroglia with IL-1β or cytokine combinations involving IL-1β as a component, led to 3- to 4-fold increase in 7.2-kb promoter induced luciferase reporter activity (Pahan et al., 2002), while a 5.5- to 8.2-fold activation was achieved in case of 8.8-kb segment construct (Jana et al., 2005).
If we are to think ‘linearly’, are we to conclude from facts presented above that transcription regulatory events of iNOS are spread across 8-kb or more of the genomic highway? While the physical span of the entire event is hard to predict, it has been however, experimentally proved that the iNOS promoter bends physically during iNOS activation by TNF-α and IFN-γ in RAW264.7 cells (Saura et al., 1999). Such a ‘loop’ formed due to bending of the DNA may help compose a spatially well-organized transcription initiation complex (Model in Fig. 1). As proposed by Saura et al. (1999), the bending of DNA is contingent on interaction of two TFs, NF-κB and IRF-1. Simultaneous binding to their consensus sequences far apart on the DNA stretch and the subsequent interaction of these TFs among themselves probably helps arc the DNA.
Speaking of TFs, several pro-inflammatory TFs in addition to NF-κB and IRF-1 have been recorded to be essential in mediating iNOS expression signals. Among them C/EBPβ, C/EBPδ, STAT-1α, and AP-1 are important transcription factors. These TFs are engaged with various degree of involvement in a context dependent fashion. For example, when astrocytes are treated with IL-1β only, AP-1 acts downstream of JNK and is essential for iNOS up-regulation. This is proved by attenuation of AP-1 reporter activity and iNOS expression upon pretreatment of cells with JNK inhibitor (Jana et al., 2005). However, the JNK inhibitor has no effect on iNOS expression in response to a combined treatment of cells with (IL-1β + IFNγ). This corroborates well with lack of AP-1 driven luciferase reporter activity upon (IL-1β + IFNγ) challenge (Jana et al., 2005) suggesting lack of AP-1 participation in the process. This suggests that AP-1 recruitment is contingent on stimuli. At this point, it must be emphasized that the exact role of AP-1 in iNOS regulation is elusive. For a detailed discussion on controversial literature in this regard, the review by Kleinert et al. (2004) is hereby recommended. In further studies to illuminate role of various TFs, we found that several pro-inflammatory TFs mediate iNOS regulatory signals induced by various pro-inflammatory cytokines with various degree of contingency. Fig. 3 summarizes our finding in this regard.
Fig. 3.
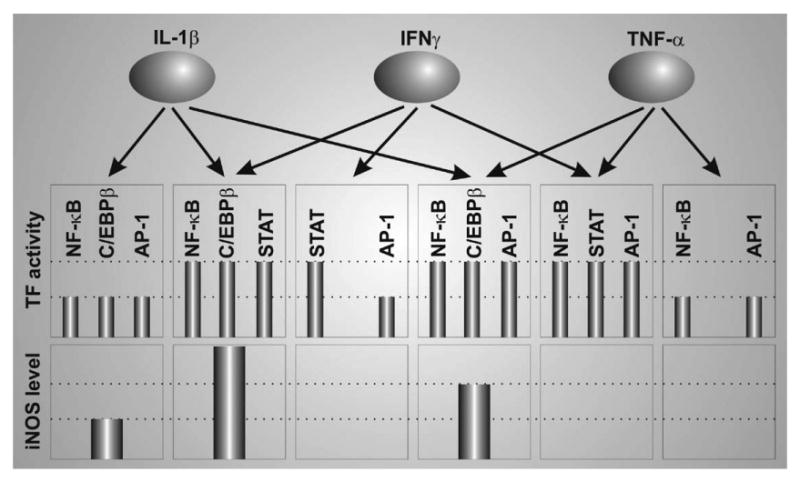
A possible model depicting various degree of dependency of human promoter iNOS on different transcription factors. The graphs are arbitrary and are solely meant to provide a comparative scale to represent the activity of various transcription factors and responsive iNOS expression after treatment with indicated cytokines alone or in combination. (Adapted from Jana et al., 2005).
Furthermore, TFs are recruited by a wide array of upstream signaling pathways, again, in a context-dependent dictum. Here are few examples. As stated already, AP-1 acts downstream of the JNK pathway (Jana et al., 2005) in response to IL-1β treatment. In response to the same treatment, AP-1 DNA binding activity is also abolished when cells are pre-treated with SB203580, the inhibitor of p38-MAPK (Hua et al., 2002). On the other hand, C/EBPβ is sensitive to inhibitors of the MEK-ERK1/2 pathway in response to HIV-Tat (Liu et al., 2002) while it is also regulated by p38-MAPK when conducting signals induced by dsRNA (Auch et al., 2004). During the dsRNA trigger, NF-κB is sensitive to inhibitors of protein kinase R but is independent of p38-MAPK regulation (Auch et al., 2004). As a matter of fact, inter-relationship of NF-κB and p38 in astrocytes is very intriguing. SB203580 does not inhibit DNA binding ability (as assessed by EMSA) of NF-κB in response to IL-1β or (IL-1β + IFNγ) treatment in human astrocytes (Hua et al., 2002, our unpublished observation). Despite similar treatment, blocking of p38-MAPK however, leads to transcriptional crippling of NF-κB as assessed by NF-κB driven luciferase reporter activity (our unpublished observation). Similar observations have also been reported in C6 glial cells (Bhat et al., 2002). Thus, p38-MAPK may regulate the transcriptional activity of a transcription factor without interfering with its DNA binding ability. The exact mechanism of such selective inhibition still remains mystic.
Transcriptional co-activators have also been suggested to be involved in astroglial iNOS expression. For example, Giri et al. (2004) have found that over-expression of p300 histone acetyltransferase (HAT) induces the expression of iNOS in rat astrocytes. However, this study does not indicate if p300 is required for LPS- and/or cytokine-mediated astroglial expression of iNOS. We have observed marked inhibition of IL-1β-and (IL-1β + IFN-γ)-induced expression of iNOS by Δp300, a dominant-negative mutant of p300, in human primary astrocytes (our unpublished observation). However, not enough research has been performed yet to negate the involvement of other HATs in the process.
6. Pondering on therapeutic opportunities: concluding thoughts
Inducible nitric oxide synthase in brain diseases serves as a double-edged sword. Too much or too little of this gaseous moiety is equally bad. As seen in EAE, the murine MS model, selective inhibition of iNOS in the CNS by intraventricular administration of antisense oligonucleotides blocks the disease process (Ding et al., 1998), suggesting that too much iNOS is one of the causal agents of the disease. In contrast, the induction of EAE leads to enhanced clinical symptoms of EAE and greater mortality rate in the iNOS (−/−) mice compared to wild type littermates (Fenyk-Melody et al., 1998) suggesting that some amount of iNOS may be beneficial in disease amelioration. Thus, partial knockdown of iNOS by pharmacological compounds to inhibit the excessive production of NO, but not complete knockout of the iNOS, is a feasible therapeutic approach for EAE. Ideally, direct application of calculated dose of non-toxic drugs capable of blocking these signaling pathways for glial expression of iNOS in treating NO-mediated neuroinflammatory and neurodegenerative diseases, seems a plausible idea. Indeed, iNOS blockers like statins and other mevalonate inhibitors, salicylates, non-steroidal anti-inflammatory drugs, ligands of PPAR, antioxidants, cAMP phosphodiesterase inhibitors, proteosome inhibitors, and natural compounds (like, Curcumin) are being considered for various neurodegenerative disorders. However, in addition to suppressing the expression of iNOS, these drugs also inhibit the expression of other inflammation-associated molecules. Thus, therapeutic efficacy of these drugs may not be necessarily due to the inhibition of iNOS.
Acknowledgments
This study was supported by grants from NIH (NS39940 and NS48923), National Multiple Sclerosis Society (RG3422A1/1) and Michael J. Fox Foundation for Parkinson Research.
References
- Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- Auch CJ, Saha RN, Sheikh FG, Liu X, Jacobs BL, Pahan K. Role of protein kinase R in double-stranded RNA-induced expression of nitric oxide synthase in human astroglia. FEBS Lett. 2004;563:223–228. doi: 10.1016/S0014-5793(04)00302-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat NR, Feinstein DL, Shen Q, Bhat AN. p38 MAPK-mediated transcriptional activation of inducible nitric-oxide synthase in glial cells. Roles of nuclear factors, nuclear factor kappa B, cAMP response element-binding protein, CCAAT/enhancer-binding protein-beta, and activating transcription factor-2. J Biol Chem. 2002;277:29584–29592. doi: 10.1074/jbc.M204994200. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Shen Q, Fan F. TAK1-mediated induction of nitric oxide synthase gene expression in glial cells. J Neurochem. 2003;87:238–247. doi: 10.1046/j.1471-4159.2003.01998.x. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco AM, Pascual M, Valles SL, Guerri C. Ethanol-induced iNOS and COX-2 expression in cultured astrocytes via NF-kappa B. Neuroreport. 2004;15:681–685. doi: 10.1097/00001756-200403220-00021. [DOI] [PubMed] [Google Scholar]
- Chen CC, Wang JK, Chen WC, Lin SB. Protein kinase C eta mediates lipopolysaccharide-induced nitric-oxide synthase expression in primary astrocytes. J Biol Chem. 1998;273:19424–19430. doi: 10.1074/jbc.273.31.19424. [DOI] [PubMed] [Google Scholar]
- Cvetkovic I, Miljkovic D, Vuckovic O, Harhaji L, Nikolic Z, Trajkovic V, Mostarica-Stojkovic M. Taxol activates inducible nitric oxide synthase in rat astrocytes: the role of MAP kinases and NF-kappaB. Cell Mol Life Sci. 2004;61:1167–1175. doi: 10.1007/s00018-004-3408-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva J, Pierrat B, Mary JL, Lesslauer W. Blockade of p38 mitogen-activated protein kinase pathway inhibits inducible nitric-oxide synthase expression in mouse astrocytes. J Biol Chem. 1997;272:28373–28380. doi: 10.1074/jbc.272.45.28373. [DOI] [PubMed] [Google Scholar]
- Dell’Albani P, Santangelo R, Torrisi L, Nicoletti VG, de Vellis J, Giuffrida-Stella AM. JAK/STAT signaling pathway mediates cytokine-induced iNOS expression in primary astroglial cell cultures. J Neurosci Res. 2001;65:417–424. doi: 10.1002/jnr.1169. [DOI] [PubMed] [Google Scholar]
- Ding M, Zhang M, Wong JL, Rogers NE, Ignarro LJ, Voskuhl RR. Antisense knockdown of inducible nitric oxide synthase inhibits induction of experimental autoimmune encephalomyelitis in SJL/J mice. J immunol. 1998;160:2560–2564. [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Dugas N, Lacroix C, Kilchherr E, Delfraissy JF, Tardieu M. Role of CD23 in astrocytes inflammatory reaction during HIV-1 related encephalitis. Cytokine. 2001;15:96–107. doi: 10.1006/cyto.2001.0896. [DOI] [PubMed] [Google Scholar]
- Feinstein DL, Galea E, Cermak J, Chugh P, Lyandvert L, Reis DJ. Nitric oxide synthase expression in glial cells: suppression by tyrosine kinase inhibitors. J Neurochem. 1994;62:811–814. doi: 10.1046/j.1471-4159.1994.62020811.x. [DOI] [PubMed] [Google Scholar]
- Fenyk-Melody JE, Garrison AE, Brunnert SR, Weidner JR, Shen F, Shelton BA, Mudgett JS. Experimental autoimmune encephalomyelitis is exacerbated in mice lacking the NOS2 gene. J immunol. 1998;160:2940–2946. [PubMed] [Google Scholar]
- Galea E, Reis DJ, Fox ES, Xu H, Feinstein DL. CD14 mediate endotoxin induction of nitric oxide synthase in cultured brain glial cells. J Neuroimmunol. 1996;64:19–28. doi: 10.1016/0165-5728(95)00143-3. [DOI] [PubMed] [Google Scholar]
- Gibson CL, Coughlan TC, Murphy SP. Glial nitric oxide and ischemia. Glia. 2005;50:417–426. doi: 10.1002/glia.20143. [DOI] [PubMed] [Google Scholar]
- Giri S, Rattan R, Singh AK, Singh I. The 15-deoxy-delta12, 14-prostaglandin J2 inhibits the inflammatory response in primary rat astrocytes via down-regulating multiple steps in Phosphatidylinositol 3-kinase-Akt-NF-kappaB-p300 pathway independent of peroxisome proliferator-activated receptor gamma. J Immunol. 2004;173:5196–5208. doi: 10.4049/jimmunol.173.8.5196. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Suzuki S, Nomura J, Ono A, Okuma Y, Akira S, Nomura Y. Bacterial DNA induced iNOS expression through MyD88-p38 MAP kinase in mouse primary cultured glial cells. Brain Res Mol Brain Res. 2004;124:159–164. doi: 10.1016/j.molbrainres.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Hua LL, Zhao ML, Cosenza M, Kim MO, Huang H, Tanowitz HB, Brosnan CF, Lee SC. Role of mitogen-activated protein kinases in inducible nitric oxide synthase and TNFalpha expression in human fetal astrocytes. J Neuroimmunol. 2002;126:180–189. doi: 10.1016/s0165-5728(02)00055-3. [DOI] [PubMed] [Google Scholar]
- Ikeda U, Yamamoto K, Ichida M, Ohkawa F, Murata M, Iimura O, Kusano E, Asano Y, Shimada K. Cyclic AMP augments cytokine-stimulated nitric oxide synthesis in rat cardiac myocytes. J Mol Cell Cardiol. 1996;28:789–795. doi: 10.1006/jmcc.1996.0073. [DOI] [PubMed] [Google Scholar]
- Jana M, Anderson JA, Saha RN, Liu X, Pahan K. Regulation of inducible nitric oxide synthase in proinflammatory cytokine-stimulated human primary astrocytes. Free Radic Biol Med. 2005;38:655–664. doi: 10.1016/j.freeradbiomed.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Takahashi H, Nomura Y, Taniguchi T. Possible involvement of Janus kinase JaK2 in interferon-gamma induction of nitric oxide synthase in rat glial cells. Eur J Pharmacol. 1996;306:297–306. doi: 10.1016/0014-2999(96)00212-9. [DOI] [PubMed] [Google Scholar]
- Kleinert H, Euchenhofer C, Ihrig-Biedert I, Forstermann U. In murine 3T3 fibroblasts, different second messenger pathways resulting in the induction of NO synthase II (iNOS) converge in the activation of transcription factor NF-kappaB. J Biol Chem. 1996;271:6039–6044. doi: 10.1074/jbc.271.11.6039. [DOI] [PubMed] [Google Scholar]
- Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. 2004;500:255–266. doi: 10.1016/j.ejphar.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Koide M, Kawahara Y, Nakayama I, Tsuda T, Yokoyama M. Cyclic AMP-elevating agents induce an inducible type of nitric oxide synthase in cultured vascular smooth muscle cells synergism with the induction elicited by inflammatory cytokines. J Biol Chem. 1993;268:24959–24966. [PubMed] [Google Scholar]
- Kong LY, McMillian MK, Maronpot R, Hong JS. Protein tyrosine kinase inhibitors suppress the production of nitric oxide in mixed glia, microglia-enriched or astrocyte-enriched cultures. Brain Res. 1996;729:102–109. [PubMed] [Google Scholar]
- Kristof AS, Marks-Konczalik J, Moss J. Mitogen-activated protein kinases mediate activator protein-1-dependent human inducible nitric-oxide synthase promoter activation. J Biol Chem. 2001;276:8445–8452. doi: 10.1074/jbc.M009563200. [DOI] [PubMed] [Google Scholar]
- Leroith D, Nissley P. Knock your SOCS off! J Clin Invest. 2005;115:233–236. doi: 10.1172/JCI24228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K. Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J Biol Chem. 2002;277:39312–39319. doi: 10.1074/jbc.M205107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltese WA. Post-translational modification of proteins by isoprenoids in mammalian cells. FASEB J. 1990;4:3319–3328. doi: 10.1096/fasebj.4.15.2123808. [DOI] [PubMed] [Google Scholar]
- Marcus JS, Karackattu SL, Fleegal MA, Sumners C. Cytokine-stimulated inducible nitric oxide synthase expression in astroglia: role of Erk mitogen-activated protein kinase and NF-kappaB. Glia. 2003;41:152–160. doi: 10.1002/glia.10168. [DOI] [PubMed] [Google Scholar]
- Muhl H, Kunz D, Pfeilschifter J. Expression of nitric oxide synthase in rat glomerular mesangial cells mediated by cyclic AMP. Br J Pharmacol. 1994;112:1–8. doi: 10.1111/j.1476-5381.1994.tb13019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Nanetti L, Vignini A, Moroni C, Pessina GP, Mazzanti L. LDL and HDL affect nitric oxide metabolism in human astrocytoma cells. Brain Res. 2004;1020:173–177. doi: 10.1016/j.brainres.2004.06.026. [DOI] [PubMed] [Google Scholar]
- Nishiya T, Uehara T, Nomura Y. Herbimycin A suppresses NF-kappa B activation and tyrosine phosphorylation of JAK2 and the subsequent induction of nitric oxide synthase in C6 glioma cells. FEBS Lett. 1995;371:333–336. doi: 10.1016/0014-5793(95)00933-z. [DOI] [PubMed] [Google Scholar]
- Pahan K, Jana M, Liu X, Taylor BS, Wood C, Fischer SM. Gemfibrozil, a lipid-lowering drug, inhibits the induction of nitric-oxide synthase in human astrocytes. J Biol Chem. 2002;277:45984–45991. doi: 10.1074/jbc.M200250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K, Liu X, Wood C, Raymond JR. Expression of a constitutively active form of phosphatidylinositol 3-kinase inhibits the induction of nitric oxide synthase in human astrocytes. FEBS Lett. 2000;472:203–207. doi: 10.1016/s0014-5793(00)01465-4. [DOI] [PubMed] [Google Scholar]
- Pahan K, Namboodiri AM, Sheikh FG, Smith BT, Singh I. Increasing cAMP attenuates induction of inducible nitric-oxide synthase in rat primary astrocytes. J Biol Chem. 1997a;272:7786–77891. doi: 10.1074/jbc.272.12.7786. [DOI] [PubMed] [Google Scholar]
- Pahan K, Raymond JR, Singh I. Inhibition of phosphatidylinositol 3-kinase induces nitric-oxide synthase in lipopolysaccharide- or cytokine-stimulated C6 glial cells. J Biol Chem. 1999;274:7528–7536. doi: 10.1074/jbc.274.11.7528. [DOI] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Khan M, Namboodiri AM, Singh I. Sphingomyelinase and ceramide stimulate the expression of inducible nitric-oxide synthase in rat primary astrocytes. J Biol Chem. 1998a;273:2591–2600. doi: 10.1074/jbc.273.5.2591. [DOI] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Namboodiri AM, Singh I. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J Clin Invest. 1997b;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Namboodiri AM, Singh I. Inhibitors of protein phosphatase 1 and 2A differentially regulate the expression of inducible nitric-oxide synthase in rat astrocytes and macrophages. J Biol Chem. 1998b;273:12219–12226. doi: 10.1074/jbc.273.20.12219. [DOI] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Namboodiri AM, Singh I. N-acetyl cysteine inhibits induction of NO production by endotoxin or cytokine stimulated rat peritoneal macrophages C6 glial cells and astrocytes. Free Radic Biol Med. 1998c;24:39–48. doi: 10.1016/s0891-5849(97)00137-8. [DOI] [PubMed] [Google Scholar]
- Pannu R, Won JS, Khan M, Singh AK, Singh I. A novel role of lactosylceramide in the regulation of lipopolysaccharide/interferon-gamma-mediated inducible nitric oxide synthase gene expression: implications for neuroinflammatory diseases. J Neurosci. 2004;24:5942–5954. doi: 10.1523/JNEUROSCI.1271-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EJ, Park SY, Joe EH, Jou I. 15d-PGJ2 and rosiglitazone suppress Janus kinase-STAT inflammatory signaling through induction of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 in glia. J Biol Chem. 2003;278:14747–14752. doi: 10.1074/jbc.M210819200. [DOI] [PubMed] [Google Scholar]
- Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res. 2004;77:540–551. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Poole AW, Jones ML. A SHPing tale: Perspectives on the regulation of SHP-1 and SHP-2 tyrosine phosphatases by the C-terminal tail. Cell Signal. 2005;17:1323–1332. doi: 10.1016/j.cellsig.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Qu CK. Role of the SHP-2 tyrosine phosphatase in cytokine-induced signaling and cellular response. Biochim Biophys Acta. 2002;1592:297–301. doi: 10.1016/s0167-4889(02)00322-1. [DOI] [PubMed] [Google Scholar]
- Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3:69–75. doi: 10.1038/ni748. [DOI] [PubMed] [Google Scholar]
- Saura M, Zaragoza C, Bao C, McMillan A, Lowenstein CJ. Interaction of interferon regulatory factor-1 and nuclear factor kappaB during activation of inducible nitric oxide synthase transcription. J Mol Biol. 1999;289:459–471. doi: 10.1006/jmbi.1999.2752. [DOI] [PubMed] [Google Scholar]
- Scumpia PO, Kelly KM, Reeves WH, Stevens BR. Double-stranded RNA signals antiviral and inflammatory programs and dysfunctional glutamate transport in TLR3-expressing astrocytes. Glia. 2005;52:153–162. doi: 10.1002/glia.20234. [DOI] [PubMed] [Google Scholar]
- Sowa G, Przewlocki R. cAMP analogues and cholera toxin stimulate the accumulation of nitrite in rat peritoneal macrophage cultures. Eur J Pharmacol. 1994;266:125–129. doi: 10.1016/0922-4106(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Taylor BS, de Vera ME, Ganster RW, Wang Q, Shapiro RA, Morris SM, Jr, Billiar TR, Geller DA. Multiple NF-kappaB enhancer elements regulate cytokine induction of the human inducible nitric oxide synthase gene. J Biol Chem. 1998;273:15148–15156. doi: 10.1074/jbc.273.24.15148. [DOI] [PubMed] [Google Scholar]
- Taylor BS, Geller DA. Molecular regulation of the human inducible nitric oxide synthase (iNOS) gene. Shock. 2000;13:413–424. doi: 10.1097/00024382-200006000-00001. [DOI] [PubMed] [Google Scholar]
- Won JS, Im YB, Singh AK, Singh I. Dual role of cAMP in iNOS expression in glial cells and macrophages is mediated by differential regulation of p38-MAPK/ATF-2 activation and iNOS stability. Free Radic Biol Med. 2004;37:1834–1844. doi: 10.1016/j.freeradbiomed.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Wymann MP, Marone R. Phosphoinositide 3-kinase in disease: timing, location, and scaffolding. Curr Opin Cell Biol. 2005;17:141–149. doi: 10.1016/j.ceb.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Xie QW, Whisnant R, Nathan C. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J Exp Med. 1993;177:1779–1784. doi: 10.1084/jem.177.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BK, Choi JW, Han BH, Kim WK, Kim HC, Ko KH. Role of MAPK/ERK1/2 in the glucose deprivation-induced death in immunostimulated astroglia. Neurosci Lett. 2005;376:171–176. doi: 10.1016/j.neulet.2004.11.077. [DOI] [PubMed] [Google Scholar]
- Zhang H, Chen X, Teng X, Snead C, Catravas JD. Molecular cloning and analysis of the rat inducible nitric oxide synthase gene promoter in aortic smooth muscle cells. Biochem Pharmacol. 1998;55:1873–1880. doi: 10.1016/s0006-2952(98)00078-1. [DOI] [PubMed] [Google Scholar]
- Zhang J, Somani AK, Siminovitch KA. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin immunol. 2000;12:361–378. doi: 10.1006/smim.2000.0223. [DOI] [PubMed] [Google Scholar]